Introduction
More than 100 types of tumors are clinically
apparent on the skin. Non-melanoma skin cancer (NMSC) is the most
common malignancy in the world, with cutaneous squamous cell
carcinomas (cSCCs) constituting ~20% of all NMSC. While cSCCs
typically behave in an indolent fashion and can be cured with local
destructive or surgical methods (1,2), if
the tumor occurs on the face, surgery could potentially reduce the
quality of life of the patient. At present, ifosfamide and
cisplatin are commonly used chemotherapy drugs for skin cancers.
They kill the skin cancer cells by suppressing the synthesis of
DNA. However, non-invasive, effective, and targeted skin cancer
chemotherapy drugs are lacking.
An essential characteristic of tumors is increased
cell proliferation, which is controlled by cell cycle progression.
Cyclin-dependent kinases (CDKs) bind to cell cycle proteins
(cyclin) to form a complex, and regulate transcription, metabolism,
neuro-physiological processes such as differentiation and
development (3–12). Disorders of CDK activity directly or
indirectly cause cell proliferation, genomic instability (increase
DNA mutations and chromosome deletions) and chromosome instability
(chromosome number changes), which are critical for the development
and progression of cancer (13,14).
Among them, CDK4/6 activity is necessary for the regulation of the
cell cycle in the G1 phase (15).
CDK4/6 have been selected as important cancer therapeutic target
proteins, as they are hallmarks of cancers.
It has been well documented that the expression
levels of CDK4/6 are significantly higher in many tumors (16–18).
In skin cancers, it has been reported that CDK4 and CDK6 levels are
overexpressed in >90% of cases (19). Therefore, CDK4/6 are important
targets for skin cancer drugs. Recently, two CDK4/6 inhibitors have
been approved for the treatment of breast cancer. However, there is
no study on CDK4/6 inhibitors which focuses on skin cancer.
Using structure-based virtual screening via
protein-ligand docking to select candidates from FDA-approved
small-molecule drugs, we previously identified two CDK2 inhibitors,
adapalene (20) and fluspirilene
(21). In the present study, we
attempted to identify drugs with the ability to specifically
inhibit both CDK4/6 at the same time without affecting CDK2. To
achieve this purpose, the free and open-source docking software
idock v2.2.1 (22,23) developed by our group, was applied to
dock small molecule compounds onto each and all of the available
CDK4/6 structures, and to predict their binding conformations, as
well as their binding affinities. The top compounds displaying the
highest average predicted binding free energy were screened for
their ability to reduce the viability of human skin cancer cells.
Among them, rafoxanide was discovered as a CDK4/6 dual-inhibitor
with the highest anticancer effects in A375 and A431 human skin
cancer cell lines. The anticancer effects were also validated in
vivo in BALB/C nude mice subcutaneously xenografted with A375
cells.
At present, rafoxanide is mainly used for
fasciola hepatica infection (24,25).
The present study is the first to indicate that rafoxanide inhibits
CDK4/6 and is a potential candidate drug for the treatment of human
skin cancer.
Materials and methods
Ethics statement
The present study, was approved by the Ethics
Committee of Yunnan University of Chinese Traditional Medicine
(Kunming, China).
Docking
We first collected five X-ray crystallographic
structures of CDK4 and 8 X-ray crystallographic structures of CDK6
in a complex with a ligand from the Protein Data Bank (PDB)
(26–27), and then the co-crystallized water
and ligand molecules were deleted manually. Subsequently, the
structures of 3,167 approved drugs were gathered from the ZINC
database (28,29). To predict the binding conformations
and the binding affinities of these drugs with the CDK4/6 proteins,
the free and popular docking software idock v2.2.1 (20,21)
was used to dock all of the compounds onto all of the CDK2/4/6
structures. Prioritized in accordance with the average predicted
binding affinity, 9 commercially available compounds were
identified, purchased and subsequently evaluated.
Chemicals and antibodies
We purchased lifibrate, nizofenone, pimozide,
trifluperidol, tosufloxacin, cloricromene, sulconazole, sertindole,
rafoxanide, oxaliplatin from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). The antibodies used in the present study, anti-cyclin D
(cat. no. ab134175), anti-CDK2/4/6 (cat. nos. ab32147, ab108357 and
ab124821), anti-Rb (cat. no. ab181616), anti-phosphorylated
(pho)-CDK2/6 (cat. nos. ab76146 and ab194871), anti-pho-Rb (cat.
no. ab184796) and GAPDH (cat. no. ab8245) were obtained from Abcam
(Cambridge, MA, USA); pho-CDK4 (cat. no. AP0593) was obtained from
ABclonal (Wuhan, China).
Cell lines and cell culture
conditions
The skin cancer A431 and A375 cell lines were
obtained from the Library of the Chinese Academy of Sciences
Committee on Type Culture Collection of Cells (Shanghai, China).
These cells were cultured in Dulbecco's modified Eagle's medium
(DMEM; GE Healthcare Life Sciences, Shanghai, China) containing 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) at 37°C, in 5% CO2 and 95% humidified
air.
Cell culture experimental
conditions
Briefly, cells were plated in 96-, 24- or 6-well
plates (Wuxi Nest Biotechnology Co., Ltd., Wuxi, China) with medium
for 24 h. Subsequently, they were treated with medium containing 8%
FBS and the test compounds at various concentrations (3, 10 and 30
µM/l) and incubation times (24, 48 and 72 h) as indicated.
MTS assay
Cell proliferative ability was analyzed at various
time-points (days 1, 3 and 6) using colorimetric
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy methoxy phenyl)-
2-(4-sulfophenyl)-2H-tetra zolium] (MTS) assay (Promega Corp.,
Beijing, China). Cells were seeded at an initial density of
9×103 cells/well in 96-well plates, and cultured under
5% CO2 at 37°C. After a 24-h incubation, the media was
replaced with fresh growth media containing testing compounds at
various concentrations (3, 10 and 30 µM/l) and incubation times
(24, 48 and 72 h) as indicated. At the end of the reactions, 20 µl
of a pre-diluted MTS solution was introduced to each well and the
cells were incubated at 37°C in the dark for 2 h. The absorbance
was recorded at 490 nm with a Synergy 2 microplate reader
(Multiskan FC; Thermo Fisher Scientific, Inc.).
Cell cycle analysis
We conducted cell cycle analysis using an EPICS XL4
Flow Cytometer (Beckman Coulter, Inc., Brea, CA, USA). The cell
cycle phase distribution was analyzed using ModFit LT 2.0 software
(Verity Software House, Topsham, ME, USA). Briefly, cells
(4×104) were seeded in 24-well plates in DMEM containing
0.125% FBS. After 24 h, the cell culture medium was replaced with
DMEM containing 10% FBS and various doses of rafoxanide (1, 3, 10
or 30 µM) for 6, 12 or 24 h, as indicated. At the end of the
experiments, cells (1×104) were fixed in ice-cold 70%
ethanol, and stained using a Coulter DNA Prep Reagent kit (Beckman
Coulter), and cellular DNA content was assessed. All data were
obtained from two separate experiments performed in triplicate.
Cell apoptosis analysis
Apoptosis was determined by staining cells with both
Annexin V and propidium iodide (PI) (Annexin V-FITC/PI Kit; 4A
Biotech Co., Ltd., Beijing, China), according to the manufacturer's
instructions to quantify the apoptotic cells. Briefly, cells were
plated on 24-well plates with DMEM containing 0.125% FBS. After 24
h, the medium was replaced with DMEM with 10% FBS, and 3, 10 or 30
µM of rafoxanide for various time-points (6, 12 and 24 h) as
indicated. At the end of the experiments, cells were trypsinized
with 0.25% trypsin in the absence of ethylenediaminetetraacetic
acid (EDTA), washed with PBS twice, and suspended in 500 µl of
binding buffer. Two microliters of Annexin V-EGFP and 5 µl of PI
were added to the suspension (2×105−106
cells/ml) and incubated for 5 to 15 min in the dark. The apoptotic
cells were analyzed by flow cytometry (CyFlow Space; Sysmex Partec,
Hamburg, Germany).
Western blot analysis
A431 and A375 cells were plated on 6-well plates in
DMEM containing 0.125% FBS. After 24 h, the cell culture media were
replaced with DMEM containing 10% FBS medium containing rafoxanide
at concentrations of 3, 10 and 30 µM. Cells were harvested after 6
h of incubation. As positive controls, CDK2/4/6siRNAs were used to
inhibit proteins CDK2,4,6 in A431 and A375 cells. The siRNA oligo
sequences were as follows: siRNA-CDK2, 5′-AGTTGTACCTCCCCTGGAT-3′;
siRNA-CDK4, 5′-CAGAUCUCGGUGAACGAUGdTdT-3′; siRNA-CDK6,
5′-GAUGUUGAUCAACUAGGAATT-3′. Briefly, when cells were treated with
40 pmol siRNA of CDK2/4/6/ they were delivered using 5 µl RNAiMAX
transfection reagent for transfection in each well of the 6-well
plates. Cells were lysed in radioimmunoprecipitation assay buffer
(Beyotime Institute of Biotechnology, Jiangsu, China), and
protein concentrations were assessed using a Bicinchoninic Acid
Protein Assay kit (Thermo Fisher Scientific, Inc.). Equal amounts
of protein (30 µg/lane) samples were resolved by 10% SDS-PAGE and
transferred onto membranes (Immobilon FL; Merck Millipore). After
blocking, the membranes were incubated first with the primary
antibodies (Abcam) at 4°C overnight, washed in TBS, and then the
secondary antibodies (1:4,000; cat. no. SE12; Beijing
Solarbio Science and Technology Co., Ltd.,Beijing, China) were
added for 1 h at room temperature. Prior to drying in the dark, the
membranes were then washed in TBS+0.1% Tween, and then
dH2O. The blots were detected using a chemiluminescence
detection system (Amersham Biosciences, Piscataway, NJ, USA)
(VisionWorks acquisition and analysis, Compatible with Microsoft
Windows 7, Vista, XP SP2 or later).
Evaluation of the therapeutic effect
of Rafoxanide in vivo in nude mice xenografted with A375 cells
In the present study, we used female BALB/C nude
mice (n=50; weighing 15 g; 4–5 weeks old; Vital River Laboratory
Animal Technology Co., Ltd., Beijing, China). The mice were housed
under specific pathogen-free conditions in an environment with a
12-h light/dark cycle and 50–80% humidity. Mice were cared for in
accordance with the guidelines of the Laboratory Animal Ethics
Committee of Kunming Medical University (Kunming, China). The
present study, was approved by the Ethics Committee of Yunnan
University of Chinese Traditional Medicine (Kunming, China). A375
cells (1×106) were suspended in 0.2 ml PBS, and injected
subcutaneously into the right flank of the mice (n=3). Tumor size
was assessed using a caliper. When the tumors grew to 80–100
mm3 (1 week after inoculation), mice were divided
randomly into different experimental groups (5 mice/group), and
intraperitoneally injected daily for 21 days with various testing
compounds. The mice were sacrificed by cervical dislocation, and
the tumors were excised, weighed, and images were captured by
camera (Canon, Inc., Tokyo, Japan). The tumor volume was calculated
using the formula V = ab2/2 (a = longest axis; b =
shortest axis).
Statistical analysis
Data were obtained from at least three experiments,
and values were expressed as the mean ± standard deviation (SD).
Statistical analysis was performed by ANOVA followed with Fisher's
least significant difference post hoc tests (version 16.0, SPSS
software). P<0.05 was considered to indicate a statistically
significant difference between values.
Results
Identification of candidate CDK4/6
inhibitors using structure-based virtual screening
A total of 3,167 drugs approved by worldwide
authorities constituted a library of compounds for screening. They
were individually docked to the ATP binding pockets of CDK4/6, and
then sorted in the ascending order of their predicted binding free
energy. The high-scoring compounds were manually examined based on
in silico estimations of binding strength, appropriate
molecular weight and other drug-like properties, and complementary
matching of molecular shape. Finally, high-scoring compounds were
selected and 9 commercially available compounds [Table I (28,30–37)]
were purchased for subsequent wet-lab validations.
 | Table I.The nine top-scoring compounds
purchased and tested in vitro. |
Table I.
The nine top-scoring compounds
purchased and tested in vitro.
| Compound name | ZINC ID | Average score
(kcal/mol) | MW (g/Mol) | Clinic usage | (Refs.) |
|---|
| Lifibrate | 537906 | −8.5 | 410.295 | Hypolipidemic
agent | (30) |
| Nizofenone | 538096 | −7.14 | 412.87 | Ameliorated
ischemic brain damage | (31) |
| Pimozide | 19796084 | −9.99 | 461.55 | Neuroleptic
drug | (32) |
| Trifluperidol | 538505 | −7.67 | 409.417 | Schizophrenia
drug | (33) |
| Tosufloxacin | 21983589 | −9.56 | 404.34 | Antibiotics | (34) |
| Cloricromene | 576842 | −7.06 | 395.88 | Coronary dilating
agent | (35) |
| Sulconazole | 601250 | −8.13 | 460.76 | Antifungal
agents | (36) |
| Sertindole | 538337 | −9.48 | 440.94 | Antipsychotic
drugs | (37) |
| Rafoxanide | 4181896 | −8.68 | 626.01 | Treatment of
Fasciola hepatica infection | (28) |
Rafoxanide treatment markedly
decreases the viability of A375 and A431 human skin cancer
cells
We first evaluated the cytotoxic effects of the nine
compounds using MTS assays in two skin cancer cell lines. All nine
compounds (lifibrate, nizofenone, pimozide, trifluperidol,
tosufloxacin, cloricromene, sulconazole, sertindole and rafoxanide)
decreased the viability of the A375 and A431 cells. The
IC50 values were calculated using GraphPad Prism 5
(GraphPad Software, Inc., La Jolla, CA, USA). Among them,
rafoxanide exhibited the lowest IC50 value (1.09 µM for
A375 and 1.31 µM for A431 cells, respectively). As displayed in
Fig. 1 rafoxanide exhibited the
strongest cytotoxic effects in A375 (Fig. 1A) and A431 (Fig. 1B) cells compared to the other 8
compounds. The growth inhibitory effect of rafoxanide was dose- and
time-dependent, with significant inhibition observed at
concentrations of ≥3 µM in A375 cells (Fig. 1C) and A431 cells (Fig. 1D).
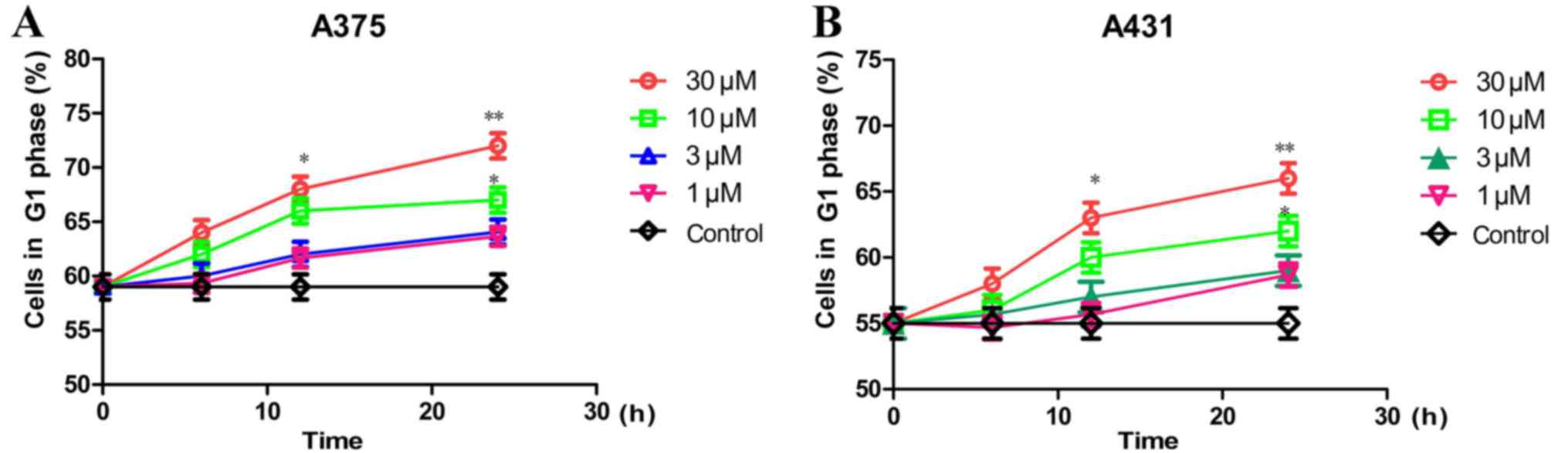
Rafoxanide treatment causes cell cycle
arrest in the G1 phase
We then assessed the effect of rafoxanide treatment
in the inhibition of cell cycle progression. The A375 and A431
cells were treated with various doses of rafoxanide (3, 10 or 30
µM) for different time-points (6, 12 or 24 h), and their effects on
cell cycle profiles were assessed using flow cytometric analysis.
After 24 h of incubation, our results indicated that rafoxanide
treatment dose- and time-dependently (P<0.05), significantly
increased the G1 phase populations, which was accompanied by
concurrent decreased S- and G2/M phase populations, in
A375 (Fig. 2A) and A431 (Fig. 2B) cells.
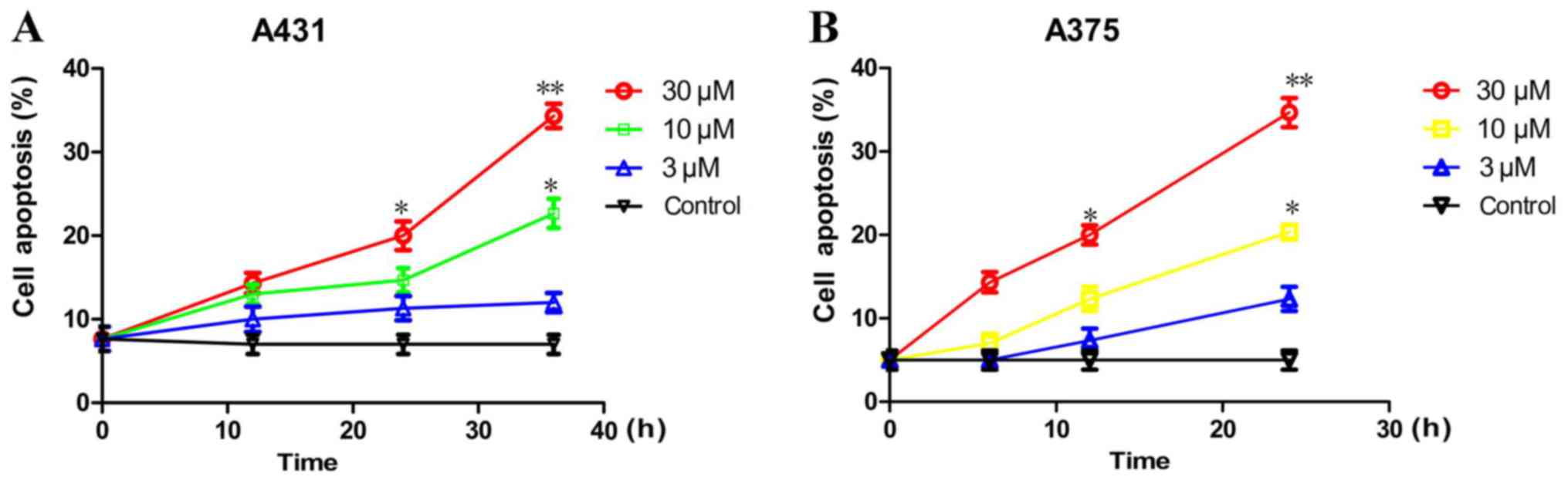
Rafoxanide treatment stimulates cell
apoptosis
In addition, we investigated whether rafoxanide
could stimulate cell apoptosis using flow cytometry with an Annexin
V-FITC/PI kit (4A Biotech Co., Ltd., Beijing, China). As displayed
in Fig. 3, rafoxanide treatment at
10 and 30 µM concentrations significantly increased the percentage
of apoptosis in A375 (Fig. 3A) and
A431 cells (Fig. 3B) in a dose- and
time-dependent manner (P<0.05).
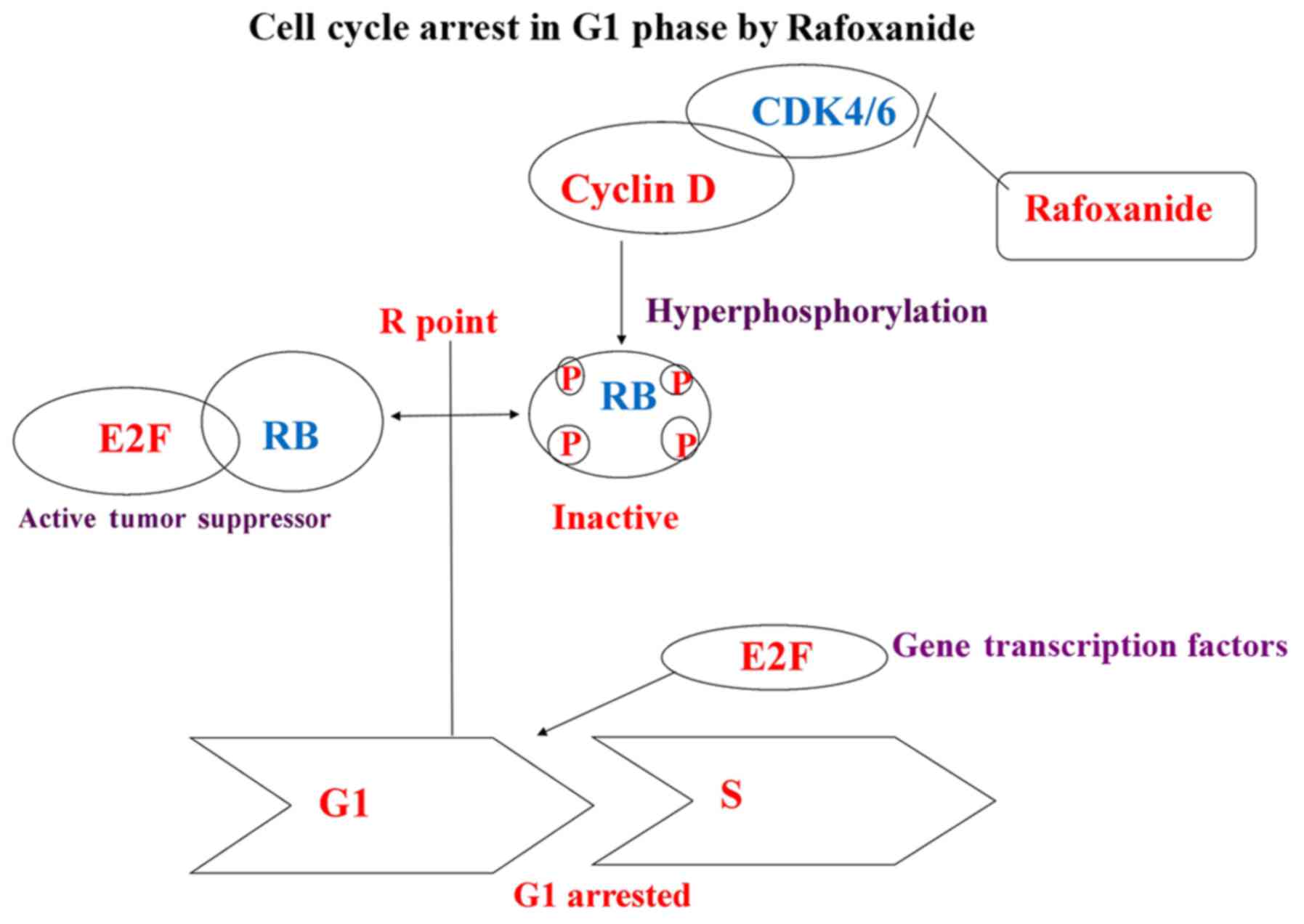
Rafoxanide decreases the expression of
CDK4/6, Rb, cyclin D, pho-CDK4/6 and pho-Rb, but not CDK2 in A375
and A431 cells
To demonstrate the mechanism of actions, we
investigated the effects of rafoxanide on the expression of key
proteins involved in cell cycle G1 phase progression, including
CDK2/4/6, cyclin D, Rb, pho-CDK2/4/6 and pho-Rb by western blot
analysis in A375 and A431 cells (Fig.
4). The results revealed that rafoxanide specifically reduced
the expression of CDK4/6, cyclin D, Rb, pho-CDK4/6, pho-Rb, in the
A375 and A431 cells. Notably, the expression level of CDK2 was not
affected. These patterns are consistent with what is expected for a
specific CDK4/6 dual inhibitor. Based on these results, we proposed
that rafoxanide reduced the activity of CDK4/6, which decreased the
binding of cyclin D and promoted degradation, suppressed Rb and Rb
phosphorylation, and inhibited the activation of E2F1-caused cell
cycle G1 arrest and suppressed cell cycle progression
from the G1 to S phases (7–12)
(Fig. 5).
 | Figure 4.Effects of rafoxanide treatment on
the expression of cyclin D, CDK4/6 and Rb. (A) A375 and A431 cells
were plated on 6-well plates with 0.125% FBS medium for 24 h and
then with 10% FBS medium containing rafoxanide at concentrations of
3, 10, 30 µM. Cells were harvested after a 6-h incubation and
proteins were analyzed by western blotting. Western blotting
results revealed that rafoxanide treatment significantly reduced
the expression of CDK4/6, pho-CDK4/6, Rb, pho-Rb, cyclin D in A375
and A431 compared to the control. (B) Graph indicated the
expression protein level of CDK2 was not affected by treatment with
rafoxanide. (C-E) Graphs revealing the dose-dependent reduction on
the protein expression level of CDK4, CDK6 and Rb in the A375 and
A431 cell lines compared to the control (*P<0.05, **P<0.01).
Based on these results, we proposed that rafoxanide reduced the
activity of CDK4/6, which resulted in decreased binding and
increased degradation of cyclin D. Furthermore, it suppressed Rb
and Rb phosphorylation, and reduced the activation of E2F1. These
functions caused cell cycle G1-arrest and reduced cell cycle
progression from the G1 to the S phase. |
Rafoxanide (i.p.) administration
reduces the growth of tumors subcutaneously xenografted with A375
cells in vivo in BALB/C nude mice
To assess the in vivo effect of rafoxanide, a
mouse model of A375 cell xenografted skin tumors was established in
BALB/C nude mice. Following the appearance of the tumors, tumor
volumes were assessed every 3–4 days. Seven days after tumor
inoculation, the tumor volume reached 80–100 mm3, and
the animals were divided into 4 groups (5 mice/group) and were
administered daily for 21 days by i.p. injection the following: i)
PBS (control group); ii) oxaplatin (5 mg/kg); iii) rafoxanide (40
mg/kg); and iv) combination of rafoxanide (40 mg/kg) and oxaplatin
(5 mg/kg). As displayed in Fig. 6A,
our results demonstrated that the potency of rafoxanide (40 mg/kg)
was similar to that of oxaplatin (5 mg/kg) in reducing tumor
weight. The combined administration of both drugs produced the
greatest therapeutic effect. In addition, the effect of rafoxanide
(40 mg/kg) was similar to that of oxaplatin (5 mg/kg) in reducing
tumor volume, with combined administration producing the greatest
therapeutic effect (Fig. 6B). We
also assessed the body weight of all the mice in the four groups.
No significant body weight changes were observed in all four groups
(P>0.05) (Fig. 6C). In addition,
we did not detect any obvious toxicity in mice during the course of
the experiments.
Discussion
The most common forms of skin cancer include basal
cell carcinoma, squamous cell carcinoma and melanoma. Early skin
cancer can be cured by surgical resection, and ~80% of cases are
treated using this method. However, in cases where skin cancer is
on the face, surgical resection would decrease the quality of life
of the patient. Thus, new drugs are urgently needed (38).
Aberrant cell cycle progression and uncontrolled
cell proliferation is an indication of cancer (39). CDK4/6 can drive cell cycle
progression from the G1 to the S phase, thus, dysregulation of CDKs
plays a central role in tumorigenesis. CDK4/6 was revealed to be
overexpressed in more than 90% of skin cancers (40,41).
The CDK4/6 activating events in skin cancer render it a potential
therapeutic target, therefore a dual-inhibitor targeting the
activities of CDK4 and CDK6 could be an effective agent to reduce
tumor cell proliferation in human neoplasms.
In the present study, we performed virtual screening
to search for a specific CDK4/6 dual inhibitor from a library of
FDA-approved small molecule drugs. We reveal in the present study
for the first time, that rafoxanide exhibited dual CDK4/6 inhibitor
activity in vitro in human skin cancer cells. Furthermore,
rafoxanide (40 mg/kg, i.p.) exhibited significant in vivo
anticancer efficacy, which was comparable to that of the clinically
used anticancer drug oxaliplatin (5 mg/kg) in nude mice
subcutaneously xenografted with A375 skin cancer cells. The
combination of rafoxanide and oxaliplatin produced the greatest
therapeutic effect.
Three CDK4/6 inhibitors: Palbociclib, ribociclib and
abemaciclib, have been tested in randomised phase II/III trials of
clinical breast cancer (BC). Tests results revealed significantly
increased progression-free survival in first- and second-line
treatment for advanced hormone receptor-positive HER2-negative BC
(42). The Food and Drug
Administration (FDA) and the European Commission (EMA) have
approved palbociclib for the treatment of patients HR+
HER2− in locally advanced or metastatic BC (aBC), in
combination with an aromatase inhibitor as initial therapy in
postmenopausal women or in combination with fulvestrant in women
who have received prior endocrine therapy. Ribociclib has been
approved by the FDA in combination with an aromatase inhibitor as
an initial therapy for postmenopausal women with HR+
HER2− in BC (43).
However, the adverse effects of these agents were neutropenia,
infections, fatigue and gastrointestinal toxicity. Furthermore, the
effect of CDK4/6 inhibitors was dependent on an intact, functional
Rb group (42,43). The incidence of Rb loss was
dependent on the clinical subtype and was more common in certain
subtypes. It has been reported that Rb loss was observed at a
percentage of 20–30% in different subtypes of cancer (44,45).
Skin cancer has been reported to have 33% of Rb loss (46). It remains to be determined whether
the effect of rafoxanide is dependent on an intact functional Rb
group.
Rafoxanide has been tested and revealed to have no
obvious toxic effects by oral administration at 6.7 mg/kg in
rabbits infected with 2-, 4-, 6- or 8-week old Fasciola
hepatica (47). The present
study did not observe any significant toxicity and change in the
body weight of the BALB/C nude mice administered (i.p.) with
rafoxanide (40 mg/kg) over 21 days. These results suggested that
i.p. injection of rafoxanide was relatively safe. The toxicity of
rafoxanide for skin cancer therapy requires further
investigations.
In addition to inhibiting CDK4/6, rafoxanide was
reported to be a potent uncoupling factor of mitochondrial
oxidative phosphorylation and exert the effects by interfering with
parasite mitochondrial ATP synthesis (48). The possible function and effect of
rafoxanide as an uncoupling agent of mitochondrial oxidative
phosphorylation, and whether it would interfere with mitochondria
ATP synthesis in skin cancer cells remain to be determined. In
addition, our results revealed that rafoxanide also promoted
apoptosis in skin cancer cells. While the mechanisms are not known,
it is possible that rafoxanide may induce apoptosis via the
E2F-p53-Bax/caspase 3 signaling pathway (49–51).
In the next phase of the study, the mechanisms of
rafoxanide-induced apoptosis in skin cancer will be identified by
microarray and other methods.
At present, rafoxanide is used for the treatment of
Fasciola hepatica infection (28,29).
As an FDA-approved drug, rafoxanide alone or in combination with
other chemotherapeutic drugs may be suitable for the treatment of
skin cancers. Further modifications may be required to improve its
efficacy and reduce its toxicity, including chemical modification
and/or packaging rafoxanide in nanoparticles or liposomes. The
possibility that rafoxanide could be considered as either an
alternative or a complementary therapy with surgery for the
treatment of skin cancer, particularly when occurring on the face,
warrants further investigations.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Hsiang-fu Kung Academician Workstation of Kunming Medical
University (no. NSFC 81272549), the Natural Science Foundation of
SZU (no. 827–000100 starting project of Shenzhen high-level
overseas talent), the Shenzhen Basic Research Project
(JCYJ20160331114230843 and JCYJ20150324141711558), the Guizhou
Science and Technology Department [no. QKHJC (2017) 1171], the
Chinese Ministry of Science and Technology (no. 2016YFC0904600),
the Vice-Chancellor's One-off Discretionary Fund, Faculty of Social
Science Postdoctoral Fellowship Scheme and Institute of Future
Cities and the Chinese University of Hong Kong and the joint
application project from the Yunnan Provincial Science and
Technology Department (2014FB060).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
MCML, HFK, KSL, MHW conceived and designed the
study. XL, LLi, LLe, QH, WC, AS and HY conducted the research. KK,
CD, YZ, YQ, YD and YHH performed the analysis of data. XS and HL
revised the manuscript and were also involved in the conception of
the study. MCML critically revised the article for important
intellectual content. All authors read and approved the manuscript
and agree to be accountable for all aspects of the research in
ensuring that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Mice were cared for in accordance with the
guidelines of the Laboratory Animal Ethics Committee of Kunming
Medical University (Kunming, China). The present study, was
approved by the Ethics Committee of Yunnan University of Chinese
Traditional Medicine (Kunming, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bikle DD, Jiang Y, Nguyen T, Oda Y and Tu
CL: Disruption of vitamin D and calcium signaling in keratinocytes
predisposes to skin cancer. Front Physiol. 7:2962016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fu T, Aasi SZ and Hollmig ST: Management
of high-risk squamous cell carcinoma of the skin. Curr Treat
Options Oncol. 17:342016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lim S and Kaldis P: CDKcyclins and CKIs:
Roles beyond cell cycle regulation. Development. 140:3079–3093.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin Dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:1–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cicenas J, Kalyan K, Sorokinas A, Jatulyte
A, Valiunas D, Kaupinis A and Valius M: Highlights of the latest
advances in research on CDK inhibitors. Cancers. 6:2224–2242. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Canavese M, Santo L and Raje N: Cyclin
dependent kinases in cancer: Potential for therapeutic
intervention. Cancer Biol Ther. l3:1–457. 2014.
|
|
7
|
Lukas J, Bartkova J and Bartek J:
Convergence of mitogenic signalling cascades from diverse classes
of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled
G1 checkpoint. Mol Cell Biol. 16:6917–6925. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hinz M, Krappmann D, Eichten A, Heder A,
Scheidereit C and Strauss M: NF-κB function in growth control:
Regulation of cyclin D1 expression and G0/G1-to-S-phase transition.
Mol Cell Biol. 19:2690–2698. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Malumbres M: Physiological relevance of
cell cycle kinases. Physiol Rev. 91:973–1007. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Choi YJ and Anders L: Signaling through
cyclin D-dependent kinases. Oncogene. 33:1890–1903. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pucci B, Kasten M and Giordano A: Cell
cycle and apoptosis. Neoplasia. 2:291–299. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cohen P: Protein kinase-the major drug
targets of the twenty-first century? Nat Rev Drug Discov.
1:309–315. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li M, Xiao A, Floyd D, Olmez I, Lee J,
Godlewski J, Bronisz A, Bhat KPL, Sulman EP, Nakano I and Purow B:
CDK4/6 inhibition is more active against the glioblastoma proneural
subtype. Oncotarget. 8:55319–55331. 2017.PubMed/NCBI
|
|
17
|
Dall'Acqua A, Sonego M, Pellizzari I,
Pellarin I, Canzonieri V, D'Andrea S, Benevol S, Sorio R, Giorda G,
Califano D, et al: CDK6 protects epithelial ovarian cancer from
platinum-induced death via FOXO3 regulation. EMBO Mol Med.
9:1415–1433. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Malumbres M: Oncogene-induced mitotic
stress: p53 and pRb get mad too. Cancer Cell. 19:691–2. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sheppard KE and McArthur GA: The cell
cycle regulator CDK4: An emerging therapeutic target in melanoma.
Clin Cancer Res. 19:5320–5328. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shi XN, Li H, Yao H, Liu X, Li L, Leung
KS, Kung HF and Lin MC: Adapalene inhibited the activity of
cyclin-dependent kinase 2 in colorectal carcinoma. Mol Med Rep.
12:6501–6508. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi XN, Li H, Yao H, Liu X, Li L, Leung
KS, Kung HF, Lu D, Wong MH and Lin MC: In silico identification and
in vitro and in vivo validation of anti-psychotic drug fluspirilene
as a potential CDK2 inhibitor and a candidate anti-cancer drug.
PLoS One. 10:e01320722015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H, Leung KS and Wong MH: Idock: A
multithreaded virtual screening tool for flexible ligand
dockingProceedings of the 2012 IEEE symposium on computational
intelligence in bioinformatics and computational biology (CIBCB).
San Diego: pp. 77–84. 2012, View Article : Google Scholar
|
|
23
|
Li H, Leung KS, Ballester PJ and Wong MH:
Istar: A web platform for large-scale protein-ligand docking. PLoS
One. 9:e856782014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ross DB: Treatment of experimental
Fasciola hepatica infection of sheep with rafoxanide. Vet Rec.
87:110–111. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elitok B, Elitok OM and Kabu M: Field
trial on comparative efficacy of four fasciolicides against natural
liver fluke infection in cattle. Vet Parasitol. 135:279–285. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Berman HM, Westbrook J, Feng Z, Gilliland
G, Bhat TN, Weissig H, Shindyalov IN and Bourne PE: The protein
data bank. Nucleic Acids Res. 28:235–242. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang Z and Wong CF: inexpensive method
for selecting receptor structures for virtual screening. J Chem Inf
Model. 56:21–34. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Irwin JJ and Shoichet BK: ZINC-a free
database of commercially available compounds for virtual screening.
J Chem Inf Model. 45:177–182. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Irwin JJ, Sterling T, Mysinger MM, Bolstad
ES and Coleman RG: ZINC: A free tool to discover chemistry for
biology. J ChemInf Model. 52:1757–1768. 2012. View Article : Google Scholar
|
|
30
|
Arnold A, McAuliff MP and Beyler AL:
Metabolic effects of a new hypolipidemic agent, ciprofibrate. J
Pharm Sci. 68:1557–1558. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matsumoto Y, Aihara K, Kamata T and Goto
N: Nizofenone, a neuroprotective drug, suppresses glutamate release
and lactate accumulation. Eur J Pharmacol. 262:157–161. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Janssen PA, Niemegeers CJ, Schellekens KH,
Dresse A, Lenaerts FM, Pinchard A, Schaper WK, van Nueten JM and
Verbruggen FJ: Pimozide, a chemically novel, highly potent and
orally long-acting neuroleptic drug. I. The comparative
pharmacology of pimozide, haloperidol, and chlorpromazine.
Arzneimittelforschung. 18:261–279. 1968.PubMed/NCBI
|
|
33
|
Bueno JR: Therapeutic evaluation of R2498
(triperidol) in hospitalized schizophrenic patients. J Bras
Psiquiatr. 14:81–91. 1965.(In Italian). PubMed/NCBI
|
|
34
|
Fujimaki K, Noumi T, Saikawa I, Inoue M
and Mitsuhashi S: In vitro and in vivo antibacterial activities of
T-3262, a new fluoroquinolone. Antimicrob Agents Chemother.
32:827–833. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dejana E, de Castellarnau C, Balconi G,
Rotilio D, Pietra A and de Gaetano G: AD 6, a coronary dilating
agent, stimulates PGI2 production in rat aorta ex vivo and in human
endothelial cells in culture. Pharmacol Res Commun. 14:719–724.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lassus A, Forström S and Salo O: A
double-blind comparison of sulconazole nitrate 1% cream with
clotrimazole 1% cream in the treatment of dermatophytoses. Br J
Dermatol. 108:195–198. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Muscatello MR, Bruno A, Micali
Bellinghieri P, Pandolfo G and Zoccali RA: Sertindole in
schizophrenia: Efficacy and safety issues. Expert Opin
Pharmacother. 15:1943–1953. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gray-Schopfer V, Wellbrock C and Marais R:
Melanoma biology and new targeted therapy. Nature. 445:851–857.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Walker GJ, Flores JF, Glendening JM, Lin
AH, Markl ID and Fountain JW: Virtually 100% of melanoma cell lines
harbor alterations at the DNAlevel within CDKN2A, CDKN2B, or one of
their downstream targets. Genes Chromosomes Cancer. 22:157–163.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Curtin JA, Fridlyand J, Kageshita T, Patel
HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, Le Boit PE, et
al: Distinct sets of genetic alterations in melanoma. N Engl J Med.
353:2135–2147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Polk A, Kolmos IL, Kümler I and Nielsen
DL: Specific CDK4/6 inhibition in breast cancer: A systematic
review of current clinical evidence. ESMO Open. 1:e0000932017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
de Groot AF, Kuijpers CJ and Kroep JR:
CDK4/6 inhibition in early and metastatic breast cancer: A review.
Cancer Treat Rev. 60:130–138. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dean JL, Thangavel C, McClendon AK, Reed
CA and Knudsen ES: Therapeutic CDK4/6 inhibition in breast cancer:
Key mechanisms of response and failure. Oncogene. 29:4018–4032.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bosco EE and Knudsen ES: RB in breast
cancer: At the crossroads of tumorigenesis and treatment. Cell
Cycle. 6:667–671. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tchernev G and Orfanos CE: Downregulation
of cell cycle modulators p21, p27, p53, Rb and proapoptotic
Bcl-2-relatedproteins Bax and Bak in cutaneous melanoma is
associated with worse patient prognosis: Preliminary findings. J
Cutan Pathol. 34:247–256. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Stammers BM: The effects of rafoxanide and
nitroxynil on the survival, growth and morphology of Fasciola
hepatica in rabbits. Z Parasitenkd. 46:153–156. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Matsubara K, Sanoh S, Ohta S, Kitamura S,
Sugihara K and Fujimoto N: An improved thyroid hormone reporter
assay to determine the thyroid hormone-like activity of amiodarone,
bithionol, closantel and rafoxanide. Toxicol Lett. 208:30–35. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xie Y, Si J, Wang YP, Li HY, Di CX, Yan
JF, Ye YC, Zhang YS and Zhang H: E2F is involved in radio
resistance of carbon ion induced apoptosis via Bax/caspase 3 signal
pathway in human hepatoma cell. J Cell Physiol. 233:1312–1320.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ma L, Yu HJ, Gan SW, Gong R, Mou KJ, Xue J
and Sun SQ: p53-Mediated oligodendrocyte apoptosis initiates
demyelination after compressed spinal cord injury by enhancing
ER-mitochondria interaction and E2F1 expression. Neurosci Lett.
644:55–61. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhan L, Huang C, Meng XM, Song Y, Wu XQ,
Miu CG, Zhan XS and Li J: Promising roles of mammalian E2Fs in
hepatocellular carcinoma. Cell Signal. 26:1075–1081. 2014.
View Article : Google Scholar : PubMed/NCBI
|