Introduction
Non-small cell lung cancer (NSCLC) is one of the
most common malignancies, leading in both the incidence and
mortality rates worldwide (1).
According to recent reports, metastatic NSCLC contributes to rising
morbidity and underlies the majority of lung cancer-related deaths
(2,3). Although significant progress has been
made in the identification of the factors involved in NSCLC
metastasis, the pathophysiological functions and mechanisms
underlying these aberrant factors remain unclear. Estrogen
receptors (ERs) are members of the nuclear steroid receptor
superfamily; ERs mediate cellular responses to the hormone estrogen
(4). Estradiol (E2), which is also
known as 17β-estradiol, is the major and most potent product
synthesized during estrogen biosynthesis, which can bind ERs and
activate rapid cytoplasmic kinase signaling (5). In previous studies, we observed that
activation of ERβ by estrogen in lung cancer significantly promotes
tumor metastasis by increasing the expression of
invasiveness-associated matrix metalloprotease 2 (MMP2) in
vitro and in vivo (6).
Consistent with our results, another clinical study that included
over 16,000 postmenopausal female patients in the Women's Health
Initiative found that higher incidence of and mortality from lung
cancer occurred in women who received daily hormone replacement
therapy (HRT) for over 5 years (7).
However, a recent randomized phase II study, which examined the
approach of coupling anti-estrogen drugs with traditional target
tyrosine kinase inhibitor (TKI) therapy, found no significant
differences from the results obtained using TKI therapy alone
(8). Therefore, understanding the
factors contributing to cancer metastasis is beneficial for the
development of effective therapeutic strategies. In this study, we
focused on revealing the unknown mechanisms and key signaling
pathways that promote E2-induced metastasis in NSCLC.
Toll-like receptors (TLRs) are a highly conserved
family of transmembrane pattern recognition receptors that bind
pathogen-associated molecular patterns (9). TLRs play a crucial role in
inflammation and innate host defense against invading
microorganisms by recognizing conserved motifs of microbial origin
(9,10). TLR4 is specifically activated by
lipopolysaccharide (LPS) from gram-negative bacterial cell walls
(11). Activation of TLR4 on the
respiratory epithelium activates myeloid differentiation primary
response 88 (myd88) adaptor protein-dependent signaling; this
phosphorylates and activates downstream signaling pathways, leading
to host defense responses, including the production of inflammatory
cytokines (12,13). TLR4 signaling plays an important
role in maintaining tissue homeostasis, a process that is
deregulated in cancer. Recent studies have shown that TLR4
activation activates mitogen-activated protein kinases (MAPKs),
such as P38MAPK, ERK1/2, and JNK, leading to augmented NSCLC cell
adhesion, migration, and metastasis in vitro and in murine
NSCLC-metastasis models (14).
Correspondingly, the knockdown of TLR4 can significantly suppress
constitutive phosphorylation of AKT and PI3K, thereby contributing
to the inhibition of human NSCLC cancer cell growth and
inflammatory cytokine secretion in vitro and in vivo
(15). In this process, the
phosphorylation of P65 nuclear factor (NF)-κB and upregulation of
MMP2 play crucial roles in promoting tumor metastasis induced by
TLR4 activation (16,17); this interaction has also been
recognized as the TLR4/myd88/NF-κB/MMP2 axis in signaling pathways
that contribute to cancer metastasis. In vivo, LPS, which is
specifically recognized by TLR4, increases the growth of
experimental metastases in murine tumor models. How exposure to LPS
affects key determinants of MMP2 overexpression has been examined
previously (16).
Activated ERβ and TLR4 may similarly enhance the
invasiveness of NSCLC tumor cells. MMP2, whose expression is
upregulated by treatment with estradiol, can also be overexpressed
by exposure to LPS. When TLR4 activates downstream signaling
pathways involving P38MAPK or ERK1/2-AKT, it also triggers the
activation of ERβ in the cytoplasm of NSCLC cells (4). In the present study, we used NSCLC
cell lines A549 and H1793 to examine the role of ERβ in the
activation of TLR4/myd88 signaling and in tumor progression and
metastasis. We also used an NSCLC model to explore the
tumor-promoting effect of the combined administration of estradiol
and LPS in vitro and in vivo. Our results will help
determine how estrogen contributes to NSCLC metastasis, which may
lead to new therapies against advanced NSCLC.
Materials and methods
Clinical samples, clinicopathological
analysis, and tissue microarray (TMA)
This study was approved by the Institutional Ethics
Committee of Tongji Medical College, Huazhong University of Science
and Technology (IRB ID no. 20141101). Paired samples of primary
NSCLC tumors and corresponding non-tumorous lung tissues from 241
Chinese patients were obtained at the time of surgical resection at
the Department of Thoracic Surgery, Affiliated Tongji Hospital of
Huazhong University of Science and Technology Tongji Medical
College (Wuhan, China) from August 2013 to September 2015. None of
the patients underwent chemotherapy or radiotherapy before surgery.
Patient demographics, including gender, age, smoking history,
pathological diagnosis, pathological tumor-node-metastasis stage
(18), and tumor differentiation
grade, were obtained from the Tongji Hospital records. Baseline
characteristics of the patients are documented in Table I.
 | Table I.Correlation of ERβ and TLR4
expression with clinicopathological features in 241 cases of
non-small cell lung carcinoma. |
Table I.
Correlation of ERβ and TLR4
expression with clinicopathological features in 241 cases of
non-small cell lung carcinoma.
|
|
| ERβ expression |
|
| TLR4
expression |
|
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| Patient
characteristics | No. of patients
(%) | High | Low | χ2 | P-value | High | Low | χ2 | P-value |
|---|
| Total patient
no. | 241 |
|
|
|
|
|
|
|
|
| Sex |
|
|
| 0.571 | 0.450 |
|
| 0.02 | 0.886 |
|
Female | 69
(28.63) | 35 | 34 |
|
| 35 | 34 |
|
|
|
Male | 172 (71.36) | 78 | 94 |
|
| 89 | 83 |
|
|
| Age (years) |
|
|
| 1.827 | 0.176 |
|
| 0.825 | 0.364 |
|
<60 | 139 (57.26) | 60 | 79 |
|
| 75 | 64 |
|
|
|
≥60 | 102 (42.74) | 53 | 49 |
|
| 49 | 53 |
|
|
| Smoking |
|
|
| 0.892 | 0.345 |
|
| 2.083 | 0.149 |
| Ex | 133 (55.19) | 66 | 67 |
|
| 74 | 59 |
|
|
|
Never | 108 (44.81) | 47 | 61 |
|
| 50 | 58 |
|
|
| T stage |
|
|
| 2.411 | 0.121 |
|
| 0.758 | 0.384 |
|
1a-2b | 190 (78.84) | 94 | 96 |
|
| 95 | 95 |
|
|
|
3–4 | 51
(21.16) | 19 | 32 |
|
| 29 | 22 |
|
|
| Lymph node
metastasis |
|
|
| 2.145 | 0.143 |
|
| 5.732 | 0.017 |
|
Yes | 118 (48.96) | 61 | 57 |
|
| 70 | 48 |
|
|
| No | 123 (51.04) | 52 | 71 |
|
| 54 | 69 |
|
|
| Metastasis |
|
|
| 1.000 | 0.585 |
|
| 0.765 | 0.471 |
|
Yes | 11 (4.57) | 5 | 6 |
|
| 5 | 6 |
|
|
| No | 230 (95.43) | 108 | 122 |
|
| 118 | 112 |
|
|
| TNM stage |
|
|
| 0.003 | 0.958 |
|
| 3.180 | 0.075 |
|
I–II | 151 (62.66) | 71 | 80 |
|
| 71 | 80 |
|
|
|
III–IV | 90
(37.34) | 42 | 48 |
|
| 53 | 37 |
|
|
| Tumor
histology |
|
|
| 0.141 | 0.707 |
|
| 10.152 | 0.001 |
|
SCC | 84
(34.85) | 38 | 46 |
|
| 55 | 29 |
|
|
|
ADC | 157 (65.14) | 75 | 82 |
|
| 69 | 88 |
|
|
| Tumor
differentiation |
|
|
| 0.025 | 0.873 |
|
| 0.552 | 0.457 |
|
Well/Moderate | 193 (80.09) | 90 | 103 |
|
| 97 | 96 |
|
|
|
Poor | 48
(19.91) | 23 | 25 |
|
| 27 | 21 |
|
|
Among the 241 patients, metastatic lymph nodes were
obtained from 30 patients with paired primary tumors; metastatic
lymph nodes were obtained via surgical resection of the primary
tumor with lymph node dissection. Patients with lymphadenitis and
primary malignancies of the lymph node were excluded. Details on
the 30 cases with IIA-IIIB NSCLC with lymph node metastasis have
been provided in a previous study from our group (6). Palliative care or surgical biopsy was
administered, after obtaining informed consent, to 32 patients with
inoperable stage IIIb–IV primary NSCLC.
Fresh tissues were immediately snap-frozen and
stored at −80°C, or formalin-fixed and embedded in paraffin.
Samples were diagnosed and confirmed by at least two lung cancer
pathologists. TMA was prepared by Outdo Biotech Co., Ltd.
(Shanghai, China). All patients had provided their written,
informed consent to tissue collection prior to sampling, and all
procedures involving human samples were conducted in accord with
the Declaration of Helsinki.
Immunohistochemical (IHC)
analyses
Sample processing and immunohistochemistry were
performed as previously described (6). Rabbit anti-human TLR4 polyclonal
antibody (Ab) (dilution 1:100, cat. no. ab13556) and rabbit
anti-human ERβ monoclonal Ab (dilution 1:100, cat. no. ab3577) were
purchased from Abcam. Protein expression levels were scored
independently by two pathologists. Immunoreactivity scores of
cancer-tissue samples were determined based on staining intensity
and area of positive staining according to the method described in
Tang et al (19). Positive
cells were scored as follows: 1, ≤20% positive cells; 2, 20–50%
positive cells; 3, 50–75% positive cells; and 4, >75% positive
cells. Staining intensity was evaluated as follows: 1, negative; 2,
weakly positive; 3, moderately positive; and 4, strongly positive.
A score of 1–16 was obtained by multiplying the staining intensity
and proportion of positive cells: (−), ≤4; (+), >4 and ≤; (++),
>8 and ≤ 12; and (+++), >12 and ≤16. A total score >12 was
defined as high expression, and a score ≤8 was defined as low
expression.
Cell lines and culture conditions
Human NSCLC cell lines PC9, A549, H1793 and H1975
were purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA), grown for 2 weeks, and passaged four times
before freezing aliquots for subsequent analyses. The cell lines
were tested and authenticated by ATCC. The normal human bronchial
epithelial (HBE) cell line was obtained from the Shanghai Cancer
Institute (Shanghai, China). PC9, A549, H1975 and HBE cells were
cultured in Roosevelt Park Memorial Institute Medium (RPMI)-1640
medium, and H1793 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM):nutrient mixture F12. All media (HyClone; GE
Healthcare) were supplemented with 10% fetal bovine serum (FBS;
Clark Bioscience). Cells were incubated in a humidified atmosphere
with 5% CO2 and 95% air at 37°C.
Cell transfection
The expression vectors and small interfering RNAs
(siRNAs) targeting ERβ were constructed as previously described
(6,20). Briefly, TLR4- or ERβ-expressing
plasmids (pcDNA3.1-TLR4 or -ERβ) and corresponding empty plasmids
were obtained from OriGene Technologies, Inc. siRNAs targeting TLR4
or ERβ (TLR4-siRNA or ERβ-siRNA Stealth siRNAs HSS103378; Life
Technologies) and control siRNAs (ctrl-siRNA or siRNA-NC; Life
Technologies) were constructed using a gene silencing vector.
Lipofectamine 3000 (Invitrogen; Thermo Fisher Scientific, Inc.) was
used during transfection following the manufacturer's instructions.
TLR4 and ERβ protein expression was analyzed by western blotting
following transfection with plasmids or siRNAs.
Drug exposure
Parental NSCLC cells and mice in the metastatic
model were exposed to E2 (Sigma-Aldrich; Merck KGaA),
4,40,400-(4-propyl-[1H]-pyrazole-l,3,5-triyl), LPS (isolated from
Escherichia coli 0111:B4; Sigma-Aldrich; Merck KGaA), fulvestrant
(Ful; an ER antagonist; Cayman Chemical), and TAK-242 (also known
as CLI-095; InvivoGen), either alone or in combination. The dosages
of each drug, used in vitro and in vivo, were used as
previously (21–24). Each group of cells was treated for
48 h and harvested for further analysis. Cell culture experiments
were performed using reagents dissolved in 100% dimethyl sulfoxide
(DMSO), and DMSO was also used as a control (vehicle).
Western blot analysis
Western blotting was performed as previously
described (6). The primary antibody
(Ab) used for western blots included rabbit anti-human ERβ
(dilution 1:1,000) from Abcam (cat. no. ab3577), rabbit anti-human
TLR4 (dilution 1:1,000) from Abcam (cat. no. ab13556), mouse
anti-human MMP-2 (dilution 1:500) from Santa Cruz Biotechnology
(cat. no. sc-53630), rabbit anti-human P38MAPK (dilution 1:500,
cat. no. AP0424)/p-P38MAPK (dilution 1:500, cat. no. BS4844), and
rabbit anti-human tAkt (dilution 1:500, cat. no. AP0485)/p-Akt
(dilution 1:500, cat. no. AP0484) from Bioworld Technology, rabbit
anti-human P65NF-κB (dilution 1:1,000, cat. no. ab16502)/p-P65NF-κB
(dilution 1:1,000, cat. no. ab86299) from Abcam, rabbit anti-human
myd88 (dilution 1:1,000) from Proteintech (cat. no. 23230-1-AP, and
mouse anti-human GAPDH (dilution 1:10,000) from Cell Signaling
Technology (cat. no. 51332). The densitometry of the western blots
was analyzed by ImageLab software (version 6.0.0; Bio-Rad
Laboratories, Inc.).
Co-immunoprecipitation
Cultured A549 cells were placed into 10-cm dishes
and transfected with empty vector pcDNA3.1-ERβ, pcDNA3.1-TLR4,
siRNA-ERβ, or siRNA-TLR4. The cells were then washed in 1X
phosphate-buffered saline (PBS) and resuspended in 1 ml of NP-40
lysis buffer (70 mM NaCl, 50 mM Tris pH 8, and 0.5% NP-40)
supplemented with phosphatase inhibitor and protease inhibitor
cocktails (Sigma-Aldrich; Merck KGaA), and the cell lysates were
ultrasonicated. After rotating at 4°C for 30 min, the cell lysates
were collected and precleared by centrifugation at 12,000 rpm
(12,800 g rcf) at 4°C for 10 min. Protein concentration was
assessed using 1 mg of total protein via the Bradford Assay
(Bio-Rad Laboratories, Inc.). For co-immunoprecipitation of
endogenous ERβ with TLR4, total protein was isolated from A549
cells as described previously (22). For each pull-down, 5 µg of anti-ERβ
Ab (dilution 1:200, cat. no. ab3577; Abcam) or anti-TLR4 Ab
(dilution 1:200, cat. no. ab30667; Abcam) was added to the
normalized lysate, and the mixture was incubated overnight at 4°C.
Normal rabbit IgG (Santa Cruz Biotechnology) was used as the
negative control. Immune complexes were then precipitated with
protein A/G plus agarose (Thermo Fisher Scientific, Inc.).
Immunoprecipitates were collected by centrifugation (2,850 × g rcf,
4°C, 3 min) and washed gently with lysis buffer. Immunoprecipitated
samples were resolved by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS/PAGE) for immunoblotting.
Wound-healing assay
NSCLC cell lines A549 and H1973 were cultured in a
6-well culture plate until 90% confluence. The cell monolayers were
then scratched using a 200 µl sterile pipette tip. Cell migration
was determined by measuring the movement of cells into the
scratched area. Representative images (×40) of wound closure were
captured at 0 and 24 h using an inverted light microscope (Olympus
Corp.).
Cell migration and invasion
assays
Transwell® Permeable Supports (inserts
6.5 mm in diameter; Corning, Inc.) were used for the cell migration
assay. For the cell invasion assay, Transwell® Permeable
Support inserts were coated with BD Matrigel™ Basement Membrane
Matrix (BD Biosciences). Cells (A549 and H1793) suspended in
serum-free media were added to the upper chamber at various
densities depending on the cell line. Migration and invasion were
evaluated based on the number of cells invading the
Transwell® membrane, and counting was performed using an
Olympus microscope (Olympus Corp.) at ×100 magnification. Four
fields were randomly selected for analysis. Detailed procedures are
described elsewhere.
Immunofluorescence analysis
After 48 h of treatment with specific drugs or
combinations of drugs, A549 and H1793 cells were seeded overnight
on coverslips in a 24-well plate. After the cells reached 50%
confluence, they were washed in 1X PBS, fixed with 4%
paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100
for 15 min, blocked in 5% goat serum at room temperature (RT) for
30 min, and incubated overnight at 4°C with the indicated primary
Ab. Next, cell-seeded coverslips were rewarmed for 1 h and
incubated with a specific secondary Ab for 2 h at 37°C. Nuclei were
stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min at RT.
Next, the samples were washed in 1X PBS for 5 min four times.
Finally, the coverslips were observed under a fluorescence
microscope (Olympus Corp.; magnification of ×200).
3D spheroid invasion assay
The 3D invasion assay was described previously
(25). The Cultrex® 3D
Spheroid Cell Invasion Assay kit (Trevigen) was utilized in this
procedure. Briefly, 1×105 cells in 500 ml fresh culture
media containing 2.5% Matrigel and 5 ng/ml spheroid formation
extracellular matrix (ECM) were plated into 24-well plates coated
with a collagen/Matrigel mixture. The spheres with protrusions were
considered positive for cell invasion. Distances between the
invasive cell frontier and the spheroid edge were measured on day
1, 3 and 6 using an Olympus IX70 (Olympus) inverted microscope
(magnification of ×100). Each experiment was repeated twice, and
each procedure was performed in triplicate.
Fluorescent gelatin degradation
assay
Coverslips were cleaned with 20% nitric acid and
coated with poly-L-lysine in a 24-well plate. Poly-L-lysine was
fixed with 0.5% glutaraldehyde before adding fluorescein
isosthiocynanate-conjugated (FITC) gelatin (QCM™ Gelatin
Invadopodia Assay kit (Green), EMD Millipore). A thin layer of
FITC-conjugated gelatin was placed onto the coverslips and was
crosslinked using glutaraldehyde on ice for 10 min. Crosslinking
was continued at RT for an additional 30 min. The coverslips were
rinsed with PBS, incubated with 5 mg/ml sodium borohydride at RT
for 3 min, rinsed again with PBS, incubated with 70% ethanol for 10
min, and dried at 37°C for 15 min in a CO2 incubator.
One hour before plating the cells, the coverslips were quenched
with RPMI-1640 containing 10% FBS at 37°C. The cells were plated on
FITC-gelatin-coated coverslips and cultured in RPMI-1640 for 24 h
to quantify formation of invadopodia. Images were visualized by
confocal microscopy (Olympus FV1200; magnification of ×200).
Gelatin zymography
After 48 h of treatment with specific drugs or
combinations of drugs, the cells were washed and incubated in
serum-free medium for 24 h. MMP2 activity was measured by gelatin
zymography. Samples were electrophoresed using 10% SDS-PAGE
containing gelatin. After electrophoresis, the gel was washed four
times with washing buffer [50 mM Tris-HCl (pH 7.5), 100 mM NaCl,
and 2.5% Triton X-100], followed by a brief rinse in washing buffer
without Triton X-100. The gel was incubated with incubation buffer
[50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM CaCl2,
0.02% NaN3, and 1 mM ZnCl2] at 37°C for 20 h.
After incubation, the gel was stained by Coomassie Blue and
destained. A clear zone of gelatin digestion in the gel indicated
the presence of MMP2 (55 kDa).
Mouse lung metastasis model
Non-obese diabetic/severe combined immunodeficiency
(NOD/SCID) female mice (4 weeks old; average weight, 15 g) were
obtained from the Experimental Animal Center of Hubei Province
(Animal Study Permit no. SCXK 2013-0004). The mice were maintained
under specific pathogen-free conditions (System Barrier Environment
no. 00127070) in the Experimental Animal Center of Tongji Hospital
of Huazhong University of Science and Technology. All experiments
were carried out according to the regulations specified by the
Ethics Committee for Tongji Hospital of Huazhong University of
Science and Technology. Mice were first intraperitoneally
anesthetized with 1% sodium pentobarbital (50 mg/kg of body weight,
Sigma-Aldrich, Merck KGaA) and received ovariectomy to eliminate
the effects of endogenous estrogen. Next, A549 cells
(5×106/100 µl) suspended in PBS were injected into the
4-week-old female NOD/SCID mice via the tail vein. Seven days after
injection of the cells, the mice were randomly divided into six
groups (n=5/group): mice exposed to E2 (0.09 mg/kg), mice
administered E2+Ful (1.46 mg/kg), mice treated with Ful alone, mice
exposed to LPS (10 mg/kg), mice exposed to a combination of E2+LPS,
and negative control mice. The above-mentioned agents and
combinations of agents were administered by subcutaneous injection
twice per week for 10 weeks.
At week 10 after exposure, the mice were humanely
euthanized by continuous inhalation with 30% CO2 for 5
min, and were observed for 5 min to ensure the vital activity
stopped. All of the experimental mice were sacrificed at the humane
endpoint 10 weeks, to ensure the tumor progression time of each
group was consistent. After that the lungs were surgically removed.
The total number of lung nodules, lung wet weights, and lung cancer
metastatic indices were statistically analyzed as previously
described (6). The carcinoma
tissues were then excised from the lungs; 10 mg of carcinoma tissue
per mouse was homogenized in 200 ml radioimmunoprecipitation assay
buffer A and subjected to western blotting.
Bioinformatics analysis
The prognostic value of ERβ/TLR4 was analyzed using
the web-based Kaplan-Meier plotter (http://www.kmplot.com/lung), which is a meta-analysis
tool for examining gene expression and survival data of 2,437
patients with lung cancer (2018 version) using multiple microarray
data.
Statistical analysis
All experiments were repeated at least twice, and
each procedure was performed in triplicate. All data were examined
at least three times. Quantitative data are expressed as means ±
SD. Statistical significance was established using the SPSS 19.0
statistical software package (SPSS, Inc.). A two-tailed P-value
<0.05 was considered statistically significant.
Results
Overexpression of ERβ in NSCLC primary
tumor tissues and metastatic lymph nodes correlates with TLR4
expression
Our previous research and recent studies revealed
that ERβ and TLR4 are overexpressed in the cytoplasm and nuclei of
NSCLC tumor tissues (15,21,22).
To identify the potential roles of ERβ and TLR4 in the development
and progression of NSCLC, we collected NSCLC samples from 241
patients with NSCLC and evaluated the expression of ERβ and TLR4
using immunohistochemistry. Our results showed that expression of
ERβ was negative in adjacent non-tumor lung tissues, but positive
in tumor tissue, and that TLR4 was highly expressed in NSCLC and
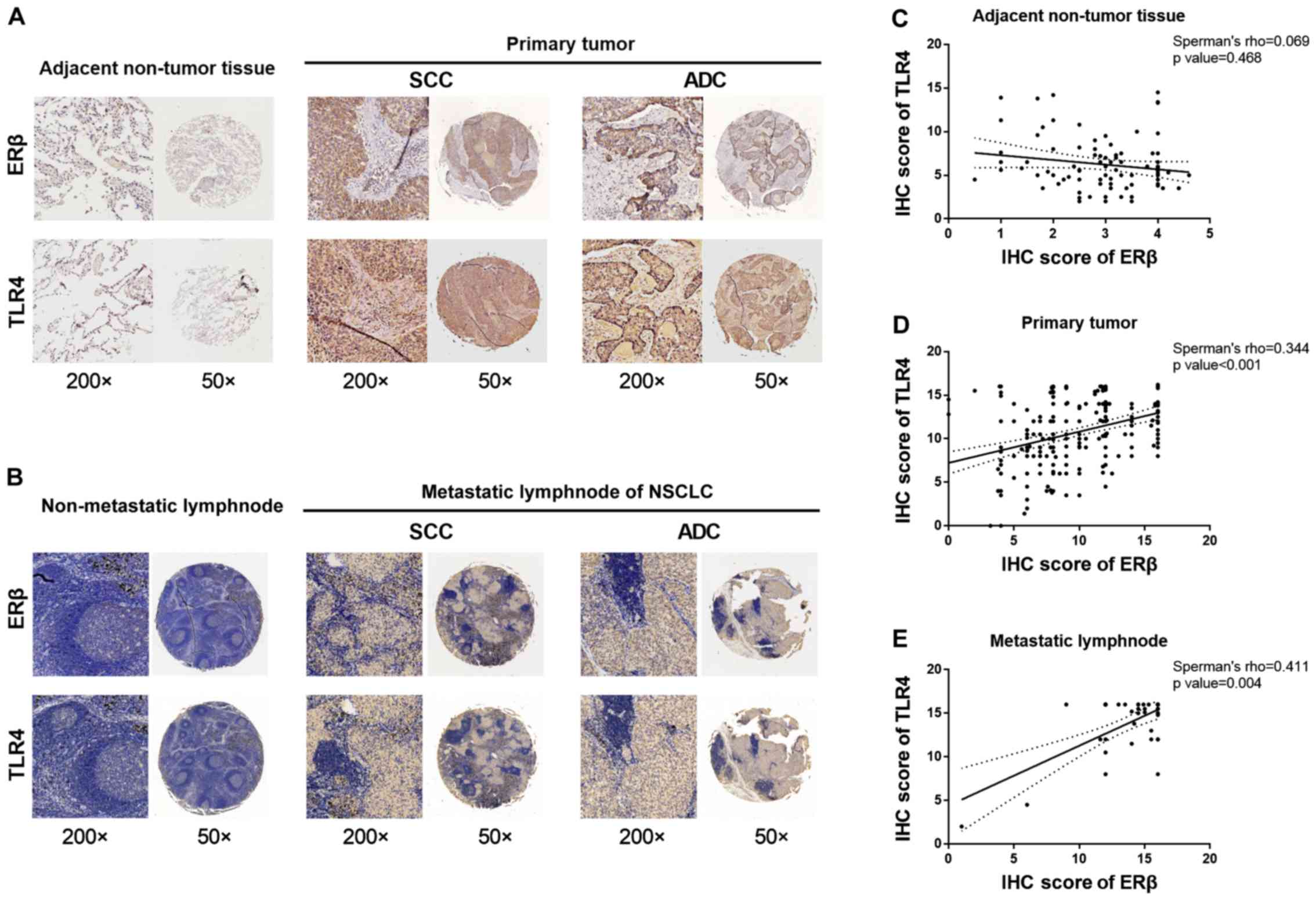
normal lung tissues (Fig. 1A).
Analysis of clinicopathological features indicated that high
expression of TLR4 was significantly correlated with lymph node
metastasis (χ2=5.732, P=0.017) and adenocarcinoma
histological type (χ2=10.152, P=0.001; Table I). However, other
clinicopathological features, including sex, age, smoking status,
tumor stage, distant metastasis, and pathological
tumor-node-metastasis stage, were not directly associated with the
expression of ERβ or TLR4 (Table
I).
We next used immunostaining to examine the
association between expression of ERβ and that of TLR4 in adjacent
non-tumor tissues, and in primary NSCLC tissues and metastatic
lymph nodes. The results showed the correlation between ERβ and
TLR4 co-expression has an increasing Spearman's rank correlation
coefficient (Spearman's ρ for short), which showed a significant
positive correlation with cancer metastasis stage (adjacent
non-tumor lymph nodes, primary tumor, and metastatic lymph nodes).
These results are shown in Fig. 1B.
The percentage of ERβ-positive stained cells was highly correlated
with the level of TLR4 overexpression in metastatic lymph nodes
(Spearman's correlation coefficient, rs=0.411,
P=0.004; Table II and Fig. 1E). However, no significant
correlation was observed between the expression levels of ERβ and
TLR4 in non-tumor lung tissue (rs=0.069, P=0.468;
Table II and Fig. 1C). A slightly stronger correlation
between the expression levels of ERβ and TLR4 was observed in
primary tumor tissue (rs=0.344, P<0.001;
Table II and Fig 1D). Detailed IHC scores for ERβ and
TLR4 expression in primary NSCLC tumors and metastatic lymph nodes
and in adjacent non-tumor tissue are shown in Fig. S1A and Table SI. Altogether, these data indicate
a positive correlation between protein expression levels of ERβ and
TLR4 in clinical NSCLC tissues.
| Table II.Correlation between ERβ expression
and TLR4 in the 241 cases of NSCLC. |
Table II.
Correlation between ERβ expression
and TLR4 in the 241 cases of NSCLC.
| Tissues | Expression |
TLR4+ |
TLR4− | P-value | Sperman's ρ |
|---|
| Adjacent
non-tumor |
ERβ+ | 19 | 2 |
0.468 | 0.069 |
|
|
ERβ− | 70 | 22 |
|
|
| Primary tumor |
ERβ+ | 79 | 34 |
<0.001 | 0.344 |
|
|
ERβ− | 45 | 83 |
|
|
| Metastatic lymph
node |
ERβ+ | 22 | 5 |
0.004 | 0.411 |
|
|
ERβ− | 10 | 11 |
|
|
Estrogen significantly upregulates the
expression of TLR4 and that of the downstream myd88/NF-κB/MMP2 axis
via ERβ signaling in NSCLC cell lines
LPS is a significant component of the outer
membranes of gram-negative bacteria; LPS triggers TLR4 signaling
(13,26). However, it remains unclear whether
and how estrogen activates the signaling of ER, TLR4, and its
downstream myd88/NF-κB/MMP2 axis. To evaluate the influence of
estrogen on the progression of NSCLC, we first exposed NSCLC cell
lines to a gradient dose (at 0, 0.1, 1, 10, 100 and 1,000 nM) and
then compared the results with those of another group treated with
100 mM E2 at successive time intervals of 0, 1, 6, 12, 24 and 48 h.
The results showed that TLR4 was markedly increased after E2
treatment in a dose- and time-dependent manner (Fig. 2A and B; Fig. S2A and B), and ERβ and
invasiveness-associated MMP2 were also increased. We then
investigated the effects of different treatments on A549 cells by
administering DMSO (vehicle/negative control), E2, E2+Ful, or Ful.
Our results indicated that the expression levels of TLR4 and MMP2
were significantly increased in the E2-treated group, reduced after
treatment with E2+Ful, and reduced after treatment with Ful alone
(P<0.05) (Fig. 2D and Fig. S2D). The baseline expression status
of ERβ, MMP2, and TLR4 in different cell lines is shown in Fig. 2C and Fig. S2C. As shown in Fig. 2D and Fig. S2D, western blotting analysis
further demonstrated an increase in the protein expression of myd88
and phosphorylated p-p65NF-κB, while the total levels of p65NF-κB
remained unchanged in the A549 cells. To support these findings, we
evaluated the expression of ERβ and TLR4 in A549 and H1793 cells
using immunofluorescence analysis. TLR4 overexpression was
associated with ERβ activation following exposure to E2 (Fig. 2E and F). Expression of TLR4 and the
activation of its signaling pathways were markedly affected by E2.
These results indicate a positive correlation between the
expression of ERβ and that of TLR4 during NSCLC metastasis.
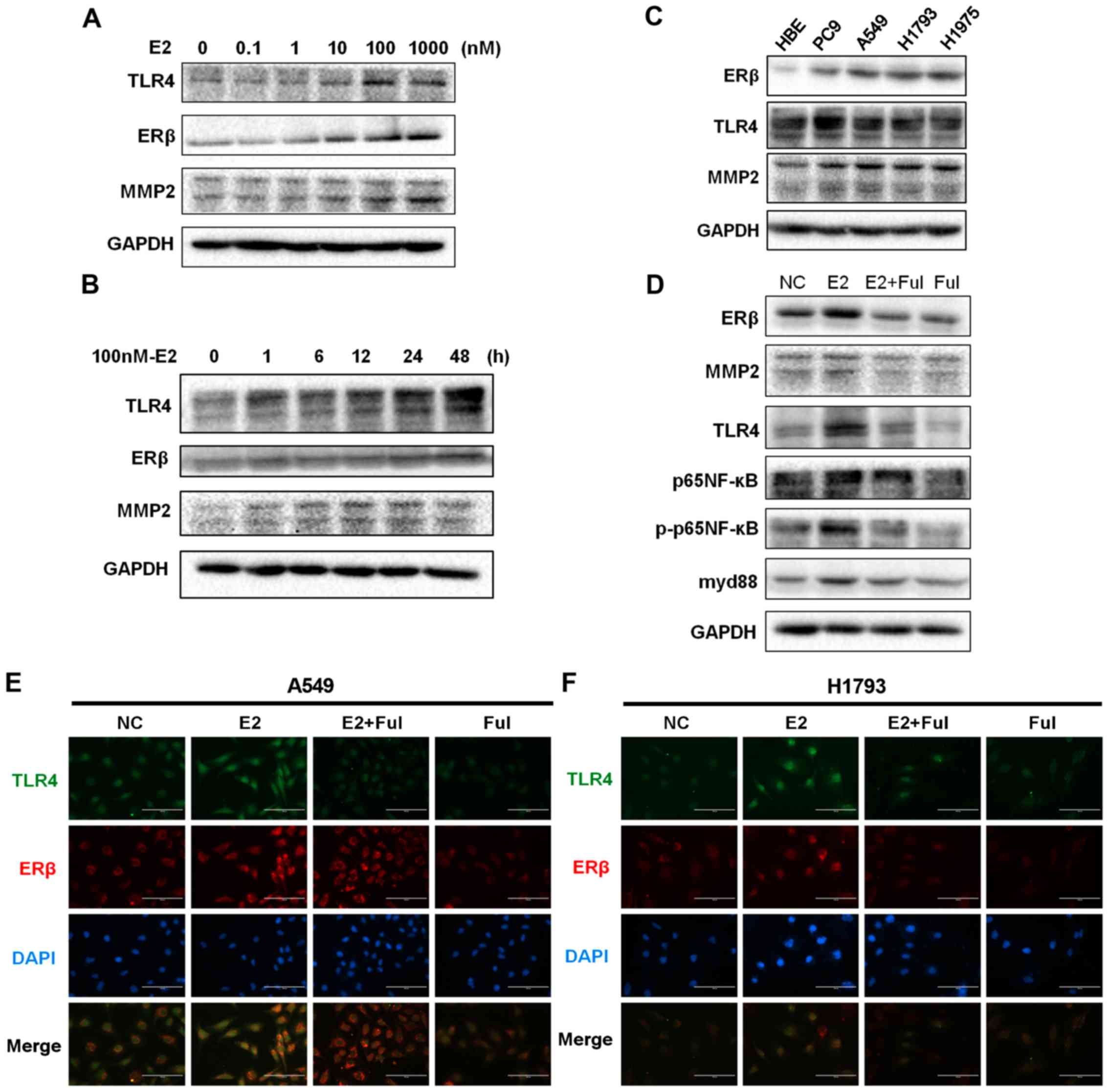 | Figure 2.Protein expression of ERβ, TLR4, and
the downstream myd88/NF-κB/MMP2 axis in NSCLC cell lines treated
with estrogen and estrogen inhibitors. (A) The synchronized cells
were exposed to E2 at different concentrations (0, 0.1, 1, 10, 100
and 1,000 nM) for 48 h. Protein expression of ERβ, TLR4, and MMP2
was analyzed using western blot analysis. The data represent mean ±
SEM from three different experiments. E2 stimulated ERβ, TLR4 and
MMP2 response in the A549 cell line in a dose-dependent manner. (B)
The synchronized cells were treated with E2 at different time
points (0, 1, 6, 12, 24 and 48 h) at concentrations of 100 nM. The
data represent means ± SEM from three different experiments. E2
stimulated ERβ, TLR4, and MMP2 response in a time-dependent manner
in the A549 cells. GAPDH expression was used as a control. (C)
Western blot analysis of ERβ, TLR4 and MMP2 expression in cultured
NSCLC cell lines (PC9, A549, H1793 and H1975) and normal bronchial
epithelial cell line (HBE). (D) Western blot analysis of ERβ, TLR4,
MMP2, p65NF-κB, phosphorylated (p)-p65NF-κB, and myd88 protein
levels at 48 h in A549 cells. Estrogen exposure significantly
upregulated the expression of TLR4 and activated the
myd88/NF-κB/MMP2 pathway, while anti-estrogen drugs showed the
opposite effect. GAPDH expression was used as a control. (E and F)
Immunofluorescence staining of ERβ and TLR4 in A549 and H1793
cells. Scale bar, 200 µm. NSCLC, non-small cell lung cancer; MMP2,
matrix metalloprotease 2; ERβ, estrogen receptor β; TLR4, Toll-like
receptor 4; myd88, myeloid differentiation primary response 88;
NF-κB, nuclear factor-κB. |
Inhibition of ERβ expression by siRNA
decreases TLR4 expression
To investigate how ERβ expression affects the
expression of TLR4, ERβ-targeting siRNA or NC siRNA was transfected
into NSCLC A549 and H1793 cells to generate a specific
ERβ-knockdown cell model. In contrast, an ERβ-overexpression cell
model was generated by transfecting an ERβ expressing pcDNA-plasmid
into the A549 and H1793 cells. The changes in ERβ and TLR4
expression were evaluated by western blot and immunofluorescence
analyses (Fig. 3A and B; Fig. S3A). Our results revealed that
removal of endogenous ERβ expression significantly suppressed the
expression of TLR4, while overexpression of ERβ significantly
promoted the expression of TLR4. In addition, inhibition of ERβ
expression significantly decreased expression of MMP2 and myd88.
These results indicate that the expression levels of ERβ and TLR4
were positively associated with NSCLC progression.
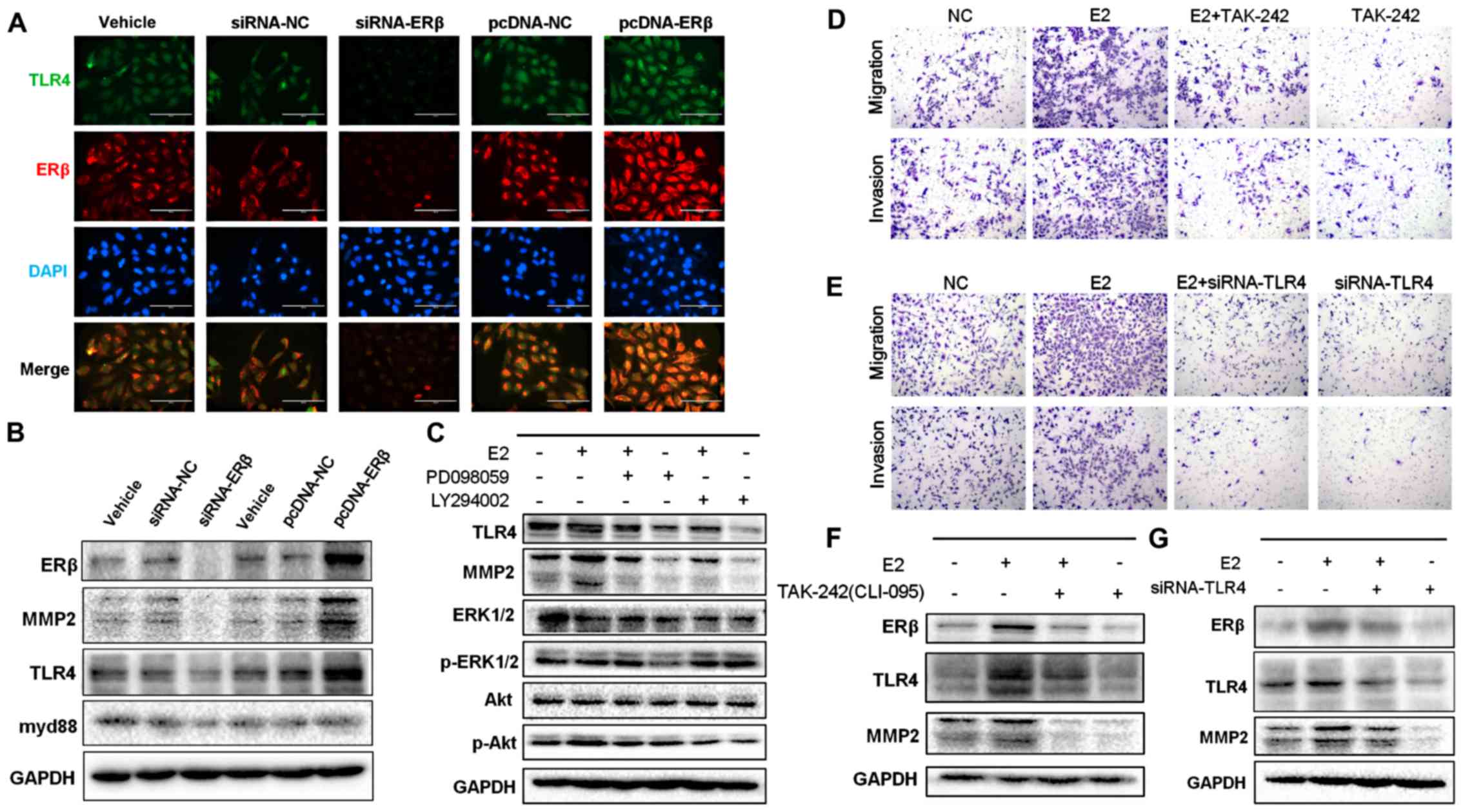 | Figure 3.Protein expression of ERβ and TLR4
under RNAi intervention or overexpression of ERβ. (A)
Immunofluorescence staining of ERβ and TLR4 in siRNA-ERβ- and
pcDNA-ERβ-treated A549 cells. Scale bar, 200 µm. (B) Decreased
protein levels of TLR4, MMP2, and myd88 in siRNA-ERβ-treated A549
cells compared with those in siRNA-NC-treated cells as evaluated by
western blotting. The panel also showed increased expression of
TLR4, MMP2, and myd88 in cells treated with pcDNA-ERβ. (C) Western
blot analysis of TLR4, MMP-2, p-p38MAPK, P38MAPK, pAKT, and AKT
protein levels in A549 cells treated with E2, PD098059, or
LY294002, in combination or alone. The upregulation of TLR4
signaling pathway by E2 was inhibited when either ERK/MAPK or
PI3K/AKT was blocked. GAPDH expression was used as a control. (D)
Representative images of Transwell assays used to assess E2-induced
NSCLC cell invasiveness and migration with or without treatment
with TAK-242, a specific antagonist of TLR4. TLR4 blocked by
TAK-242 impaired the invasiveness of NSCLC cells treated with E2.
(E) Representative images of Transwell assays used to assess
E2-induced NSCLC cell invasion and migration with or without
siRNA-TLR4 interference. TLR4 knockdown by siRNA impaired the
invasiveness of A549 cells treated with E2. (F) Representative blot
showing decreased protein expression of MMP2 when TLR4 expression
was blocked by TAK-242. (G) Representative blot showing decreased
protein expression of MMP2 in TLR4 knockdown cells induced by
siRNA. NSCLC, non-small cell lung cancer; MMP2, matrix
metalloprotease 2; ERβ, estrogen receptor β; TLR4, Toll-like
receptor 4. |
Suppression of downstream signaling
pathways impairs the effect of TLR4 upregulation induced by
treatment with estrogen
After treatment with estrogen, cytoplasmic kinase
signaling is rapidly activated via ERβ signaling in seconds to
minutes. This rapid signaling is termed non-genomic and occurs via
non-nuclear ERs located in the membrane or cytoplasm of the cell
(27). Our previous research showed
that MMP2 upregulation is affected by ERK/P38MAPK and PI3K/AKT
signaling pathways activated by estrogen (6). Therefore, we next investigated how the
expression of ERβ affects that of TLR4 and downstream signaling
pathways. For this, several inhibitors were used to determine the
effect of estrogen during blockade of the expression of TLR4/myd88,
ERK/P38MAPK, or PI3K/AKT. TAK-242 is a selective inhibitor that
suppresses the interaction between TLR4 and its adaptor molecules;
this suppression occurs via the intracellular Cys747 residue of
TLR4 (23,28). Our results showed that the
upregulated expression of TLR4 and MMP2 induced by treatment with
estrogen was reversed by treatment with TAK-242 (Figs. 3F and S3E) or with TLR4 siRNA (Figs. 3G and S3F). Next, to support these findings, we
performed Transwell migration/invasion assays. Our results
demonstrated that invasiveness and aggressiveness of the cells,
induced by treatment with E2, were impaired by blocking (Figs. 3D and S3C) or silencing (Figs. 3E and S3D) TLR4 signaling. We then used the pAkt
inhibitor LY294002 and the pERK inhibitor PD098059 to treat A549
cells and found that TLR4 expression was significantly abrogated
when expression of ERK/P38MAPK and PI3K/AKT was suppressed
(P<0.05); this effect was sustained even after exposure to E2
(Figs. 3C and S3B). Altogether, these findings confirm
that promotion of NSCLC cell metastasis induced by E2 may be
reversed by inhibiting the signaling of TLR4. Our results also
showed that activation of the ERK/P38MAPK and PI3K/AKT pathways was
required for ERβ-mediated TLR4 upregulation and was necessary to
enhance migration and invasiveness of NSCLC cells.
ERβ co-localizes with TLR4 in the
NSCLC cell lines
We next used immunofluorescence analysis to evaluate
the potential molecular mechanisms underlying the positive
association between the expression of ERβ and that of TLR4. For
this, A549 cells were treated with 100 nM E2 for 48 h. Indirect
immunofluorescence analysis indicated that endogenous expression of
ERβ and TLR4 was primarily co-localized in the cytoplasm, while the
nuclear region showed sparse co-expression (Fig. 4A). Similar results were observed in
the cytoplasm of another NSCLC cell line, H1793. In this cell line,
co-localization was particularly visible under higher magnification
and at a higher resolution. These data indicate that ERβ and TLR4
proteins likely form a complex or interact with each other.
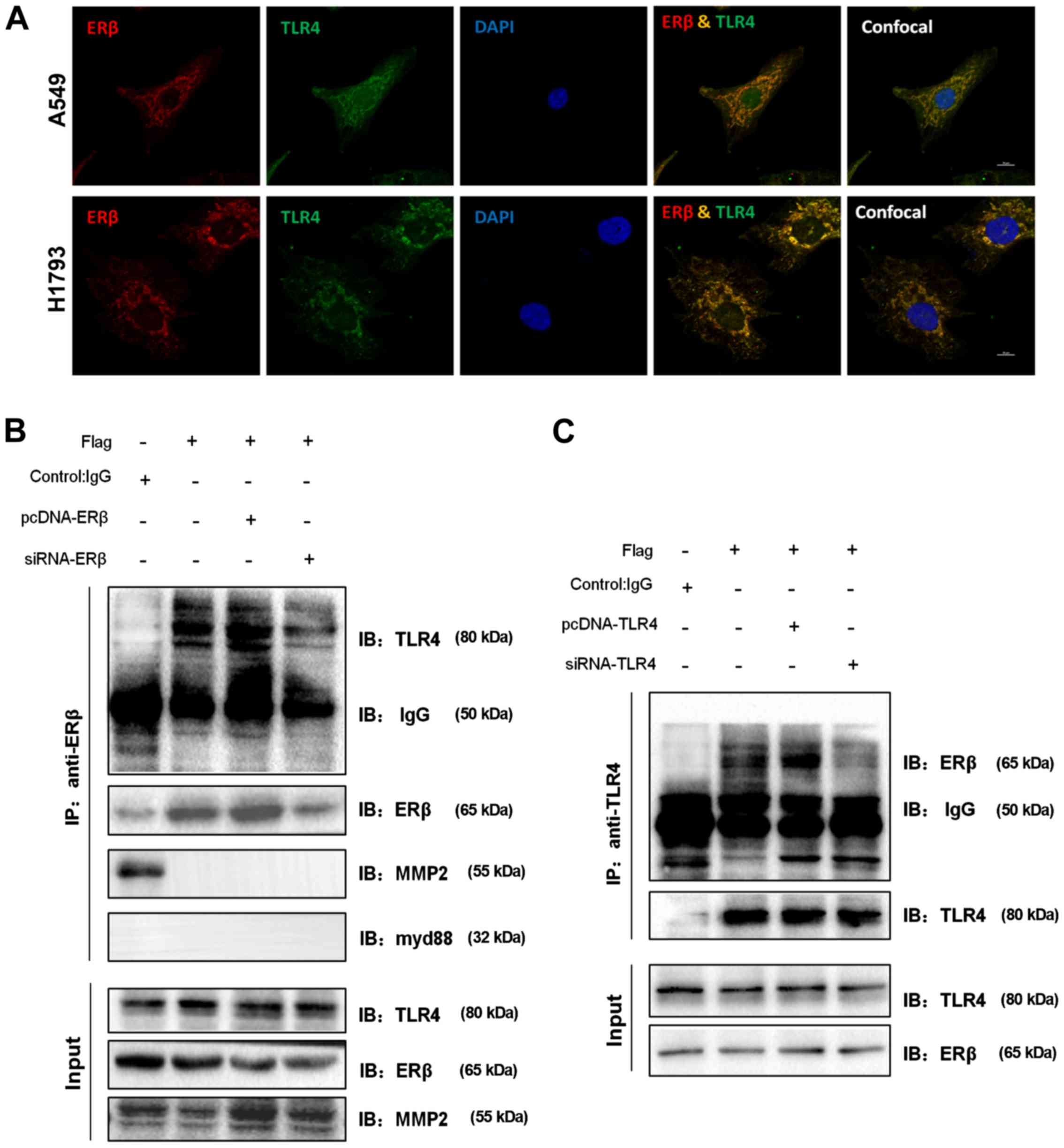 | Figure 4.ERβ co-localizes and interacts with
TLR4 in NSCLC cell lines. (A) Representative confocal images of
cells treated with E2 (100 nM, 48 h) showing co-localization (in
yellow) of endogenous ERβ with TLR4 in A549 and H1793 cells. Scale
bar, 10 µm. (B) ERβ co-immunoprecipitates with endogenous TLR4 but
not with myd88 or MMP2. Cell lysates were prepared from A549 cells
and immunoprecipitated (IP) with rabbit ERβ or control IgG
antibodies. Precipitates were resolved by 10% SDS-PAGE, followed by
immunoblotting (IB) with rabbit TLR4, rabbit myd88, and rabbit
MMP2. Protein levels of TLR4, MMP2, and ERβ in 1% of total input
cell lysate are also shown. (C) TLR4 co-immunoprecipitates with
endogenous ERβ as assessed by co-immunoprecipitation analysis.
NSCLC, non-small cell lung cancer; MMP2, matrix metalloprotease 2;
ERβ, estrogen receptor β; TLR4, Toll-like receptor 4; myd88,
myeloid differentiation primary response 88. |
Novel ERβ-TLR4 interaction
Previous studies have shown that the amino-terminal
region of ER enables it to dimerize or bind to co-activators,
co-repressors, regulators, and ligands. This binding is responsible
for the promotion of enhanced tumorigenesis (27,29).
Our previous findings indicated a positive signaling association
and interaction between ERβ and TLR4. This prompted us to examine
whether there are direct physical and molecular interactions
between ERβ and TLR4. To determine whether endogenous ERβ and TLR4
interact with each other, we co-immunoprecipitated ERβ with TLR4
using cell lysates isolated from A549 cells. TLR4 was readily
detected in ERβ immunoprecipitates (Fig. 4B), while ERβ was found in TLR4
immunoprecipitates (Fig. 4C).
However, the interaction of endogenous myd88 or MMP2 with ERβ was
not detected (Fig. 4B). Hence, our
results indicate that there is an interaction between ERβ and TLR4.
These data suggest that the interaction of ERβ and TLR4 in cellular
cytoplasm contributes to NSCLC metastasis.
Activation of ERβ and TLR4 expression
synergistically promotes migratory and invasive abilities of the
NSCLC cells
After confirming that exposure to E2 induced
endogenous interaction of ERβ and TLR4 and increased TLR4 protein
levels, we assessed the extent to which the metastatic
aggressiveness of the human NSCLC A549 and H1793 cell lines was
mediated by exposure to E2, LPS, and combined administration of
E2+LPS. The wound-healing assay revealed that cells treated with
E2+LPS significantly increased their rate of lateral migration into
wounds introduced on confluent cellular monolayer, compared with
the rate of lateral migration shown by the single-drug-treated
groups and inhibitor-treated groups (A549 cells: Fig. 5A and B, P<0.05; H1793 cells:
Fig. S4A and B, P<0.05). To
detect and quantify the aggressiveness of NSCLC cells, we next
performed Transwell migration\invasion assays. The highest number
of invading cells was found in the E2+LPS treated group. These
results indicate that treatment with E2+LPS increased the
metastatic abilities of the NSCLC cells (A549 cells: Fig. 5C and D, P<0.05; H1793 cells:
Fig. S4C and D, P<0.05).
Western blotting and immunofluorescence staining were used to
determine the expression levels of different proteins involved in
the ERβ/TLR4 interaction and downstream signaling pathways (A549
cells: Figs. 5E and F and S4E; H1793 cells: Figs. 5G and S5C and D). The results of the
densitometric analysis showed that the levels of ERβ, TLR4, myd88,
and MMP2 proteins and phosphorylation of p65NF-κB (p-p65NF-κB)
induced by the treatment with E2+LPS were significantly higher than
were those induced by treatment with E2 or LPS. Taken together, our
findings indicate that activation of ERβ and TLR4 synergistically
promoted the metastatic abilities of the NSCLC cells.
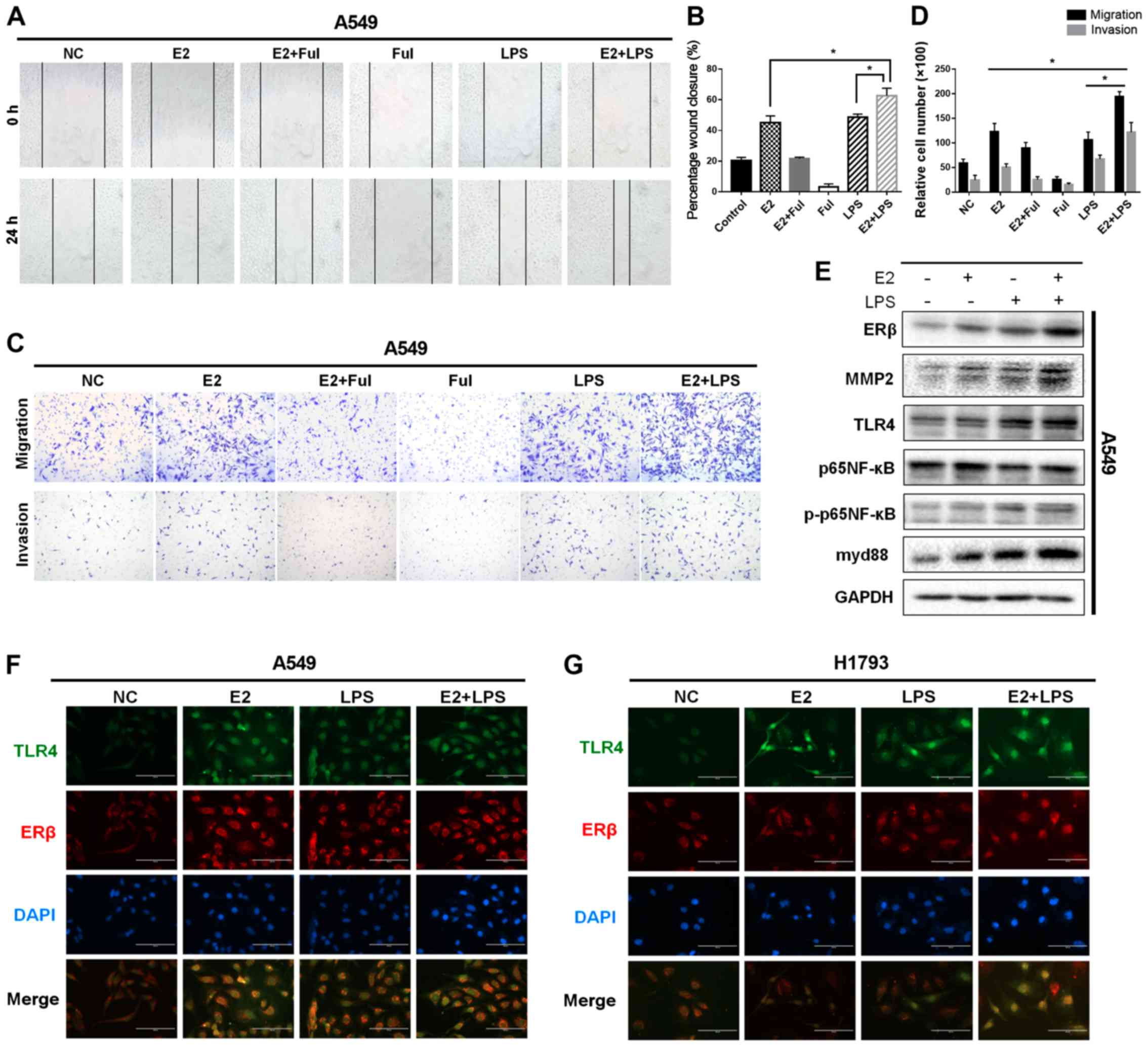 | Figure 5.Activation of ERβ and TLR4
synergistically promotes migratory/invasive abilities of NSCLC
cells. (A) Migration in a wound-healing assay of A549 cells after
treatment with DMSO (negative control, NC) or E2 (10 nM), Ful (1
µM), E2+Ful, LPS (10 µg/ml), and E2+LPS for 24 h (magnification
×40). (B) Effect on wound closure (shown as a percentage) in A549
cells. (C and D) Matrigel Transwell assays were performed to
determine the invasiveness of A549 cells in different treatment
groups. Treatment with E2 and LPS enhanced cell invasion, while the
combination of E2 and LPS increased cell invasiveness more than did
treatment with either agent alone. (E) Western blot analysis of
ERβ, TLR4, MMP2, p65NF-κB, phosphorylated (p)-p65NF-κB, and myd88
protein levels at 48 h in A549 cells, respectively. Protein levels
of ERβ, TLR4, myd88, MMP2, and phosphorylation of P65NF-κB, induced
by the combined treatment of E2 and LPS, were much higher compared
with those in cells treated with E2 or LPS alone. (F and G)
Immunofluorescence staining of ERβ and TLR4 expression in A549 and
H1793 cells. Scale bar, 200 µm. *P<0.05 indicates statistical
significance. NSCLC, non-small cell lung cancer; LPS,
lipopolysaccharide; Ful, fulvestrant (ER antagonist); MMP2, matrix
metalloprotease 2; ERβ, estrogen receptor β; TLR4, Toll-like
receptor 4; myd88, myeloid differentiation primary response 88;
NF-κB, nuclear factor-κB. |
Combination of estrogen and LPS
accelerates invadopodium formation in the NSCLC cells
Increased cell invasion in metastatic progression is
induced by invadopodium formation followed by extracellular matrix
(ECM) degradation (30,31). Hence, we explored whether the
combined exposure to E2 and LPS accelerates invadopodium formation
and ECM degradation. We performed in vitro Matrigel 3D
spheroid invasion assays using A549 and PC9 cells administered E2,
LPS, E2+LPS, and DMSO (vehicle) to evaluate invasiveness in the
cell lines. Density and length of cell-invasion structures were
evaluated on days 1, 3 and 6. Compared with negative control
treatment, exposure to E2 or LPS alone significantly increased the
invasive capability of cells, which formed invadopodia on day 6.
The group treated with E2+LPS showed accelerated formation of
invadopodia on day 3 (A549 cells: Fig.
6A and B). The H1793 cells failed to form spheroids in Matrigel
media; therefore, we used PC9 NSCLC cells for this procedure (PC9
cells: Fig. S5A and B).
Invadopodia appear as accumulations of F-actin associated with dark
areas of fluorescent gelatin degradation (31,32).
To fully determine whether the combination of E2 and LPS enhanced
the function of invadopodia, we conducted a fluorescent gelatin
degradation assay and gelatin zymography analysis. The
co-localization of F-actin with gelatin degradation induced in
cells treated with estrogen or LPS related-drugs compared with that
in unstimulated cells is shown in Fig.
6C. A549 cells treated with E2+LPS showed over 400
µm2 dark degradation area/cell, while
single-drug-treated groups of cells showed barely 200
µm2 degraded area/cell (Fig.
6C and D), and we repeated this procedure with H1793 cells
(Fig. S6A and B). Consistently,
the results of gelatin zymography also showed that exposure to
E2+LPS induced more ECM degradation in the cells than did exposure
to the single agents (Fig. 6E and
F). Together, these data indicate that the combination of
estrogen and LPS strongly accelerated and enhanced invadopodia
function in NSCLC cells.
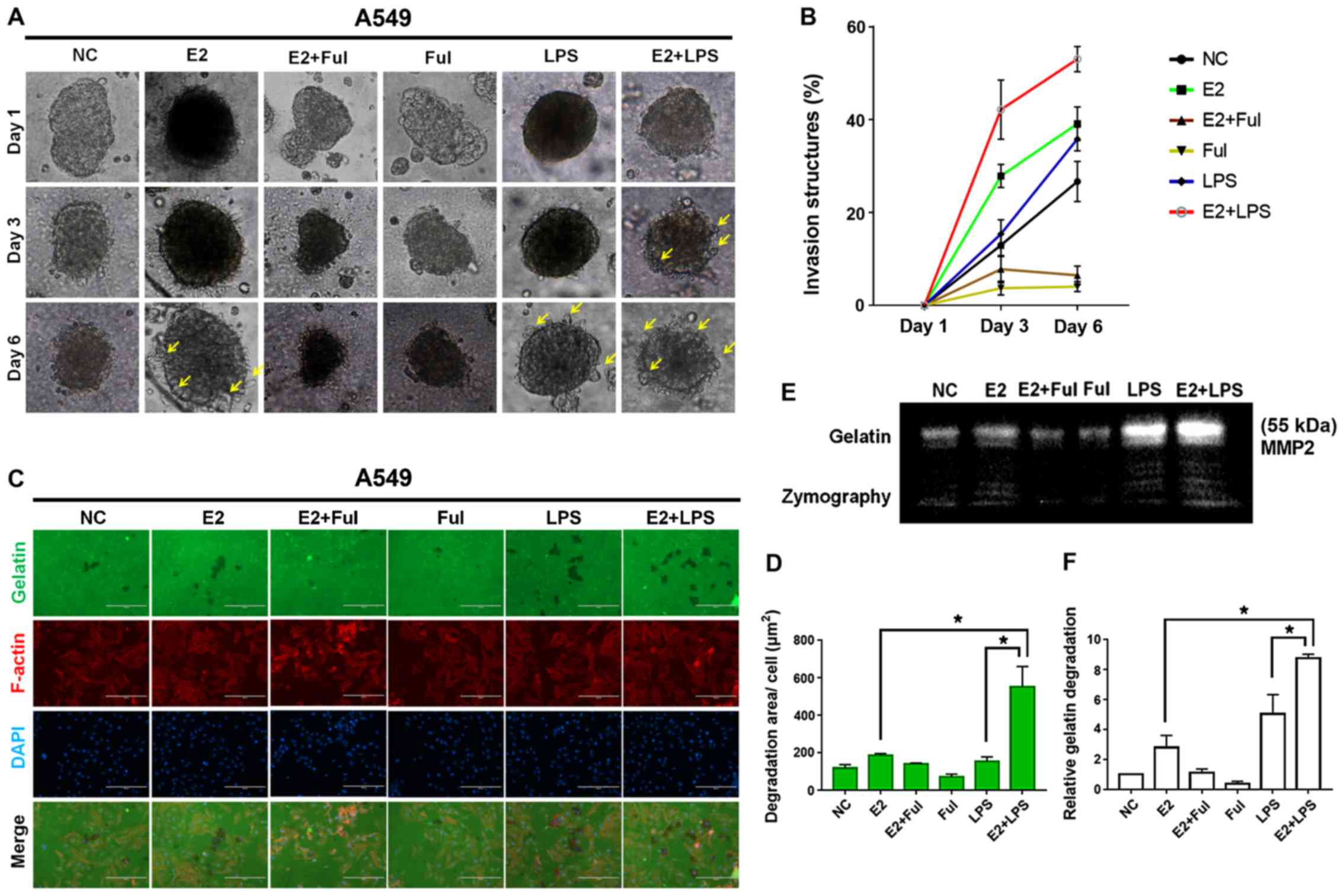 | Figure 6.Combined treatment with estrogen (E2)
and LPS accelerates invadopodium formation in NSCLC A549 cells. (A)
3D spheroid cell invasion assay of lung adenocarcinoma A549 cells
treated with DMSO (negative control, NC), E2 (10 nM), Ful (1 µM),
E2+Ful, LPS (10 µg/ml), and E2+LPS. Representative images were
acquired on day 1, 3 and 6 via light microscopy (×100). Combined
treatment of E2+LPS accelerated invadopodium formation (yellow
arrow) compared with other treatments. (B) Quantification of 3D
spheroid cell invasion assays. Quantification was carried out by
measuring the distance between the invasive cell frontier and the
spheroid edge. (C) To confirm the invasive activity of cancer
cells, slides were coated with FITC-conjugated gelatin (green).
NSCLC cells were cultured on the FITC-gelatin-coated coverslips for
36 h. To visualize F-actin, phalloidin (red) and DAPI (blue) were
used to stain cytoplasm and nuclei, respectively. Punctuate black
areas indicate representative degraded gelatin regions. Scale bar,
200 µm. (D) Quantification of degradation level in FITC-gelatin as
assessed by degradation assay (area per cell, µm2). (E
and F) The activity of MMP2 was assessed by gelatin zymography. The
aggressiveness of NSCLC cells was higher in the E2+LPS-treated
group than in the other treatment groups. *P<0.05 indicates
statistical significance. NSCLC, non-small cell lung cancer; LPS,
lipopolysaccharide; Ful, fulvestrant (ER antagonist); MMP2, matrix
metalloprotease 2. |
Combination of estrogen and LPS
synergistically enhances the tumorigenicity and metastatic ability
in a metastatic mouse model
The effect of combined exposure to E2 and LPS in
cancer metastasis, which was demonstrated in vitro, was
further confirmed in an animal model. For this, female
ovariectomized NOD/SCID mice were injected with A549 cells into the
tail vein. After 65 days of treatment, mice from each group (n=5)
were sacrificed, and the number of metastatic nodules in the lungs
was examined at the indicated time points. As expected, mice
exposed to the combination of E2 and LPS showed higher lung wet
weights, numbers of lung metastasis lesions, and metastatic indices
than did mice injected with normal saline (negative control) or
other treatments (Fig. 7A, and
C-E). Hematoxylin and eosin staining for metastatic nodules is
shown in Fig. 7F. We then used
western blotting and immunohistochemistry to assess protein levels
in each group of mice. As shown in Fig.
7G and H, the expression of ERβ and TLR4 in the E2+LPS-treated
group was significantly higher than that in groups exposed to
either of E2 or LPS alone, and expression levels were decreased in
the E2+Ful- and Ful-treated groups. Consistently,
invasiveness-associated overexpression of MMP2 was higher in the
group treated with E2+LPS than that in the groups subjected to
other treatments. Additionally, the immunohistochemical staining of
ERβ and TLR4 in murine metastatic lung tissues is shown in Fig. 7B. Altogether, our findings showed
that the combination of E2+LPS synergistically increased lung
metastasis in the mouse tumor model.
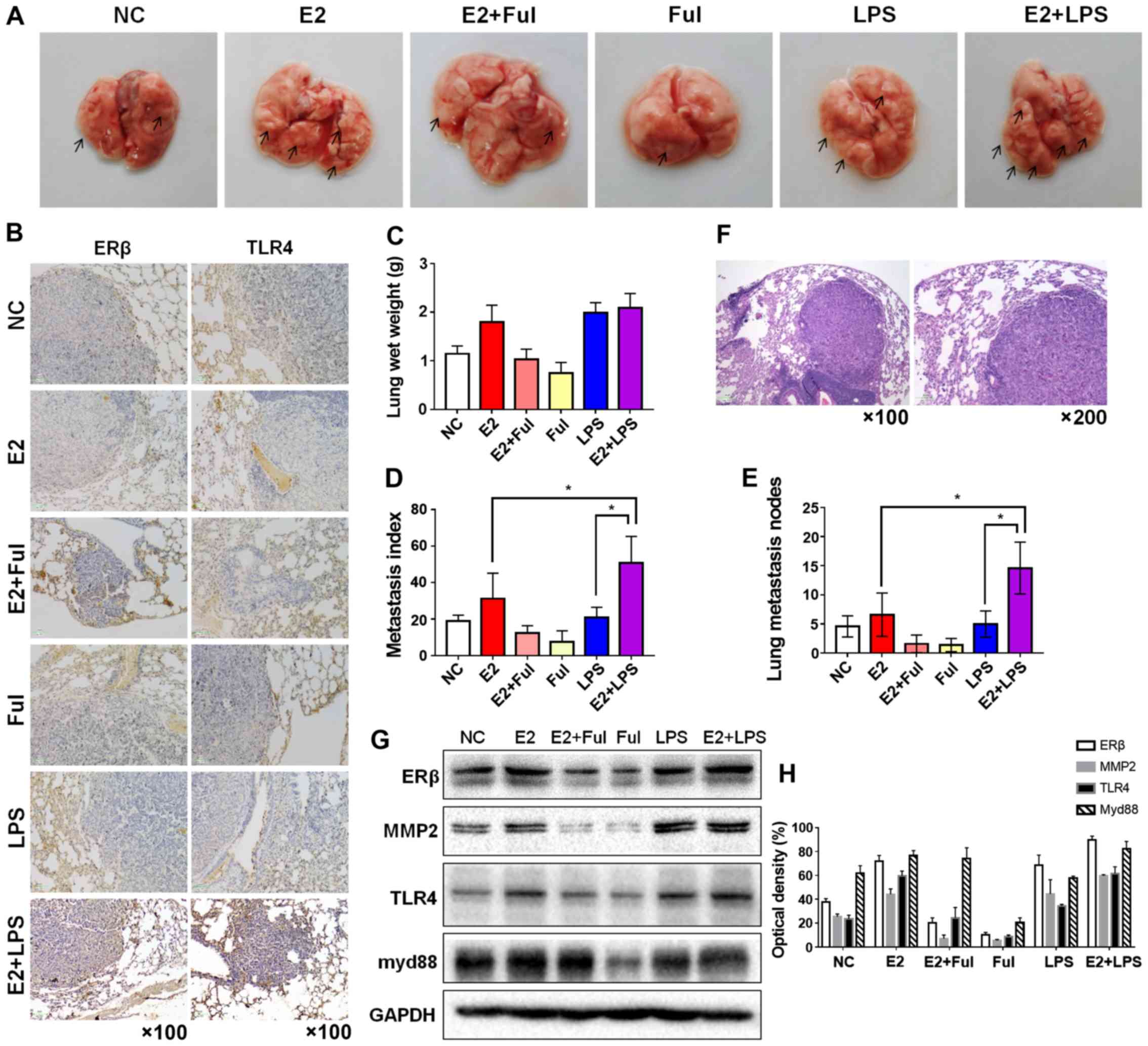 | Figure 7.Combination of estrogen (E2) and LPS
synergistically enhances tumorigenicity and metastatic ability in a
metastatic mouse model. (A) Bilateral ovariectomy was performed in
4-week-old female NOD/SCID mice. Then, NSCLC A549 cells
(5×106/100 µl) suspended in PBS were injected into the
4-week-old female NOD/SCID mice via the tail vein, and the mice
were randomly divided into six groups (n=5/group) as follows:
negative control, E2-treated (0.09 mg/kg), E2+Ful-treated (1.46
mg/kg), Ful+LPS treated (10 mg/kg) and E2+LPS-treated groups. The
lungs were removed after 6 weeks of drug treatment. Gross
appearance of metastatic lung tumor nodes in different treatment
groups is indicated by arrows. (B) IHC staining of ERβ and TLR4
expression in murine lung metastatic nodes in each group
(magnification ×100). (C) Mean lung wet weight of each group, (D)
metastatic indices in the different groups (definition mentioned in
the ‘Materials and methods’) and (E) the number of metastatic
nodules in the lungs (number of lung nodules in every mouse of each
group). Mice treated with E2 and LPS showed the highest lung wet
weight, highest number of lung metastatic lesions, and the highest
metastasis index values, compared with mice injected with normal
saline (negative control) and the other treatment groups. (F)
Hematoxylin and eosin staining of murine lung metastatic nodes
acquired at a magnification of ×100 and ×200. (G) Protein
expression of ERβ, MMP2, TLR4, and myd88 in murine lung metastatic
nodes was analyzed using western blot analysis and (H) analysis of
optical density. *P<0.05 indicates statistical significance.
NSCLC, non-small cell lung cancer; LPS, lipopolysaccharide; Ful,
fulvestrant (ER antagonist); MMP2, matrix metalloprotease 2; ERβ,
estrogen receptor β; TLR4, Toll-like receptor 4; myd88, myeloid
differentiation primary response 88. |
Discussion
Metastatic dissemination and disease relapse are
significant causes of poor clinical outcome in patients with
non-small cell lung cancer (NSCLC). The vast majority of
cancer-related deaths are due to metastasis rather than to the
influence of the primary cancer (2,3).
Recently, estrogen has been found to play a crucial role in
promoting malignant cancers, including in target organs of breast
cancer metastasis and non-target cancers (5,29). We
previously revealed that estrogen promotes NSCLC tumor metastasis
via estrogen receptor β (ERβ) signaling and upregulation of matrix
metalloprotease 2 (MMP2) expression. However, we did not determine
the exact mechanisms involved in this process (6). ERs can regulate Toll-like receptor
(TLR) signaling pathways in the immune system (33), but how and whether ERβ triggers TLRs
in NSCLC remains unknown. In the present study, we performed in
vitro and in vivo investigations to determine the
mechanisms that regulate the activity of TLR4 and how it affects
the downstream myeloid differentiation primary response 88
(myd88)/nuclear factor (NF)-κB /MMP2 signaling axis. We found that
treatment with estrogen and the combined activation of ERβ and TLR4
significantly enhanced cell motility, invasiveness, and
extracellular matrix (ECM) degradation. Additionally, we observed
increasing overexpression of ERβ and TLR4 at metastatic stages, in
particular, within lymph nodes, but not in the primary site of
human NSCLC tumor tissues. These considerations prompted us to
further investigate estradiol (E2) and lipopolysaccharide (LPS)
exposure of NSCLC in vitro.
TLR4 is a member of the Toll-like receptor family
and was initially identified as the specific receptor for LPS
(9,10). TLR4 is expressed in normal
epithelial cells and immune cells and plays a physiologically
relevant role in innate immunity as the first line of host defense
(34). TLRs are considered
significant factors implicated in chronic inflammatory-driven
carcinogenesis (35). Activation of
TLR4 promotes NSCLC metastasis in vitro and in vivo
(14–17). Chow et al (14) demonstrated that enhanced adhesion
and migration abilities of the NSCLC cell line A549 decreased when
the expression of TLR4s, P38MAPK, and ERK1/2 was blocked with
specific inhibitors; TLR4 blockade was also found to abrogate
hepatic metastasis in the NSCLC murine cell line H59. Li et
al also showed that knockdown of TLR4 markedly inhibited the
growth of human NSCLC cancer cells (15). Consistently, we demonstrated
increased cell migration and invasion of A549 and H1793 cells
following exposure to LPS (Fig. 5).
We also observed higher numbers of murine lung metastatic nodules
in the LPS-exposure group of mice than in the control mice
(Fig. 7). In addition, the
phosphorylation of p38MAPK and pAkt was evaluated by western
blotting, which is also consistent with previous reports (16,17).
Altogether, these results indicate that the TLR4/myd88/NF-κB/MMP2
axis plays a crucial role in promoting NSCLC metastasis.
ERβ was previously found to significantly increase
NSCLC cell invasiveness by upregulating the expression of MMP2
(6). Thus, we investigated the
relationship between TLR4 and ERβ. To clarify the association
between TLR4 and ERβ, we analyzed the expression levels of TLR4 and
ERβ in NSCLC primary tumors, metastatic lymph nodes, and adjacent
non-tumor lung tissues. Interestingly, our results showed that the
association of ERβ and TLR4 promoted metastasis in a
grade-dependent manner. As shown in Table II, the Spearman's rank correlation
coefficient was higher for metastatic lymph nodes than for primary
tumors, while no statistical difference was detected in non-tumor
tissue. In a previous study from our group, we reported that
patients with lung cancer express high protein levels of ERβ in
metastatic lymph nodes compared with these levels in primary tumor
tissues (6). In this previous
investigation, we found significant levels of co-expression of ERβ
and MMP2 in primary tumors, but no co-expression in lymph nodes.
This raised the question of whether estrogen-induced ERβ can
regulate MMP2 expression directly during metastasis. The results of
our present study indicate that exposure to E2, or upregulation of
ERβ, increased the expression levels of both TLR4 and MMP2 in NSCLC
cells. To determine whether MMP2 overexpression was induced mainly
by activation of TLR4, we used RNA interference to knock down TLR4
in A549 cells while treating the cells with E2. Our results showed
that MMP2 overexpression and increased cell invasiveness induced by
treatment with E2 were attenuated by the silencing of TLR4.
Furthermore, ERβ was found to immunoprecipitate with TLR4, and TLR4
was found to immunoprecipitate with ERβ, demonstrating a direct
physical interaction between ERβ and TLR4 proteins. The
co-immunoprecipitation assay showed no direct binding of ERβ with
MMP2 or myd88. The results of our confocal micrography were
consistent with the above results. Still, these findings do not
exclude the possibility that this interaction may require other
proteins. This finding may explain why ERβ shows no correlation
with MMP2 in metastatic lymph nodes, but is strongly associated
with TLR4, although a significant correlation between ERβ-TLR4 and
ERβ-MMP2 could be detected in primary tumor tissues. Taken
together, our data indicate that activated ERβ and TLR4 interact
with each other in the cell plasma, contributing to NSCLC
metastasis.
Unfortunately, the results of our prognostic
analysis were not included in this study due to a significant loss
to follow-up (overall loss to follow-up rate = 28.63%; disease-free
survival follow-up loss ratio = 38.15%). However, using the
Kaplan-Meier survival analysis of expression data from 1,928
patients with NSCLC (http://www.kmplot.com/lung), we found that high
co-expression of ERβ and TLR4 was significantly associated with
poor overall survival, with a hazard ratio (HR) = 1.21 (log-rank
test P=0.0031). These results indicate that a high co-expression of
ERβ and TLR4 can predict poor survival in patients with NSCLC, and
overexpression of ERβ and TLR4 may contribute to tumor development
(Fig. S1B-D). Our prognostic data
for this patient population will be further evaluated.
Multiple studies have shown that the effect of
estrogen on the progression of lung cancer may be associated with
other signaling pathways (4,5).
Importantly, malignant tumor carcinogenesis progresses rapidly and
dramatically when ERβ is activated and synergistically cooperates
with other cancer-promoting signaling pathways. Previously, we
showed a synergistic cooperation between ERβ and IGF-1 in NSCLC
cells (19). In the present study,
the in vitro analysis showed that the combined
administration of E2 and LPS markedly upregulated protein
expression levels of ERβ and TLR4. Furthermore, we observed the
co-localization and direct bingding of ERβ and TLR4 in cell
experiments. These results spurred us to investigate whether the
interaction between ERβ and TLR4 synergistically enhances the
metastatic aggressiveness of lung cancer cells. The results of our
in vitro and in vivo analyses showed that the
combined administration of E2 and LPS enhanced the mobility and
invasiveness of NSCLC cells more than did exposure to each agent
alone. Using a wound-healing assay, we showed that both A549 and
H1793 cells treated with E2+LPS generated a significantly narrower
wound closure than did those treated with E2 or LPS alone; similar
responses were observed in the Transwell assay. Our results
obtained using in vivo studies showed the highest number of
metastatic lung nodules, wet lung weights, and metastasis indices
in the group exposed to the combination of E2 and LPS. These
results indicate that estrogen synergistically promoted the
metastasis of NSCLC via the ERβ and TLR4 signaling pathways. These
results also indicate that ERβ and TLR4 are two key signaling
molecules involved in the synergistic activity of the two pathways
mediating the progression of NSCLC.
However, in our mouse experiments, endogenous
estrogen also affected the activation status of ERβ and TLR4 in the
internal environment. To avoid the physiological hormone, we
treated mice with a necessary large dose of estradiol/E2 (0.09
mg/kg) (19) and LPS (10 mg/kg)
(24) to show the effect of
promoting NSCLC metastasis. These doses are high enough to ignore
the effect cause by murine endogenous hormone. On the other hand,
in human tissues, it has been confirmed that primary NSCLC tissues
are equipped to secret E2 (27),
thus the endogenous estrogen in blood does not reflect the local
hormone effect of NSCLC tissue. That was the reason why we used
immunostaining to examine the association between expression of ERβ
and TLR4 to analyze the tumor-promoting effect of E2 and LPS.
Additionally, our cell culture experiment also confirmed the
increased promoting effect of NSCLC metastasis following exposure
to E2 and LPS.
During metastasis, cancer cells dissociate from a
primary tumor mass by degrading the extracellular matrix (ECM) to
locally invade the tumor stroma and adjacent tissues (36). Cancer cells achieve detachment,
degrade the ECM, and migrate by forming unique membrane structures
called invadopodia (30,31). In the present study, we demonstrated
that stimulation with E2, LPS, or both E2 and LPS provided NSCLC
cells with increased ability to degrade the surrounding
FITC-gelatin ECM after a 24 h incubation, and accelerated the
formation of invadopodia in the 3D spheroid invasion model. As
expected, the analysis revealed higher aggressiveness in NSCLC
cells exposed to the combination of E2 and LPS than in those
exposed to either individual agent. Thus, the interaction and
cooperation of ERβ and TLR4 pathways promote ECM degradation by
NSCLC cells and accelerate the formation of invadopodia, thereby
enhancing invasiveness. One of the most critical functions of
mature invadopodia is enhancing the activity of excretive MMP2.
Functional enzymes localized at the invadopodium surface membranes
enable proteolytic activity and promote MMP2 activation to initiate
the MMP activation cascade, which involves the conversion of
proMMP2 to active MMP2 (37,38).
ProMMP2 cannot efficiently degrade the ECM unless it is activated.
In the present study, we observed that overexpression of the MMP2
protein was induced by combined exposure to E2+LPS. This result
suggests that stimulation with estrogen may not only upregulate
MMP2 but may also play a role in MMP2 activation.
Even though our study mainly focused on the protein
levels of many relevant moleculars and utilized different types of
experiments for analysis, there are still some limitation to our
research. The genomic response of each gene of ERβ and TLR4 was not
analyzed or how the interaction between these two receptors could
cause the synergistic tumor metastasis-promoting effect, which both
would still be another interesting research topic. The exact gene
function and deeper mechanistic understanding of the upregulation
of TLR4 require more investigation in our future study. In
addition, the activation of another estrogen receptor
G-protein-coupled estrogen receptor (GPER), may also be triggered
by estrogen (39) and was confirmed
to promote NSCLC proliferation in our previous study (22); however, there is still no evidence
to suggest that GPER signaling is able to trigger TLR4 signaling in
non-genomic pathways to date.
In summary, our study highlights the novel role and
regulatory mechanisms of estrogen in promoting NSCLC metastasis via
ERβ by upregulating the expression of TLR4 and activating its
downstream myd88/NF-κB/MMP2 signaling axis. Importantly, in the
present study, we revealed the interaction and correlation between
the ERβ and TLR4 signaling pathways. Additionally, we showed the
combined effect of E2 and LPS in promoting NSCLC cell invasiveness
and in accelerating the formation of invadopodia. Understanding how
the multiple activities of ERβ contribute to invasive phenotypes of
cancer cells will be the focus of our future studies. Targeting of
ERβ may provide novel strategies for treating patients with
advanced metastatic lung cancer.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
We all deeply thank Mrs. Huifang Liang of the
Hepatic Surgery Center of Tongji Hospital for her technical
assistance. The cell lines used in this study were provided by the
American Type Culture Collection (ATCC).
Funding
This study was funded by the National Natural
Science Foundation of China (NSFC) (grant nos. 81272590 and
81572277).
Availability of data and materials
All relevant data are included in the manuscript and
in the Supplementary materials files.
Authors' contributions
SF and YL designed and conceived the study with the
professional help of LL and XC. SF, WQ, QH and HX performed the
experiments. CL, DL, SZ, BA, and HL carried out the technical
aspects of the research and materially supported the experiments.
SF and YL wrote the manuscript. The manuscript was edited and
revised with the help of LL and XC. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The study was approved by the Ethics or
Institutional Review Board of Tongji Hospital (Wuhan, Hubei, China)
(no. 20141101), and written, informed consent was obtained from all
subjects in accordance with the Declaration of Helsinki. All animal
experiments were carried out according to the regulations specified
by the Ethics Committee for Tongji Hospital of Huazhong University
of Science and Technology.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar
|
|
2
|
Langer CJ, Besse B, Gualberto A, Brambilla
E and Soria JC: The evolving role of histology in the management of
advanced non-small-cell lung cancer. J Clin Oncol. 28:5311–5320.
2010. View Article : Google Scholar
|
|
3
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
4
|
Blaustein JD: Steroid hormone receptors:
Long- and short-term integrators of the internal milieu and the
external environment. Horm Metab Res. 44:563–568. 2012. View Article : Google Scholar
|
|
5
|
Nair S and Sachdeva G: Estrogen matters in
metastasis. Steroids. 138:108–116. 2018. View Article : Google Scholar
|
|
6
|
Fan S, Liao Y, Liu C, Huang Q, Liang H, Ai
B, Fu S and Zhou S: Estrogen promotes tumor metastasis via estrogen
receptor beta-mediated regulation of matrix-metalloproteinase-2 in
non-small cell lung cancer. Oncotarget. 8:56443–56459. 2017.
|
|
7
|
Chlebowski RT, Schwartz AG, Wakelee H,
Anderson GL, Stefanick ML, Manson JE, Rodabough RJ, Chien JW,
Wactawski-Wende J, Gass M, et al Women's Health Initiative
Investigators, : Oestrogen plus progestin and lung cancer in
postmenopausal women (Women's Health Initiative trial): A post-hoc
analysis of a randomised controlled trial. Lancet. 374:1243–1251.
2009. View Article : Google Scholar :
|
|
8
|
Garon EB, Siegfried JM, Stabile LP, Young
PA, Marquez-Garban DC, Park DJ, Patel R, Hu EH, Sadeghi S, Parikh
RJ, et al: Randomized phase II study of fulvestrant and erlotinib
compared with erlotinib alone in patients with advanced or
metastatic non-small cell lung cancer. Lung Cancer. 123:91–98.
2018. View Article : Google Scholar :
|
|
9
|
Brown M and O'Reilly S: Innate immunity
and Toll-like receptor signaling in the pathogenesis of
scleroderma: Advances and opportunities for therapy. Curr Opin
Rheumatol. 30:600–605. 2018. View Article : Google Scholar
|
|
10
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar
|
|
11
|
Janeway CA Jr and Medzhitov R:
Introduction: The role of innate immunity in the adaptive immune
response. Semin Immunol. 10:349–350. 1998. View Article : Google Scholar
|
|
12
|
Bhattacharya D and Yusuf N: Expression of
toll-like receptors on breast tumors: Taking a toll on tumor
microenvironment. Int J Breast Cancer. 2012:7165642012. View Article : Google Scholar
|
|
13
|
Anthoney N, Foldi I and Hidalgo A: Toll
and Toll-like receptor signalling in development. Development.
145:1452018. View Article : Google Scholar
|
|
14
|
Chow SC, Gowing SD, Cools-Lartigue JJ,
Chen CB, Berube J, Yoon HW, Chan CH, Rousseau MC, Bourdeau F,
Giannias B, et al: Gram negative bacteria increase non-small cell
lung cancer metastasis via Toll-like receptor 4 activation and
mitogen-activated protein kinase phosphorylation. Int J Cancer.
136:1341–1350. 2015. View Article : Google Scholar
|
|
15
|
Li D, Jin Y, Sun Y, Lei J and Liu C:
Knockdown of toll-like receptor 4 inhibits human NSCLC cancer cell
growth and inflammatory cytokine secretion in vitro and
in vivo. Int J Oncol. 45:813–821. 2014. View Article : Google Scholar
|
|
16
|
Harmey JH, Bucana CD, Lu W, Byrne AM,
McDonnell S, Lynch C, Bouchier-Hayes D and Dong Z:
Lipopolysaccharide-induced metastatic growth is associated with
increased angiogenesis, vascular permeability and tumor cell
invasion. Int J Cancer. 101:415–422. 2002. View Article : Google Scholar
|
|
17
|
Lv W, Chen N, Lin Y, Ma H, Ruan Y, Li Z,
Li X, Pan X and Tian X: Macrophage migration inhibitory factor
promotes breast cancer metastasis via activation of HMGB1/TLR4/NF
kappa B axis. Cancer Lett. 375:245–255. 2016. View Article : Google Scholar
|
|
18
|
Rusch VW, Chansky K, Kindler HL, Nowak AK,
Pass HI, Rice DC, Shemanski L, Galateau-Sallé F, McCaughan BC,
Nakano T, et al IASLC Staging Prognostic Factors Committee,
advisory boards, participating institutions, : The IASLC
Mesothelioma Staging Project: Proposals for the M Descriptors and
for Revision of the TNM Stage Groupings in the Forthcoming (Eighth)
Edition of the TNM Classification for Mesothelioma. J Thorac Oncol.
11:2112–2119. 2016. View Article : Google Scholar
|
|
19
|
Tang H, Liao Y, Xu L, Zhang C, Liu Z, Deng
Y, Jiang Z, Fu S, Chen Z and Zhou S: Estrogen and insulin-like
growth factor 1 synergistically promote the development of lung
adenocarcinoma in mice. Int J Cancer. 133:2473–2482. 2013.
View Article : Google Scholar
|
|
20
|
Huang Q, Zhang Z, Liao Y, Liu C, Fan S,
Wei X, Ai B and Xiong J: 17β-estradiol upregulates IL6 expression
through the ERβ pathway to promote lung adenocarcinoma progression.
J Exp Clin Cancer Res. 37:1332018. View Article : Google Scholar :
|
|
21
|
Tang H, Liao Y, Chen G, Xu L, Zhang C, Ju
S and Zhou S: Estrogen upregulates the IGF-1 signaling pathway in
lung cancer through estrogen receptor-β. Med Oncol. 29:2640–2648.
2012. View Article : Google Scholar
|
|
22
|
Liu C, Liao Y, Fan S, Tang H, Jiang Z,
Zhou B, Xiong J, Zhou S, Zou M and Wang J: G protein-coupled
estrogen receptor (GPER) mediates NSCLC progression induced by
17β-estradiol (E2) and selective agonist G1. Med Oncol. 32:1042015.
View Article : Google Scholar
|
|
23
|
Hernandez H, Medina-Ortiz WE, Luan T,
Clark AF and McDowell CM: Crosstalk between transforming growth
factor beta-2 and Toll-like receptor 4 in the trabecular meshwork.
Invest Ophthalmol Vis Sci. 58:1811–1823. 2017. View Article : Google Scholar :
|
|
24
|
Shaaban AA, El-Kashef DH, Hamed MF and
El-Agamy DS: Protective effect of pristimerin against LPS-induced
acute lung injury in mice. Int Immunopharmacol. 59:31–39. 2018.
View Article : Google Scholar
|
|
25
|
Vinci M, Box C and Eccles SA:
Three-dimensional (3D) tumor spheroid invasion assay. J Vis Exp.
99:e526862015.
|
|
26
|
Del Pozo JL: Primers on molecular
pathways: Lipopolysaccharide signaling - potential role in
pancreatitis and pancreatic cancer. Pancreatology. 10:114–118.
2010. View Article : Google Scholar
|
|
27
|
Siegfried JM and Stabile LP: Estrongenic
steroid hormones in lung cancer. Semin Oncol. 41:5–16. 2014.
View Article : Google Scholar
|
|
28
|
Matsunaga N, Tsuchimori N, Matsumoto T and
Ii M: TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like
receptor (TLR) 4 signaling, binds selectively to TLR4 and
interferes with interactions between TLR4 and its adaptor
molecules. Mol Pharmacol. 79:34–41. 2011. View Article : Google Scholar
|
|
29
|
Thomas C and Gustafsson JA: The different
roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer.
11:597–608. 2011. View Article : Google Scholar
|
|
30
|
Weaver AM: Invadopodia: Specialized cell
structures for cancer invasion. Clin Exp Metastasis. 23:97–105.
2006. View Article : Google Scholar
|
|
31
|
Saykali BA and El-Sibai M: Invadopodia,
regulation, and assembly in cancer cell invasion. Cell Commun
Adhes. 21:207–212. 2014. View Article : Google Scholar
|
|
32
|
Goertzen CG, Dragan M, Turley E, Babwah AV
and Bhattacharya M: KISS1R signaling promotes invadopodia formation
in human breast cancer cell via β-arrestin2/ERK. Cell Signal.
28:165–176. 2016. View Article : Google Scholar
|
|
33
|
Kovats S: Estrogen receptors regulate
innate immune cells and signaling pathways. Cell Immunol.
294:63–69. 2015. View Article : Google Scholar :
|
|
34
|
Mai CW, Kang YB and Pichika MR: Should a
Toll-like receptor 4 (TLR-4) agonist or antagonist be designed to
treat cancer? TLR-4: Its expression and effects in the ten most
common cancers. Onco Targets Ther. 6:1573–1587. 2013.
|
|
35
|
Rich AM, Hussaini HM, Parachuru VP and
Seymour GJ: Toll-like receptors and cancer, particularly oral
squamous cell carcinoma. Front Immunol. 5:4642014. View Article : Google Scholar :
|
|
36
|
Chiang AC and Massagué J: Molecular basis
of metastasis. N Engl J Med. 359:2814–2823. 2008. View Article : Google Scholar :
|
|
37
|
Alaseem A, Alhazzani K, Dondapati P,
Alobid S, Bishayee A and Rathinavelu A: Matrix Metalloproteinases:
A challenging paradigm of cancer management. Semin Cancer Biol.
56:100–115. 2019. View Article : Google Scholar
|
|
38
|
Brown GT and Murray GI: Current
mechanistic insights into the roles of matrix metalloproteinases in
tumour invasion and metastasis. J Pathol. 237:273–281. 2015.
View Article : Google Scholar
|
|
39
|
Prossnitz ER and Barton M: The
G-protein-coupled estrogen receptor GPER in health and disease. Nat
Rev Endocrinol. 7:715–726. 2011. View Article : Google Scholar :
|