Introduction
Liver cancer is the fifth most common cancer in men
and the seventh in women. Hepatocellular carcinoma (HCC), which
accounts for >85% of primary liver cancers, has a poor prognosis
with 5-year overall survival rates of <12% (1,2). Since
its poor prognosis has been reported to be closely associated with
HCC recurrence and metastasis, it is essential to determine the
possible underlying mechanisms which mediate tumor invasion and
metastasis. EMT has been demonstrated to be involved in the
progression of various cancers, including liver, prostate and
breast cancer (3–5), and functions as a main step toward
tumor metastasis. To date, increasing studies have been directed at
uncovering the possible signaling pathways in EMT of HCC.
IGF-1 has been demonstrated to be upregulated in
many different tumor cell lines compared with normal cells and
involved in tumorigenesis and progression, which is mediated
through the activation of multiple signal transduction pathways,
including the JNK, MAPK, PI3K/Akt pathways (6). IGF-1 was found to elevate the
expression of transmembrane glycoprotein MUC1 in MCF-7 cells for
the initiation of EMT in a PI3K/Akt signaling pathway-dependent
manner (7). Furthermore, it was
reported that IGF-1 could promote the growth and metastasis of HCC
cell lines via the upregulation of cathepsin B expression (8). As a member of the inhibitor of
apoptosis protein (IAP) family, survivin is overexpressed in some
tumor specimens, including HCC, while it is nearly negative in
normal tissue (9). In human sacral
chondrosarcoma, SDF-1/CXCR4 signaling could upregulate the
expression of survivin via the MEK/ERK and PI3K/AKT pathways,
leading to cell cycle and EMT occurrence (10). Overexpression of survivin in HCC
cells was revealed to suppress the ability of migration via
upregulation of glucose-regulated protein 78 (GRP78) and reduce the
EMT marker, vimentin (11).
Previous studies have revealed that high levels of survivin
exhibited anti-apoptotic and pro-metastatic potential in cancer
cell lines but not in normal cells (12).
Although both IGF-1 and survivin could mediate
metastasis in cancer cells, the mechanisms by which they
co-regulate metastasis have not been uncovered. In the present
study, we used various molecular and cellular methods to
investigate the existence and significance of the relationship
between the IGF-1 and survivin proteins. Our data elicited a new
mechanism in which IGF-1 induced EMT through regulation of survivin
and a downstream pathway, and this can be targeted to treat HCC
patients.
Materials and methods
Antibodies and reagents
Monoclonal rabbit antibodies against survivin
(1:5,000; cat. no. 2808), Akt (1:2,000; cat. no. 2920), p-Akt
(1:5,000; cat. no. 96115), Snail (1:500; cat. no. 3879), vimentin
(1:1,000; cat. no. 5741), E-cadherin (1:1,000; cat. no. 14472),
N-cadherin (1:1,000; cat. no. 13116) and GAPDH (1:5,000; cat. no.
5174) were obtained from Cell Signaling Technology, Inc. (Danvers,
MA, USA). IGF-1 was purchased from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). DAPI was purchased from Invitrogen; Thermo
Fisher Scientific, Inc. (Carlsbad, CA, USA).
Cell culture
Human HCC SMMC7721 cells were cultured in RPMI-1640
medium (Hyclone Laboratories; GE Healthcare, Chicago, IL, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), 100 U/ml penicillin and 100
µg/ml streptomycin at 37°C with 5% CO2.
Transfection of survivin siRNA
SMMC7721 cells were seeded in a 6-well plate and
adjusted to a density of 5×105 cells/well and incubated
at 37°C in a CO2 incubator until the cells reached
60–80% confluence. Survivin siRNA or control siRNA was provided by
Shanghai GenePharma Co., Ltd., (Shanghai, China). The survivin or
control siRNAs were subjected to Opti-MEM with Lipofectamine 2000
(Invitrogen) for transfection. Following a 6-h incubation, the
medium was replaced with fresh DMEM (10% FBS) and cells were
collected for further experiments after 72 h of culture.
Wound healing assay
HCC cell migration was examined by a wound-healing
assay. SMMC7721 cells were cultured to a confluent monolayer in a
6-well plate. A scratch (wound) was introduced in the confluent
cell layer using a pipette tip. The cells were washed three times
with PBS to remove detached cells. The cells were then incubated
with different doses of IGF-1 for 24 h, and images of a defined
wound spot were captured with a phase-contrast microscope (Olympus
Corp., Tokyo, Japan) at 0 and 24 h. The width of the wound was
assessed using ImageJ software (National Institutes of Health,
Bethesda, MD, USA). The distance of the wound was calculated as:
Distance of the wound = distance at 0 h - distance at 24 h.
Transwell filter cell migration and
invasion assays
Boyden chambers containing polycarbonate filters
with 8-µm pore size (Costar Group, Bodenheim, Germany) were
employed. Cells were seeded at a density of 5×105
cells/ml. To initiate the migration assay, cells (5×104)
in 10 µl of DMEM without FBS were added to the upper chamber, and
the lower chamber was filled with 600 µl of DMEM with 10% fetal
calf serum (FCS). For the invasion assay, Matrigel was introduced
in DMEM (4:3) in the inner chamber. IGF-1 was used as an inducer of
cell migration and the cells were allowed to migrate for 12 h at
37°C. Cells on the filter were first stained with crystal violet,
and the cells that remained on the upper surface of the filter were
removed using a cotton swab. The cells that had migrated onto the
lower surface of the filter were examined using a microscope
(Olympus Corp.) after mounting them onto a slide. A total of six
random fields (magnification, ×100) per filter were photographed.
Experiments were carried out in triplicate with consistent
results.
RNA extraction and quantitative
real-time PCR
RNA extraction was prepared using TRIzol reagent
(Takara Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer's instructions. Total RNA (1 µg) was used to
synthesize the first strand of cDNA using Bestar qPCR RT kit (DBI
Bioscience, Ludwigshafen, Germany). The mRNA expression was
evaluated by real-time qPCR on the Stratagene Mx3000P real- time
PCR platform (Agilent Technologies, Santa Clara, CA, USA) with
SYBR-Green PCR core reagents. The PCR reaction system contained 10
µl of Bestar® SYBR Green qPCR Master Mix, 1.0 µl of
forward and reverse primers (10 µM), 1 µl of cDNA template, and 8
µl of ddH2O. GAPDH was applied as a reference. The
following primers were synthesized and applied: survivin forward,
5′-TCAAGGACCACCGCATCT-3′ and reverse, 5′-CGCACTTTCTCCGCAGTT-3′;
Snail forward, 5′-TCCTTCGTCCTTCTCCTCTAC-3′ and reverse,
5′-TGTGGCTTCGGATGTGC-3′; E-cadherin forward,
5′-CCGATCTTCAATCCCACC-3′ and reverse, 5′-CCCACGCCAAAGTCCTC-3′;
vimentin forward, 5′-CGCCAGATGCGTGAAAT-3′ and reverse,
5′-CACGAAGGTGACGAGCC-3′; N-cadherin forward,
5′-GGATCAAAGCCTGGAACAT-3′ and reverse, 5′-CTTGGAGCCTGAGACACGA-3′;
GAPDH forward, 5′-TGTTCGTCATGGGTGTGAAC-3′ and reverse,
5′-TGTTCGTCATGGGTGTGAAC-3′. The reaction procedure was initiated
with denaturation at 94°C for 2 min and followed by 40 repeated
cycles (denaturation at 94°C for 20 sec, annealing at 58°C for 20
sec and extension at 72°C for 20 sec). The Ct-value for each sample
was calculated with the ΔΔCq-method, and the results were expressed
as 2−ΔΔCq to analyze the fold change (13).
Western blotting
HCC cells were lysed with lysis buffer containing
135 mM NaCl, 20 mM Tris (pH 7.5), 25 mM β-glycerophoshate, 2 mM
EDTA, 2 mM sodium pyrophosphate, 2 mM DTT, 10% glycerol, 1% Triton
X-100, 10 mM NaF, 1 mM sodium orthovanadate and 1 mM PMSF
supplemented with a complete protease inhibitor cocktail (Roche
Diagnostics GmbH, Mannheim, Germany) at 4°C. Lysates were
centrifuged (15,000 × g) at 4°C for 15 min. Equal amounts of the
soluble protein were denatured in SDS, resolved on 12%
SDS-polyacrylamide gel, and transferred onto polyvinylidene
fluoride (PVDF) membranes (EMD Millipore, Bedford, MA, USA), and
incubated with blocking buffer (5% non-fat dry milk in TBST)
overnight at 4°C. Immunoblotting was performed with a primary
antibody (1:1,000) followed by the appropriate horseradish
peroxidase (HRP)-conjugated goat anti-rabbit antibody (1:15,000;
Cell Signaling Technology, Inc., Danvers, MA, USA). Immunodetection
was performed with an enhanced chemiluminescence system (ECL;
Pierce Biotechnology, Rockford, IL, USA) using hydrogen peroxide
and luminol as a substrate.
Immunofluorescence staining
Briefly, after experimental treatment, SMMC7721
cells were incubated with a primary antibody at 4°C overnight.
After being washed three times with PBS, the cells were incubated
with Alexa Fluor 488-labeled secondary goat anti-rabbit antibody
(1:10,000; cat. no. 4412; Cell Signaling Technology, Inc.) for 1 h
at 37°C. The cells were then stained with DAPI to visualize cell
nuclei and observed under a confocal microscope (Nikon A1+; Nikon
Corp., Tokyo, Japan).
Statistical analysis
GraphPad Prism version 5.0 (GraphPad Software, Inc.,
La Jolla, CA, USA) was used to analyze the experimental data, all
the results were presented as the mean ± standard deviation (SD)
from three independent experiments. The statistical analysis was
performed using one-way analysis of variance (ANOVA), followed by
Dunnett's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
IGF-1 enhances the invasive and
migratory abilities of HCC cells
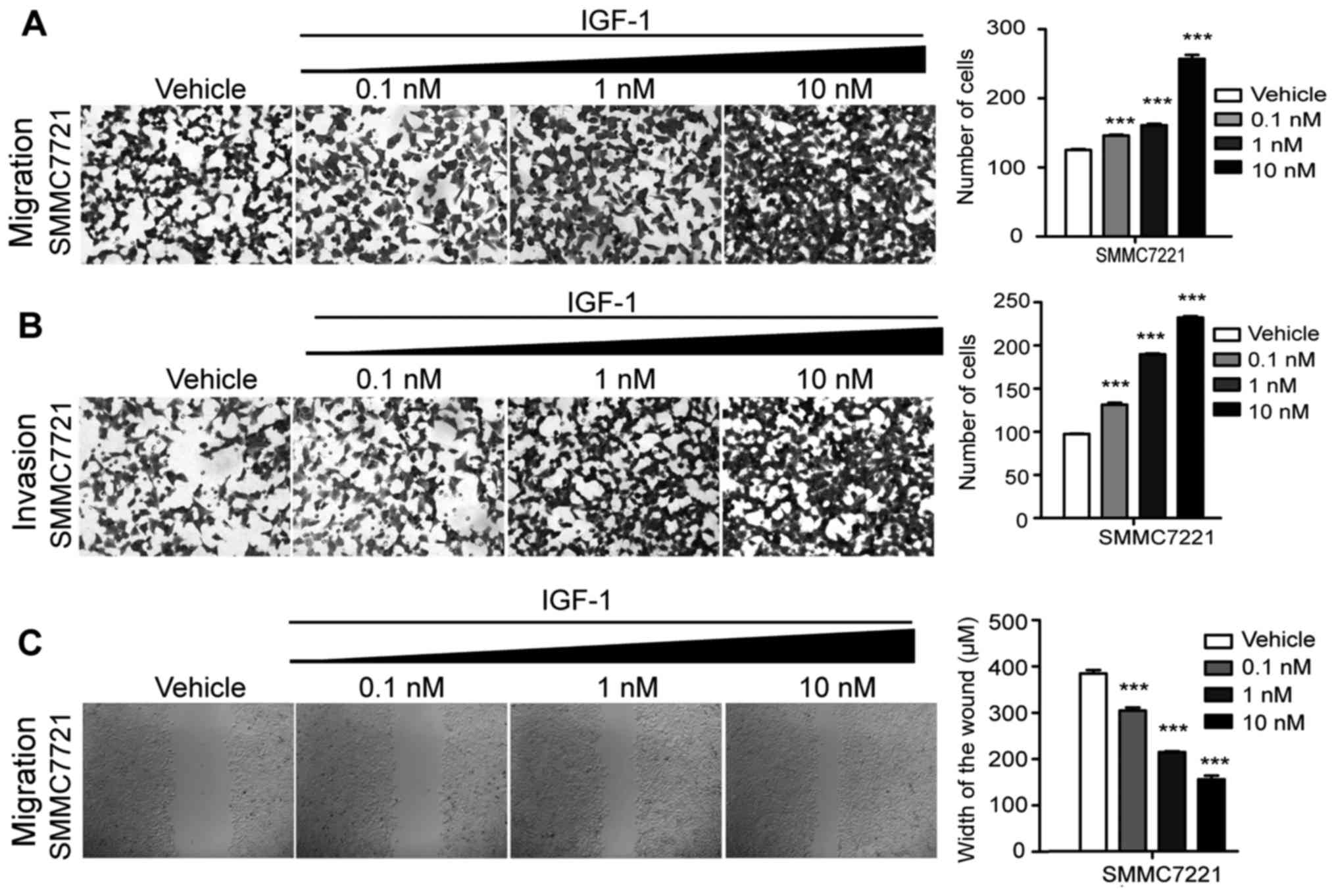
To determine the effect of IGF-1 on the invasive and
migratory abilities of HCC cells, different concentrations of
IGF-1, including 0.1, 1 and 10 nM, were added to perform the wound
healing assay and Transwell filter cell migration and invasion
assays (Fig. 1). Statistical
analysis revealed that in both the invasive and migratory assays
the number of cells were significantly increased with the
increasing doses of IGF-1, in comparison with the vehicle group
(Fig. 1A and B). In addition, the
distance of the wound healing was markedly decreased in the
IGF-1treatment groups in a dose-dependent manner (Fig. 1C). These results demonstrated that
cellular invasive and migratory abilities were highly promoted by
IGF-1, indicating that IGF-1 may act as an epigenetic activator of
invasion and migration of HCC cells.
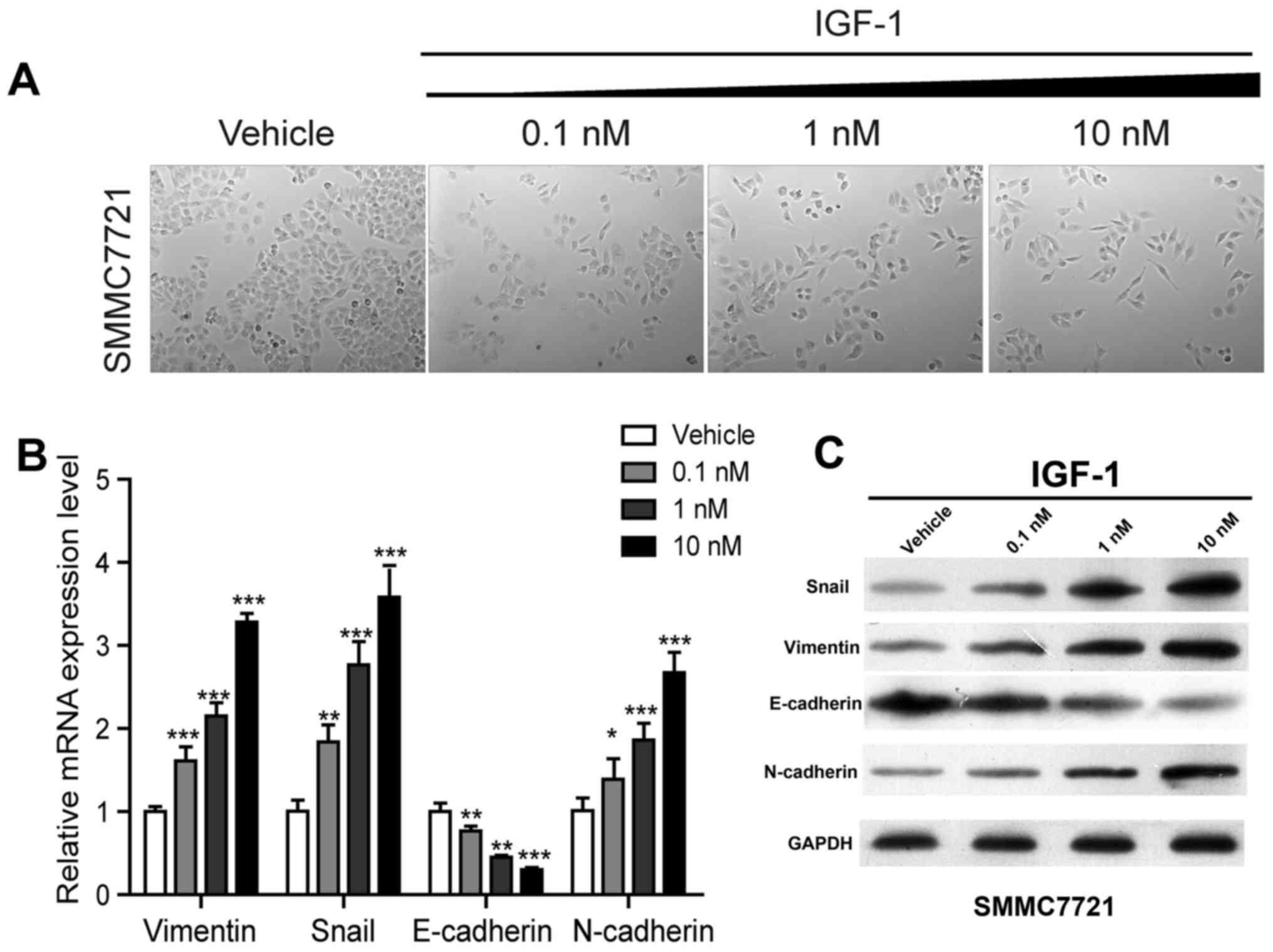
IGF-1 activates EMT in HCC cells
Losing cohesive ability and epithelial phenotype and
acquiring spindle or fiber-like shapes are classic morphological
changes in the process of EMT (14,15).
Similar morphological changes as in EMT were observed in HCC cells
after treatment with IGF-1 (Fig.
2A), hence it was speculated that IGF-1 enhances the invasive
and migratory potential via activation of the EMT-related pathway.
To confirm this hypothesis, we compared the mRNA and protein
expression patterns of EMT-associated genes of IGF-1-treated cells
to that of control cells at different concentrations. As shown in
Fig. 2B and C, the expression
levels of vimentin, Snail and N-cadherin were notably increased
while E-cadherin was significantly decreased at both transcription
levels and protein levels in a dose-dependent manner, indicating
that EMT was activated in SMMC7221 cells after treatment with
IGF-1.
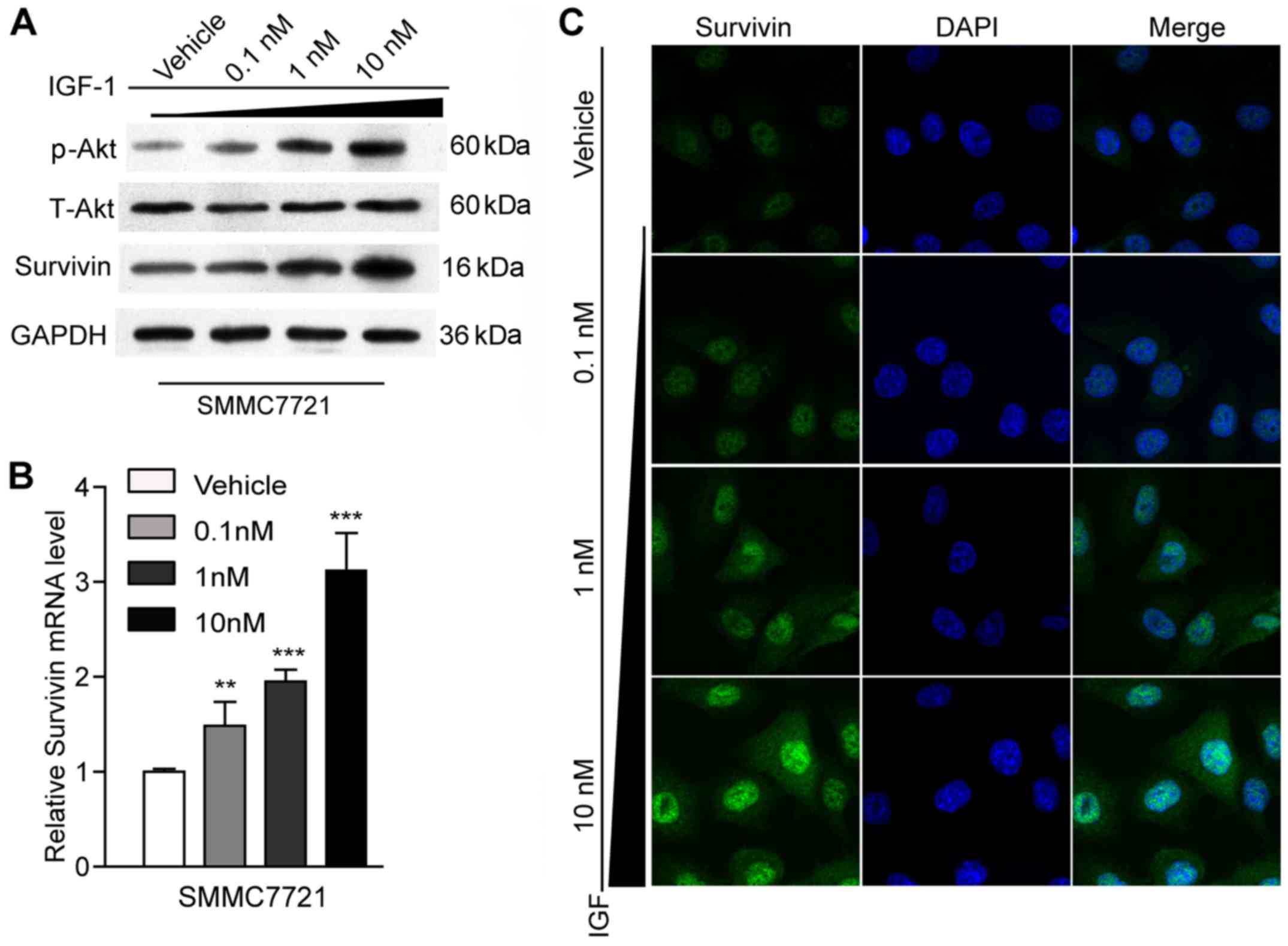
Effect of IGF-1 on survivin
expression
It was reported that survivin could be mediated by
IGF-1/mTOR signaling (4) and that
survivin participated in the activation of EMT in colon cancer
cells (16). It could be speculated
from these facts that IGF-1 may activate EMT by mediating survivin
expression. Thus, we assessed the ability of IGF-1 to affect the
expression level of survivin using RT-qPCR, western blot analysis
and immunofluorescence staining. With the increase in the
concentration of IGF-1 from 0.1 to 10 nM, the expression pattern of
survivin significantly increased at both the mRNA and protein
levels (P<0.01, P<0,001) (Fig. 3A
and B). A similar tendency was observed in the
immunofluorescence staining results (Fig. 3C). These results implied that
survivin may be involved in the IGF-1-induced EMT process.
In addition, it is well-known that IGF-1
participates in the activation of different signal transduction
pathways, including the PI3K/AKT/mTOR pathway (17,18).
In the present study, our western blot results also revealed that
AKT was activated and the p-AKT expression levels were
significantly elevated in the IGF-1-treated groups in comparison
with the vehicle group (P<0.05) (Fig. 3A).
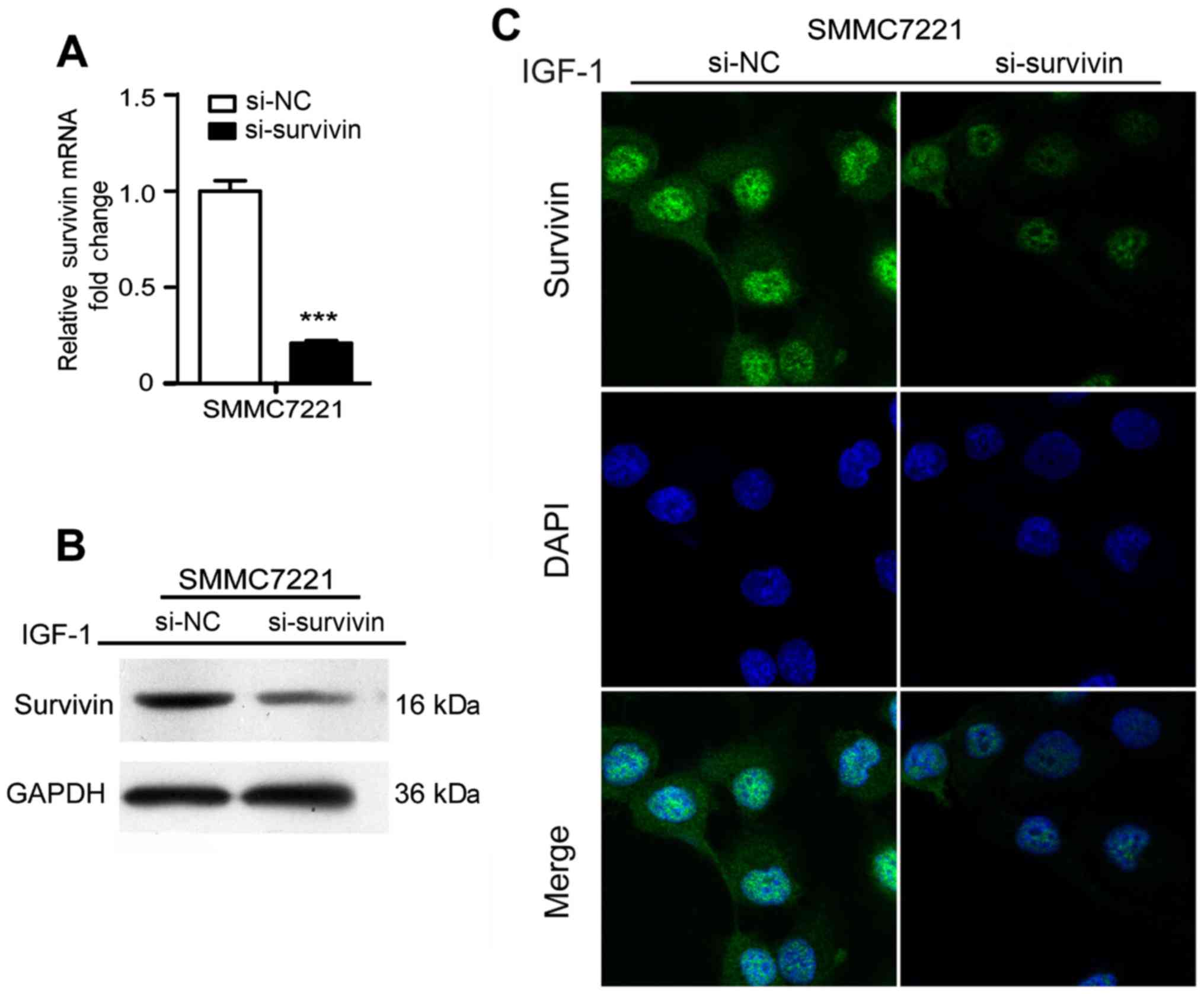
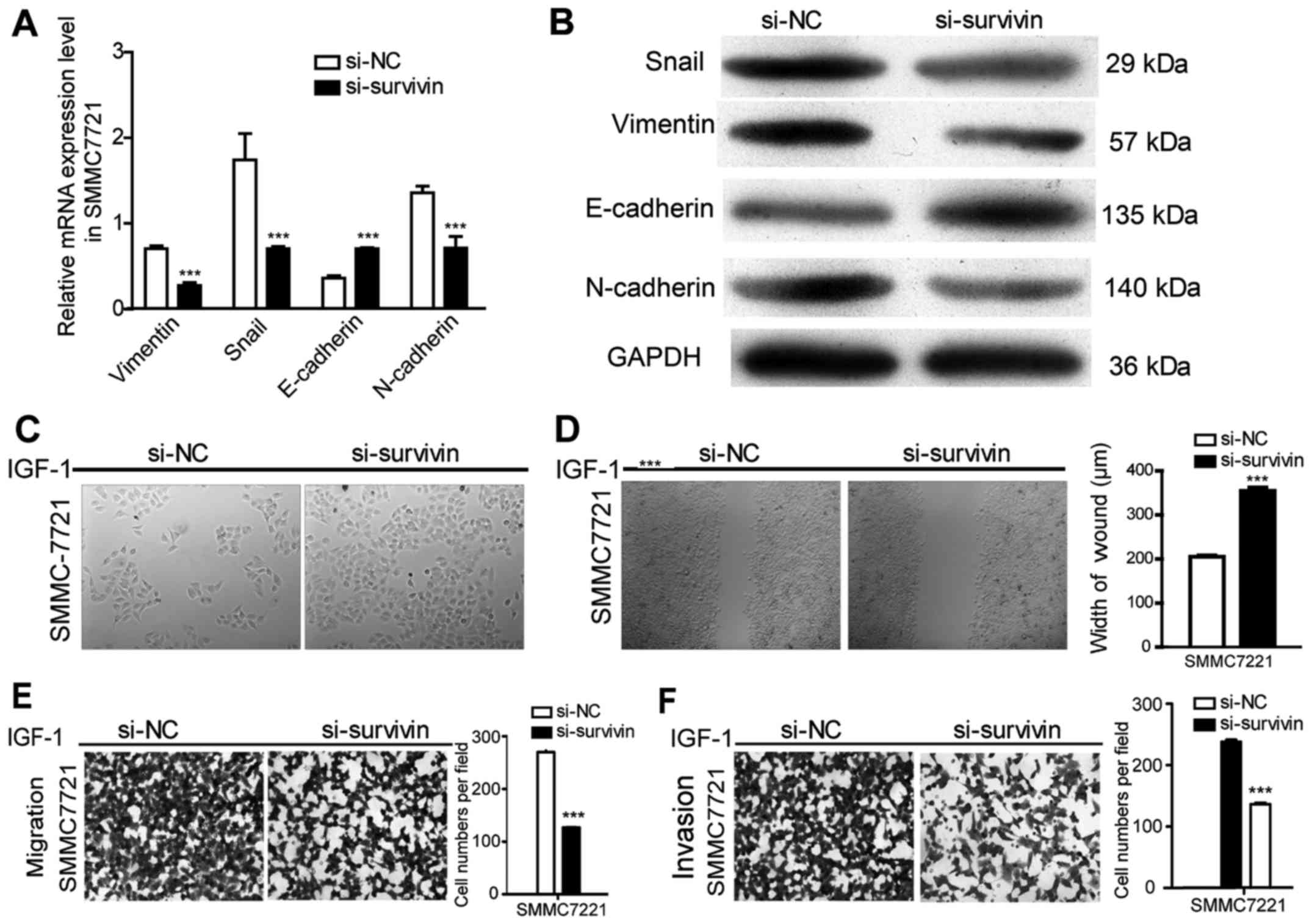
Survivin knockdown eradicates
IGF-1-induced EMT in HCC cells
To further confirm the effect of survivin on the
IGF-1-induced EMT process, survivin siRNA was transfected to
silence survivin expression promoted by IGF-1 at a concentration of
10 nM and the results revealed that survivin expression at the mRNA
and protein levels were significantly reduced (P<0.001)
(Fig. 4). After silencing survivin
expression in HCC cell lines, there was a significant decrease in
cell invasion and migration (P<0.001) (Fig. 5E and F). Furthermore, EMT events
were evaluated by observing cell morphological changes and assessed
the expression levels of EMT markers in mRNA and protein. As shown
in Fig. 5A-D, there was a
changeover in EMT marker expression and cell phenotype in the
survivin-siRNA cells compared with the control-siRNA cells. These
results indicated that silencing of survivin recovered the EMT
process of HCC induced by IGF-1.
Discussion
EMT is a reversible process in which epithelial
cells lose their cell polarity and cell-cell adhesion and acquire
mesenchymal features. During carcinogenesis, EMT enables tumor
cells to become invasive via downregulation of epithelial-specific
markers including E-cadherin, thyroid transcription factor-1
(TTF-1) and ZEB and upregulation of mesenchymal markers including
vimentin, α-SMA, N-cadherin and transcription factor Snail
(19–26). EMT is found to be a major event in
tumor metastasis by changing the cohesive ability of cells and
enhancing the invasive and migratory potential (27) and different biomarkers mediating the
EMT process have been identified. In addition, various
tumor-associated growth factors have been identified to be involved
into EMT, such as VEGF, EGF and TGF-β (28).
The important effect and involvement of IGF-1 in
metastasis have been well elucidated. IGF-1 affects cell invasion
by suppression of PTEN phosphorylation and interaction with the
PI3K/PTEN/Akt/NF-кB signaling pathway in pancreatic cancer
(29). In addition, IGF-1, together
with latent TGF-β can activate metalloproteinase activity (MMP) and
then result in EMT in MCF-7 breast cancer cells (30). IGF-1 was found to elevate the
expression of transmembrane glycoprotein MUC1 in MCF-7 cells for
the initiation of EMT in a PI3K/Akt signaling pathway-dependent
manner (7). IGF-1 was also reported
to activate PI3K/AKT/mTOR signaling to increase the expression of
survivin and control the expression of EMT biomarkers in the
development of gastric, prostate and colon cancer cells (4,16,31).
However, the relationship between IGF-1 and survivin in HCC was
unclear. In the present study, we also found that AKT was activated
with the upregulation of survivin in response to the treatment with
IGF-1. In line with previous research, the downregulation of
E-cadherin and the upregulation of N-cadherin, snail and vimentin
induced by IGF-1 in HCC were positively associated with invasion
and migration in a dose-dependent manner. On the basis of these
findings, our study suggests that IGF-1 may induce EMT involving
E-cadherin, N-cadherin, Snail and vimentin dysregulation, thus
facilitating the invasion and metastasis of HCC.
Survivin protein is overexpressed in most human
tumors and promotes tumor cell proliferation and viability
(4,32). As a nodal protein, survivin
interfaced with multiple signals involved in mitosis and apoptosis
and functionally integrated proliferation, cell death and cellular
homeostasis. Exploring strategies to lower the expression level of
survivin has been viewed as effective cancer therapy. Usually,
positive correlation between IGF and survivin can be observed in
tumor cells. As a target of IGF-1, survivin protein translation can
be driven by IGF-1 signaling in prostate cancer cells. Binding of
IGF-1 to its receptor activates downstream kinases, mammalian
target of rapamycin (mTOR) and p70S6 protein kinase (p70S6K), which
modulates survivin mRNA translation to increase the apoptotic
threshold (4). In the present
study, we focused on the role IGF-1/survivin in metastasis of HCC,
which has not been reported to date. However, target-therapeutic
functions have been found in other cancers. Sato et al also
indicated that IGF-1 can induce expression of survivin in renal
cell carcinoma (32). Furthermore,
we demonstrated that survivin depletion could recover the cohesive
ability and epithelial phenotype of HCC cells lost with IGF-1
treatment. Collectively, these data confirmed that survivin
participated in the IGF-1-mediating EMT process in HCC. To the best
of our knowledge, this is the first study to elucidate the
IGF-1/survivin cascade in HCC metastasis in vitro.
In conclusion, our results displayed indirect
evidence that IGF-1 had an effect on metastasis of HCC by mediating
the EMT process and activating survivin. AKT was also activated
with the upregulation of survivin in response to treatment with
IGF-1. These data provided a new insight for the molecular therapy
of HCC patients in clinical treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Key Research
and Development Plan in Shandong Province (2015GGH318017).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
FL, YS, HZ, HG, XZ and HC conceived and designed the
study. FL, YS, BL, JL and HL performed the experiments. FL, HZ and
HC wrote the manuscript. HG and XZ reviewed and edited the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Knox JJ, Cleary SP and Dawson LA:
Localized and systemic approaches to treating hepatocellular
carcinoma. J Clin Oncol. 33:1835–1844. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van Zijl F, Zulehner G, Petz M, Schneller
D, Kornauth C, Hau M, Machat G, Grubinger M, Huber H and Mikulits
W: Epithelial-mesenchymal transition in hepatocellular carcinoma.
Future Oncol. 5:1169–1179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vaira V, Lee CW, Goel HL, Bosari S,
Languino LR and Altieri DC: Regulation of survivin expression by
IGF-1/mTOR signaling. Oncogene. 26:2678–2684. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Haisa M: The type 1 insulin-like growth
factor receptor signalling system and targeted tyrosine kinase
inhibition in cancer. J Int Med Res. 41:253–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liao G, Wang M, Ou Y and Zhao Y:
IGF-1-induced epithelial-mesenchymal transition in MCF-7 cells is
mediated by MUC1. Cell Signal. 26:2131–2137. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lei T and Ling X: IGF-1 promotes the
growth and metastasis of hepatocellular carcinoma via the
inhibition of proteasome-mediated cathepsin B degradation. World J
Gastroenterol. 21:10137–10149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Su C: Survivin in survival of
hepatocellular carcinoma. Cancer Lett. 379:184–190. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang P, Wang G, Huo H, Li Q, Zhao Y and
Liu Y: SDF-1/CXCR4 signaling up-regulates survivin to regulate
human sacral chondrosarcoma cell cycle and epithelial-mesenchymal
transition via ERK and PI3K/AKT pathway. Med Oncol. 32:3772015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tai CJ, Chin-Sheng H, Kuo LJ, Wei PL, Lu
HH, Chen HA, Liu TZ, Liu JJ, Liu DZ, Ho YS, et al:
Survivin-mediated cancer cell migration through GRP78 and
epithelial-mesenchymal transition (EMT) marker expression in
Mahlavu cells. Ann Surg Oncol. 19:336–343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khan S, Aspe JR, Asumen MG, Almaguel F,
Odumosu O, Acevedo-Martinez S, De Leon M, Langridge WH and Wall NR:
Extracellular, cell-permeable survivin inhibits apoptosis while
promoting proliferative and metastatic potential. Br J Cancer.
100:1073–1086. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hollier BG, Evans K and Mani SA:
Identication of optimal topography of the barotropic ocean model in
the North Atlantic by variational data assimilation. Mol Cell
Endocrinol. 138:41–50. 2009.
|
|
15
|
Onoue T, Uchida D, Begum NM, Tomizuka Y,
Yoshida H and Sato M: Epithelial-mesenchymal transitions induced by
the stromal cell-derived factor-1/CXCR4 system in oral squamous
cell carcinoma cells. Int J Oncol. 29:1133–1138. 2006.PubMed/NCBI
|
|
16
|
Fang YJ, Lu ZH, Wang GQ, Pan ZZ, Zhou ZW,
Yun JP, Zhang MF and Wan DS: Elevated expressions of MMP7, TROP2,
and survivin are associated with survival, disease recurrence, and
liver metastasis of colon cancer. Int J Colorectal Dis. 24:875–884.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
DeNardo BD, Holloway MP, Ji Q, Nguyen KT,
Cheng Y, Valentine MB, Salomon A and Altura RA: Quantitative
phosphoproteomic analysis identifies activation of the RET and
IGF-1R/IR signaling pathways in neuroblastoma. PLoS One. 8:e82513.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen J, Alberts I and Li X: Dysregulation
of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum
disorders. Int J Dev Neurosci. 35:35–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Onder TT, Gupta PB, Mani SA, Yang J,
Lander ES and Weinberg RA: Loss of E-cadherin promotes metastasis
via multiple downstream transcriptional pathways. Cancer Res.
68:3645–3654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Canel M, Serrels A, Frame MC and Brunton
VG: E-cadherin-integrin crosstalk in cancer invasion and
metastasis. J Cell Sci. 126:393–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ivaska J, Pallari HM, Nevo J and Eriksson
JE: Novel functions of vimentin in cell adhesion, migration, and
signaling. Exp Cell Res. 313:2050–2062. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cosgrove BD, Mui KL, Driscoll TP, Caliari
SR, Mehta KD, Assoian RK, Burdick JA and Mauck RL: N-cadherin
adhesive interactions modulate matrix mechanosensing and fate
commitment of mesenchymal stem cells. Nat Mater. 15:1297–1306.
2016. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Amsellem V, Dryden NH, Martinelli R,
Gavins F, Almagro LO, Birdsey GM, Haskard DO, Mason JC, Turowski P
and Randi AM: ICAM-2 regulates vascular permeability and N-cadherin
localization through ezrin-radixin-moesin (ERM) proteins and Rac-1
signalling. Cell Commun Signal. 12:122014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen YS, Huang WL, Chang SH, Chang KW, Kao
SY, Lo JF and Su PF: Enhanced filopodium formation and stem-like
phenotypes in a novel metastatic head and neck cancer cell model.
Oncol Rep. 30:2829–2837. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sigurdsson V, Hilmarsdottir B,
Sigmundsdottir H, Fridriksdottir AJ, Ringnér M, Villadsen R, Borg
A, Agnarsson BA, Petersen OW, Magnusson MK, et al: Endothelial
induced EMT in breast epithelial cells with stem cell properties.
PLoS One. 6:e238332011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ma J, Sawai H, Matsuo Y, Ochi N, Yasuda A,
Takahashi H, Wakasugi T, Funahashi H, Sato M and Takeyama H: IGF-1
mediates PTEN suppression and enhances cell invasion and
proliferation via activation of the IGF-1/PI3K/Akt signaling
pathway in pancreatic cancer cells. J Surg Res. 160:90–101. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Walsh LA and Damjanovski S: IGF-1
increases invasive potential of MCF-7 breast cancer cells and
induces activation of latent TGF-β1 resulting in epithelial to
mesenchymal transition. Cell Commun Signal. 9:102011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li C, Li J, Wu D and Han G: The
involvement of survivin in insulin-like growth factor-1-induced
epithelial-mesenchymal transition in gastric cancer. Tumour Biol.
37:1091–1096. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sato A, Oya M, Ito K, Mizuno R, Horiguchi
Y, Umezawa K, Hayakawa M and Murai M: Survivin associates with cell
proliferation in renal cancer cells: Regulation of survivin
expression by insulin-like growth factor-1, interferon-gamma and a
novel NF-κB inhibitor. Int J Oncol. 28:841–846. 2006.PubMed/NCBI
|