Introduction
In 2018, >1.27 million new cases and 35,800
deaths as a result of prostate cancer were recorded worldwide
(1). Prostate cancer accounts for
13.5% of cancers diagnosed in men, which is only lower than lung
cancer and is the fifth-leading cause of cancer-related mortality
in men. Androgen deprivation therapy (ADT), the main treatment for
locally advanced or metastatic androgen-dependent prostate cancer,
can alleviate or stabilize symptoms in >80% of patients.
However, most patients become non-responsive to this treatment and
inevitably progress to castration-resistant prostate cancer (CRPC)
following treatment for 12–18 months (2). The serum level of prostate-specific
antigen (PSA), one of the most important target genes of androgen
receptor (AR) and an important biomarker for the diagnosis and
prognosis of prostate cancer, is elevated in patients with CRPC
after ADT, suggesting that the AR signaling pathway is reactivated
in CRPC cells (3–5). Two major mechanisms are involved in the
reactivation of AR in CRPC, including AR upregulation and AR splice
variant (AR-V) expression.
AR full-length (AR-FL) is a member of the steroid
receptor subfamily that belongs to the nuclear receptor family and
consists of four structural domains: Amino-terminal domain,
DNA-binding domain, small hinge region and ligand-binding domain
(LBD). Activation of the AR signaling pathway mainly depends on
androgen, an AR ligand, in prostate cancer cells (5). After binding with its ligand, the AR
protein is translocated from the cytoplasm to the nucleus. Two AR
molecules are induced to homodimerize by the D-box in the nucleus,
and the dimer binds to specific androgen response elements to
promote target gene expression (6).
AR-Vs are named based on the majority that lack
C-terminal LBD (7) and have
constitutive transcriptional activity without androgen binding
(3,8).
Several drugs targeting AR reactivation, such as the FDA-approved
abiraterone and enzalutamide (MDV3100), can prolong the overall
survival of patients with CRPC. However, the majority of patients
progress to drug-resistant disease partly because of AR-V
expression, which is unaffected by these drugs; therefore, it is
necessary to develop novel drugs targeting both AR-FL and AR-Vs to
combat CRPC.
Erastin was first discovered to kill cancer cells
overexpressing H-ras in engineered tumorigenic cells by synthetic
lethal high-throughput screening of >20,000 compounds in 2003
(9). When erastin induces cell death,
neither chromatin fragmentation nor caspase activation is detected,
and the levels of intracellular reactive oxygen species (ROS) are
upregulated and erastin-induced death can be prevented by iron
chelation. In 2012, Dixon et al (10) proposed the term ferroptosis to
describe this new death pattern. Erastin can induce ferroptosis via
different mechanisms, such as directly targeting mitochondrial
voltage-dependent anion channels (11), inhibiting the activity of the xCT
light chain of the cystine/glutamate transporter (also known as
system XC-) (12,13), playing a relevant role in the MAPK
pathway (14) and increasing heme
oxygenase-1 (15,16). Furthermore, Hasegawa et al
(17) found that mucin 1 C-terminal
subunit/xCT can downregulate erastin-induced ferroptosis in
triple-negative breast cancer. As an inducer of ferroptosis,
erastin has been demonstrated to inhibit cancer cell proliferation
in acute myeloid leukemia, and hepatocellular, breast, ovarian, and
head and neck cancer (16–18), although the mechanisms attributed to
cell death differ among these cancers. Although a considerable
number of studies have sought to determine whether erastin inhibits
prostate cancer, it is necessary to investigate the specific
mechanism of erastin, such as the activation of the AR signaling
pathway in prostate cancer cells, specifically in CRPC.
Materials and methods
Prostate cancer cell lines and
reagents
LNCaP, PC3, 22Rv1, C4-2, C4-2B, Du145 and 293T cells
were obtained from the American Type Culture Collection. These
cells were cultured in RPMI-1640 (Gibco; Thermo Fisher Scientific,
Inc.) medium supplemented with 10% fetal bovine serum (FBS; Clark
Bioscience) at 37°C in a humidified atmosphere with 5%
CO2. LNCaP95 cells were provided by Dr Alan Meeker at
the Johns Hopkins University (Baltimore, MD, USA) and were cultured
in RPMI-1640 medium supplemented with 10% charcoal-stripped FBS
(Gibco; Thermo Fisher Scientific, Inc.). Erastin, docetaxel (DTX),
ferrostatin-1, liproxstatin-1, ZVAD-FMK, necrosulfonamide and
chloroquine were purchased from Selleck Chemicals.
Sulforhodamine B (SRB) cell viability
assay
After being treated with erastin at different
concentrations (0, 2.5, 5.0, 10.0, 20.0 or 40.0 µM) for 24, 48 or
72 h, cell monolayers of 22Rv1 or LNCaP95 in 96-well plates were
fixed using 20% (wt/vol) trichloroacetic acid (100 µl/well) at room
temperature for at least 3 h, and then stained for 30 min using SRB
(Shanghai YuanYe Biotechnology Co., Ltd.) at room temperature.
Excess dye was removed by washing repeatedly with 1% (vol/vol)
acetic acid. The protein-bound dye was dissolved in 10 mM Tris base
solution and then the OD was measured at 565 nm using a Multiscan
Spectrum spectrophotometer. In order to obtain more samples in the
subsequent experiments, the doses of erastin were determined based
on the cell inhibition rate of 30%. Thus, 22Rv1 and LNCaP95 cells
were treated with 10 or 20 µM erastin, while PC3 cells were treated
with 1 µM erastin.
In order to verify whether the effect caused by
erastin was ferroptosis, the cells were treated with erastin (20
µM). Meanwhile, the cells were added with or without ferroptosis
inhibitors (1 µM ferrostatin-1 or 1 µM liproxstatin-1), apoptosis
inhibitor (10 µM ZVAD-FMK), necroptosis inhibitor (1 µM
necrosulfonamide), or autophagy inhibitor (25 µM chloroquine).
After treatment for 48 h, the cell viability was detected by SRB as
described above.
In order to calculate the combination index (CI)
between DTX and erastin, 22Rv1 and LNCaP95 cells were treated with
these two drugs at different concentrations (0, 2.5, 5.0 or 10.0 µM
erastin and 0, 5, 10 nM DTX). After treatment for 48 h, cell
viability was detected by SRB as described above.
Western blot analysis
Western blotting was conducted as previously
described (19), and proteins were
visualized using an Odyssey Infrared Imaging System (LI-COR
Biosciences), following the manufacturer's protocols.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for
normalization of the densitometry signals. The following antibodies
were used in this study: Anti-N-terminal AR antibody (cat. no.
5153; 1:1,000; Cell Signaling Technology, Inc.), anti-phospholipid
hydroperoxide glutathione peroxidase (GPX4; cat. no. ab125066;
1:1,000; Abcam) and anti-GAPDH (cat. no. A00227; 1:1,000; Wuhan
Boster Biological Technology, Ltd.). IRDye® 800CW goat
anti-human IgG (H+L) (cat. no. 926-32232; 1:10,000; LI-COR
Biosciences).
ROS assay
After treatment with erastin for 24 h, the cells
were incubated with 1 µl DCFH-DA with 1 ml phosphate-buffered
saline (PBS) for 1 h at 37°C in the dark to assess the cytosolic
ROS levels. Samples were centrifuged at 860 × g at room temperature
for 3 min, and the pellets were resuspended in 1 ml PBS.
Measurements were performed on a FACSCalibur™ (BD Biosciences) flow
cytometer using FlowJo software 7.6 (FlowJo LLC). All experimental
results are reported as represented by three replicates.
Glutathione (GSH) and lipid
peroxidation assays
The GSH and malondialdehyde (MDA) contents in cell
lysates were assessed using GSH (cat. no. A061-1-2) and lipid
peroxidation (cat. no. A003-4-1) assay kits, respectively,
according to the manufacturer's instructions. Both kits were
purchased from Nanjing Jiancheng Bioengineering Institute.
Dual-luciferase reporter gene
assay
Three luciferase reporter plasmids were used in this
research, and transfection was performed with TurboFect
Transfection Reagent (cat. no. R0531; Thermo Fisher Scientific,
Inc.). The androgen-responsive element-luciferase plasmid contains
three repeat ARE regions ligated in tandem to a luciferase reporter
(ARR3-Luc), which was provided by Dr Robert Matusik at Vanderbilt
University School of Medicine (Nashville, TN, USA), and was used to
reflect the AR-FL trans-activation activity. The
ubiquitin-conjugating enzyme E2C-luciferase plasmid (UBE2C-luc) is
driven by a minimal promoter and three repeats of an AR-V-specific
promoter element. Thus, it was used to reflect AR-V7
trans-activation activity. pGL4-ARpro8.0 is driven by an 8.0 kb
fragment of the 5′-flanking region of the human AR gene. The
transfected cells, including 22Rv1, LNCaP95, PC3 and LNCaP cells,
were divided equally into 24-well plates (1×105
cells/well) and cultivated in serum-free medium for 24 h.
Subsequently, the cells were exposed to charcoal-stripped FBS with
or without 1 nM R1881 (Sigma-Aldrich; Merck KGaA) at 37°C, which is
a type of synthetic androgen, and erastin to detect AR-FL or AR-V7
activity. After 24 h, the cells were lysed with 100 µl reporter
lysis buffer (Promega Corporation), and the luciferase activity was
assayed using the Luciferase Assay System (Promega Corporation) and
normalized based on the protein concentrations for each sample.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA of cells was extracted and collected
using an E.Z.N.A.® Total RNA Kit I (Omega Bio-Tek, Inc.)
and quantified with a NanoDrop™ spectrophotometer (Thermo Fisher
Scientific, Inc.). Reverse transcription was performed with an RNA
reverse transcription kit (Takara Bio, Inc.), and qPCR was
performed using TransStart® Green qPCR SuperMix
(TransGen Biotech Co., Ltd.), according to the manufacturer's
instructions for 45 cycles of 5 sec at 94°C and 30 sec at 60°C.
36B4 served as the normalization control for these qPCR assays. The
following primers were used: AR-FL sense,
5′-GTACAGCCAGTGTGTCCGAA-3′ and anti-sense,
5′-TTGGTGAGCTGGTAGAAGCG-3′; AR-V7 sense, 5′-AAAAGAGCCGCTGAAGGGAA-3′
and anti-sense, 5′-GCCAACCCGGAATTTTTCTCC-3′; PSA sense,
5′-CTCAGGCCAGGTGATGACTC-3′ and anti-sense,
5′-GTCCAGCACACAGCATGAAC-3′; transmembrane protease serine 2
(TMPRSS2) sense, 5′-ACACACCGATTCTCGTCCT-3′ and anti-sense,
5′-TGGCCTACTCTGGAAGTTCA-3′; UBE2C sense, 5′-TTCCCCAGTGGCTACCCTTA-3′
and anti-sense, 5′-CAGGGCAGACCACTTTTCCT-3′; transcription factor
E2F7 (E2F7) sense, 5′-TTCTGTTGCTCAGACGGACC-3′ and anti-sense,
5′-ATCCCTCTCTGACCCTGACC-3′; and 36B4 sense,
5′-CGACCTGGAAGTCCAACTAC-3′ and anti-sense,
5′-ATCTGCTGCATCTGCTTG-3′. mRNA expression levels were quantified
using the 2−ΔΔCq method (20).
Lentiviral packaging and
lentivirus-infected cells
Three plasmids were used as lentiviral vectors:
VSVG, ∆8.2, pLVX-AR-FL or pLVX-AR-V7. VSVG, ∆8.2, and oLVX were
provided by the National Engineering Laboratory for AIDS Vaccine
(Jilin University, Changchun, China). The preparation of the
lentiviral particles and lentiviral infections were performed as
previously described (21). When
lentiviral particles (collected at 48 h after transfection) were
used to infect 22Rv1 cells in 6-well plates (1×106
cells/well), polybrene was added at a final concentration of 6
µg/ml, fresh medium was replaced within 6 h, and the protein
expression was determined by western blotting.
22Rv1 tumor xenograft model
A total number of 12 male BALB/c nude mice (age, 6–8
weeks old; weight, 15–20 g) were purchased from Beijing Vital River
Laboratory Animal Technology Co., Ltd. All mice were housed at a
constant temperature and constant humidity in a specific
pathogen-free environment with free access to food and water. The
present study was approved by the Animal Ethics Committee of Basic
Medical College of Jilin University (approval no. 2016045). After 1
week of adaptation, 5×106 22Rv1 cells suspended in 50%
Matrigel and PBS were injected subcutaneously into the right dorsal
flank. Tumor formation was strictly monitored, and tumor volume was
calculated by the modified ellipsoidal formula: Tumor volume = 0.52
× length × width2. When the tumor size reached ~50
mm3, the mice were randomly allocated into two groups
(n=6/per group) and treated with or without erastin. The dose of
erastin administered to the mice was 20 mg/kg, which was injected
intraperitoneally twice every other day. After being treated for 2
weeks, all mice were injected with pentobarbital sodium at a dose
of 20 mg/kg intraperitoneally. Then, 0.5 ml mouse blood was
collected from the orbital vein to detect serum prostate-specific
antigen (PSA) levels using a human KLK3 ELISA Kit (cat. no.
KIT10771; Sino Biological, Inc.), tumors were removed for molecular
analysis, and some organs were removed for a safety evaluation at
the end of the experiment. Finally, all mice were sacrificed by
intraperitoneal injection with pentobarbital sodium at a dose of
150 mg/kg. Erastin was dissolved in 3.3% dimethyl sulfoxide (DMSO)
and 96.7% β-cyclodextrin (20%).
Histopathology assay
Hematoxylin and eosin (H&E) staining was
performed as described in a previous study (22). Briefly, the tissues of the rats were
fixed in 10% buffered formalin, and then decalcified in 10%
ethylenediaminetetraacetic acid. The tissues were then treated with
ethanol and xylene for dehydration. After being embedded in
paraffin and sliced, several 4-µm thick histological slices were
stained with H&E. The images were subsequently acquired using a
light microscope (BX51; Olympus Corporation) at ×200
magnification.
Immunohistochemistry (IHC)
IHC staining was performed as described previously
(23). Briefly, histological slices
from tumor tissues were stained with anti-N-terminal AR antibody
(1:200; cat. no. 5153; Cell Signaling Technology, Inc.).
Histological images were captured by a microscope (BX51; Olympus
Corporation) with an objective magnification of ×200.
Statistical analysis
The experiments were repeated three times
independently. Statistical analysis was performed for multiple
comparisons using one factor analysis of variance (ANOVA) followed
by Dunnett's or Bonferroni's post hoc tests. All data were analyzed
with the statistical software SPSS 11.0 (SPSS, Inc.), and the
results are expressed as the mean ± SD. P<0.05 was considered to
indicate a statistically significant difference. SPSS software was
also used to calculate the CI, a CI value of >1, 1 and <1
denotes antagonism, additivity and synergism, respectively.
Results
Erastin inhibits the growth of CRPC
cells
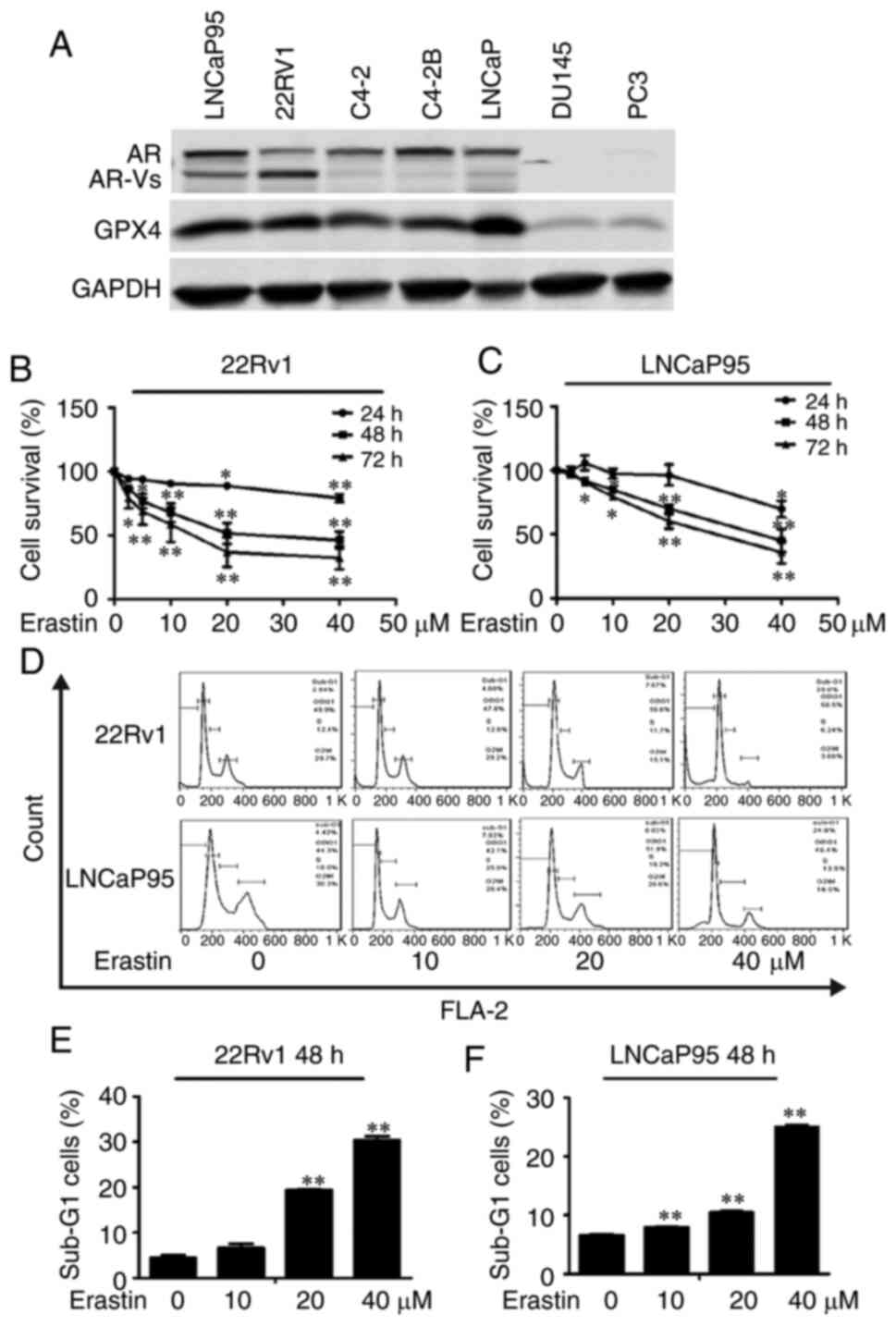
Western blotting was employed to detect the
expression of AR, AR-Vs and GPX4 in several prostate cancer cell
lines including LNCaP95, LNCaP, Du145, C4-2B, 22Rv1, C4-2 and PC3
cells. The results showed that both 22Rv1 and LNCaP95 cell lines
expressed AR, AR-Vs and GPX4 (Fig.
1A). Conversely, PC3 cells did not express AR or AR-Vs and only
showed a low expression of GPX4. Thus, 22Rv1, LNCaP95 and PC3 cell
lines were chosen for the subsequent experiments.
The effect of erastin on the proliferation of 22Rv1
and LNCaP95 cells was detected by the SRB assay. As presented in
Fig. 1B and C, erastin inhibited the
proliferation of these two CRPC cell lines in a dose-dependent
manner. Then, PI flow cytometry was employed to examine the effect
of erastin on the cell cycle. The results showed that erastin could
increase the proportion of cells in sub-G1 phase and cause cycle
arrest (Fig. 1D-F).
Erastin induces ferroptosis in CRPC
cells
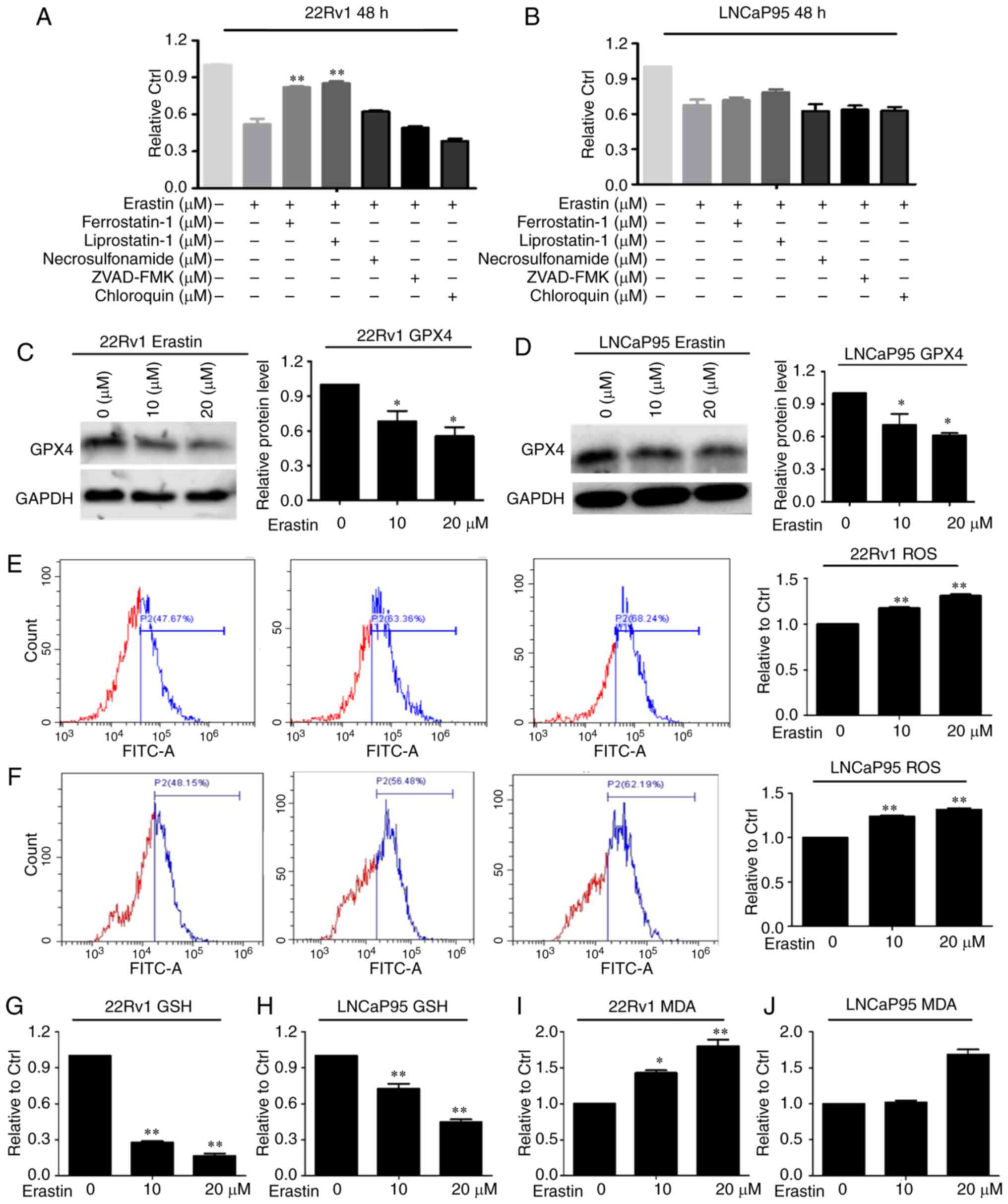
To verify that the effect caused by erastin is
ferroptosis, the cells were treated with erastin with or without
ferroptosis inhibitors (ferrostatin-1 and liproxstatin-1),
apoptosis inhibitor (ZVAD-FMK), necroptosis inhibitor
(necrosulfonamide) and autophagy inhibitor (chloroquine). The SRB
results showed that in both cell lines, ZVAD-FMK, necrosulfonamide
and chloroquine had no effect on erastin-induced cell death
(Fig. 2A and B). The death of the
22Rv1 cells induced by erastin was only reversed by ferrostatin-1
and liproxstatin-1.
Then, the expression of the ferroptosis marker
protein GPX4 was measured. The results showed that erastin
downregulated the protein expression of GPX4 in both cell lines
(Fig. 2C and D). Given that
ferroptosis is characterized by lipid peroxidation, the level of
GSH, a key regulator that maintains cellular redox homeostasis, was
investigated. The results showed that ROS levels in both cell lines
were increased with erastin treatment compared with the control
group (Fig. 2E and F). Moreover, GSH
levels were downregulated after erastin treatment in these two cell
lines (Fig. 2G and H). MDA, an end
product of lipid peroxidation, was notably increased following
erastin treatment, as expected (Fig. 2I
and J). According to the aforementioned results, it was
hypothesized that ferroptosis was initiated in these two cell lines
after erastin treatment.
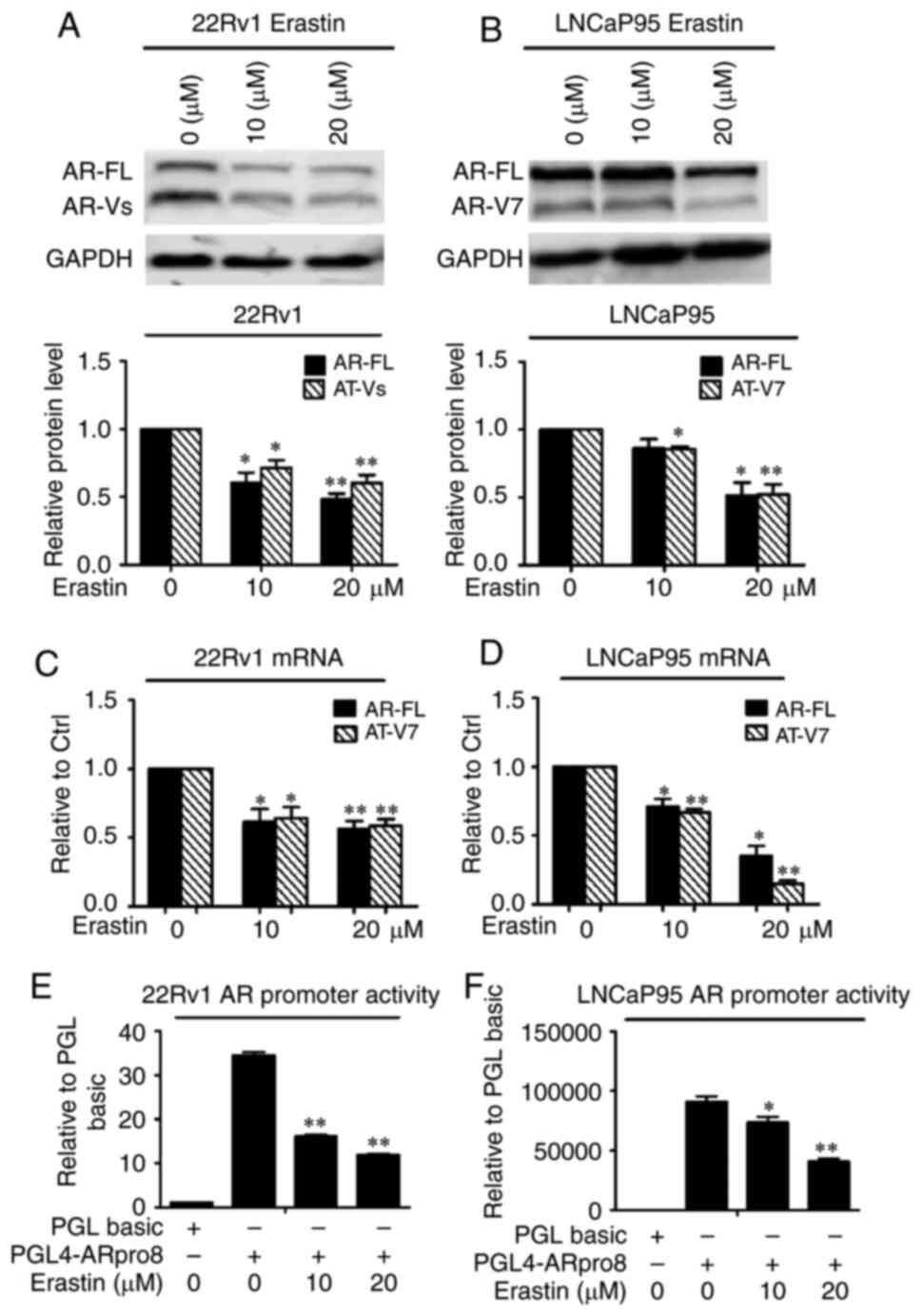
Erastin downregulates AR protein
expression by inhibiting the transcription of the AR gene
Given that AR-FL and AR-Vs play important roles in
the development of CRPC, 22Rv1 and LNCaP95 CRPC cells that express
AR-FL and AR-Vs were chosen in the present study to explore whether
erastin can inhibit AR expression. In 22Rv1 cells, AR-Vs include
AR-V7 (also called AR3), AR-V1 (also called AR4) and AR-V4 (also
called AR5) (8,24), whereas LNCaP95 cells express only
AR-V7 (25). Among these splice
variants, AR-V7 has been found to be associated with the
development of CRPC and is recognized as a biomarker of poor
prognosis for patients with CRPC (26); thus, AR-V7 was selected as the
representative of AR-Vs in the present study. The expression of
AR-FL and AR-V proteins was detected by western blotting, and the
results showed that erastin downregulated both AR-FL and AR-V
protein expression levels (Fig. 3A and
B).
To investigate how erastin decreases the protein
levels of AR-FL and AR-V, AR-FL and AR-V7 mRNA expression levels
were measured by RT-qPCR. As expected, erastin significantly
reduced the levels of AR-FL and AR-V7 mRNA (Fig. 3C and D). Then, AR promoter activity
was detected using dual-luciferase reporter plasmid pGL4-ARpro8.0.
Erastin treatment resulted in the significant inhibition of AR
promoter activity in both cell lines (Fig. 3E and F). Taken together, the data
indicated that erastin had the ability to inhibit the transcription
of the AR gene in CRPC cells.
Erastin downregulates AR-FL and AR-V
transactivation
To evaluate the effects of erastin on AR
transactivation, AR-FL and AR-V transcriptional activity and their
target genes were measured by a reporter gene assay and RT-qPCR,
respectively. First, 22Rv1 cells were transfected with the ARR3-luc
luciferase construct, which contained three tandem repeats of
androgen response elements. The results showed that erastin
treatment induced a reduction in luciferase activity in 22Rv1 cells
(Fig. 4A). Considering that the
ARR3-luc construct can be regulated not only by AR-FL, but also by
AR-Vs, the LNCaP cell line was selected for transfection with a
ARR3-luc luciferase construct because these cells only express
AR-FL. The results in LNCaP cells clearly showed that erastin
inhibited AR-FL trans-activating activity (Fig. 4B). To specifically assess the effect
of erastin on AR-V transcriptional activity, 22Rv1 and LNCaP95
cells were transfected with the UBE2C-luc construct in which the
luciferase gene is driven by an AR-V-specific promoter element of
the UBE2C gene (27). Most of the
AR-Vs identified to date display constitutive activity, when AR-V
transcriptional activity was measured (28); therefore, these cells were cultured
with 10% charcoal-stripped FBS. As shown in Fig. 4C and D, erastin caused the effective
inhibition of AR-V trans-activating activity.
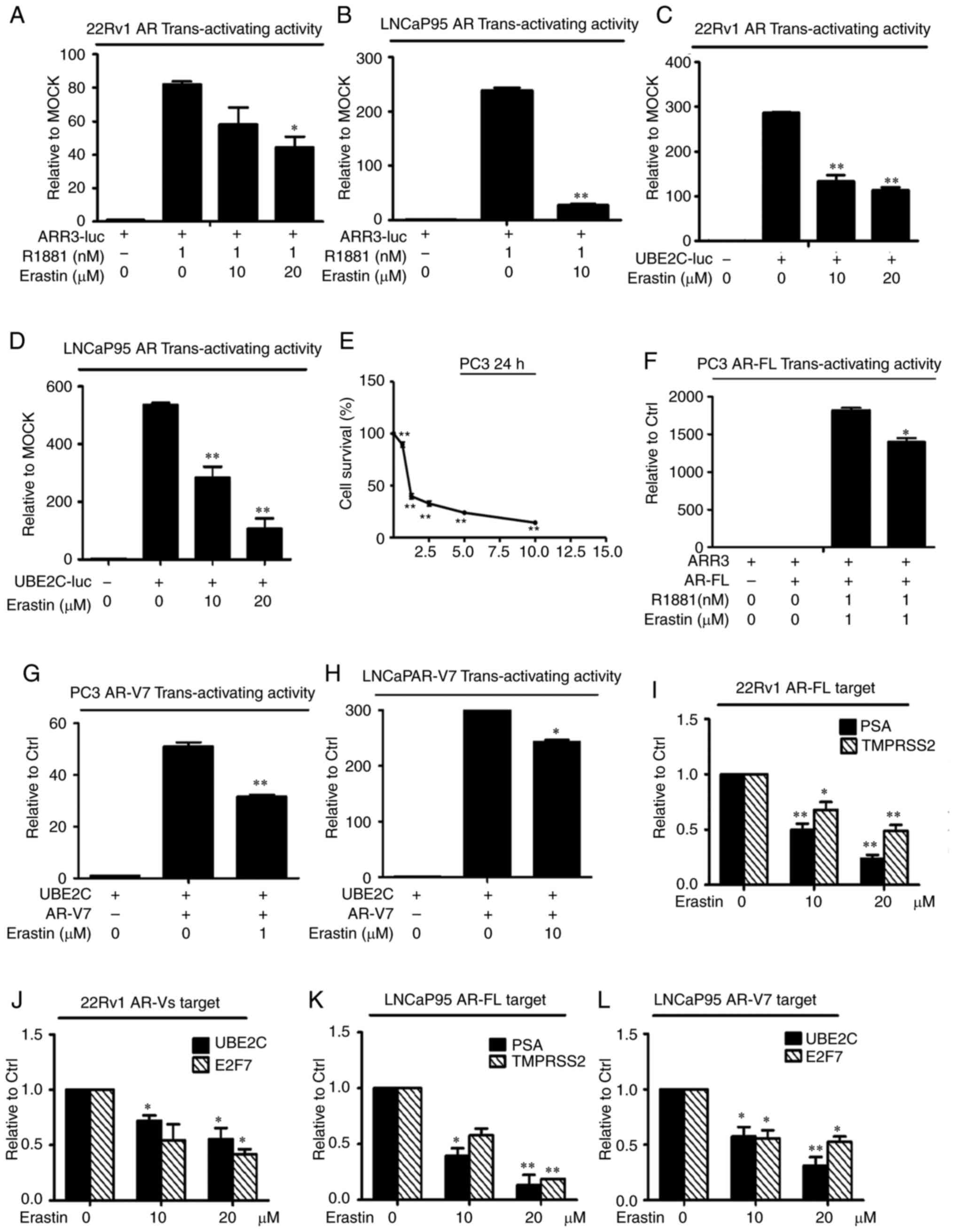 | Figure 4.Erastin downregulates AR-FL and AR-V
transactivation. A luciferase assay showed that erastin inhibited
endogenous (A and B) AR-FL and (C and D) AR-V transcriptional
activity. Cells transfected with the ARR3-luc or UBE2C-luc
construct were treated with erastin. (E) Sulforhodamine B assays
showed that erastin inhibited the growth of PC3 cells in a
dose-dependent manner. Then, erastin inhibited exogenous (F) AR-FL
and (G and H) AR-V transcriptional activity in PC3 and LNCaP cells.
Reverse transcription-quantitative PCR analysis showed that erastin
decreased the levels of (I and K) AR-FL target genes PSA and
TMPRSS2 and (J and L) AR-V target genes UBE2C and E2F7. Statistical
analysis was performed using ANOVA followed by Dunnett's post hoc
test. *P<0.05 and **P<0.01 vs. control. AR, androgen
receptor; AR-V, AR splice variant; AR-FL, AR full-length; PSA,
prostate-specific antigen; TMPRSS2, transmembrane protease serine
2; UBE2C, ubiquitin-conjugating enzyme E2C; E2F7, transcription
factor E2F7; luc, luciferase plasmid; ARR3, arrestin-C. |
Considering that erastin had a significant
inhibitory effect on endogenous AR-FL and AR-V transcriptional
activity, it was speculated that erastin had an inhibitory effect
on exogenous AR activity. To test this hypothesis, the effect of
erastin on exogenously expressed AR-FL and AR-V7 trans-activating
activity was evaluated in the PC-3 cell line (null-AR). The results
showed that PC3 cells were significantly inhibited and the
exogenous AR-FL and AR-V trans-activating activity was evidently
decreased after the treatment of 1 µM erastin (Fig. 4E-G). The exogenous AR-V7
trans-activating activity was further tested in LNCaP cells
(without AR-Vs), and the results were consistent with those of the
PC3 cells (Fig. 4H).
Consistently, the mRNA levels of the AR-FL target
genes PSA and TMPRSS2 and the AR-V-specific target genes UBE2C and
E2F7 were significantly downregulated by erastin in both the 22Rv1
and LNCaP95 cell lines (Fig. 4I-L).
Collectively, these findings indicated that erastin could
downregulate AR-FL and AR-V trans-activation.
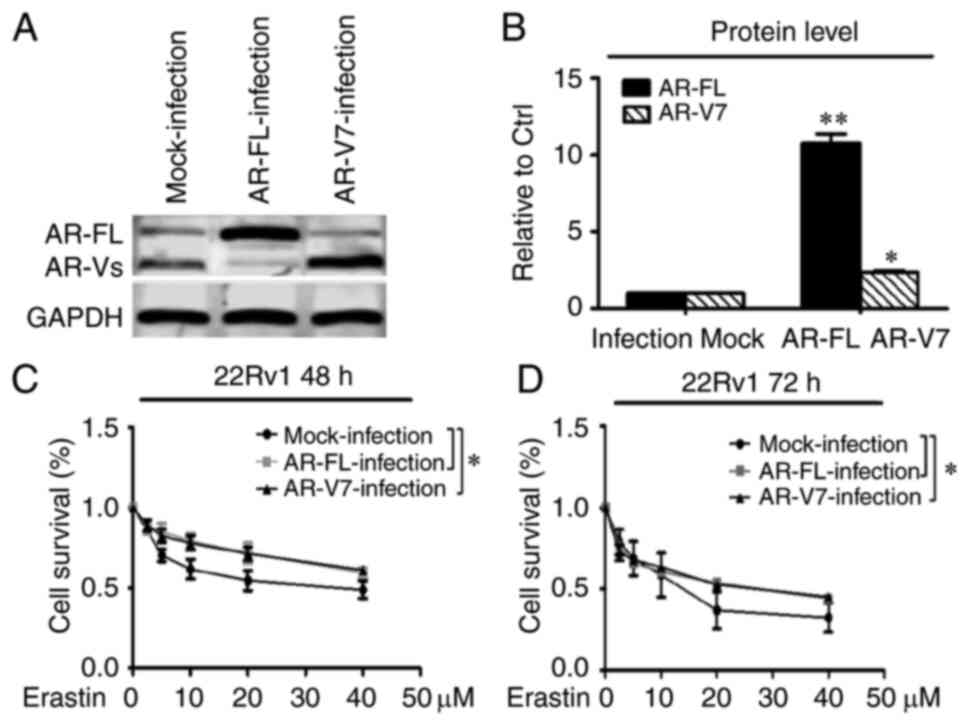
Upregulation of AR-FL and AR-V7
expression reverses the growth inhibition of erastin in 22Rv1
cells
To further test the importance of the roles of AR-FL
and AR-V in the action of erastin in CRPC cells, AR-FL and AR-V7
were overexpressed in the 22Rv1 cell line using lentiviral
infections that delivered AR-FL and AR-V7 RNA expression
constructs, and growth inhibition was evaluated using the SRB
method. As shown in Fig. 5A and B,
the expression of AR-FL and AR-V7 proteins in the 22Rv1 cells was
significantly upregulated, especially the AR-FL protein. The
overexpression of AR-FL and AR-V7 significantly promoted the
resistance to high concentrations of erastin after 48 and 72 h
compared with controls in the 22Rv1 cells. However, there was no
significant difference between the overexpression of AR-FL and
AR-V7 protein (Fig. 5C and D).
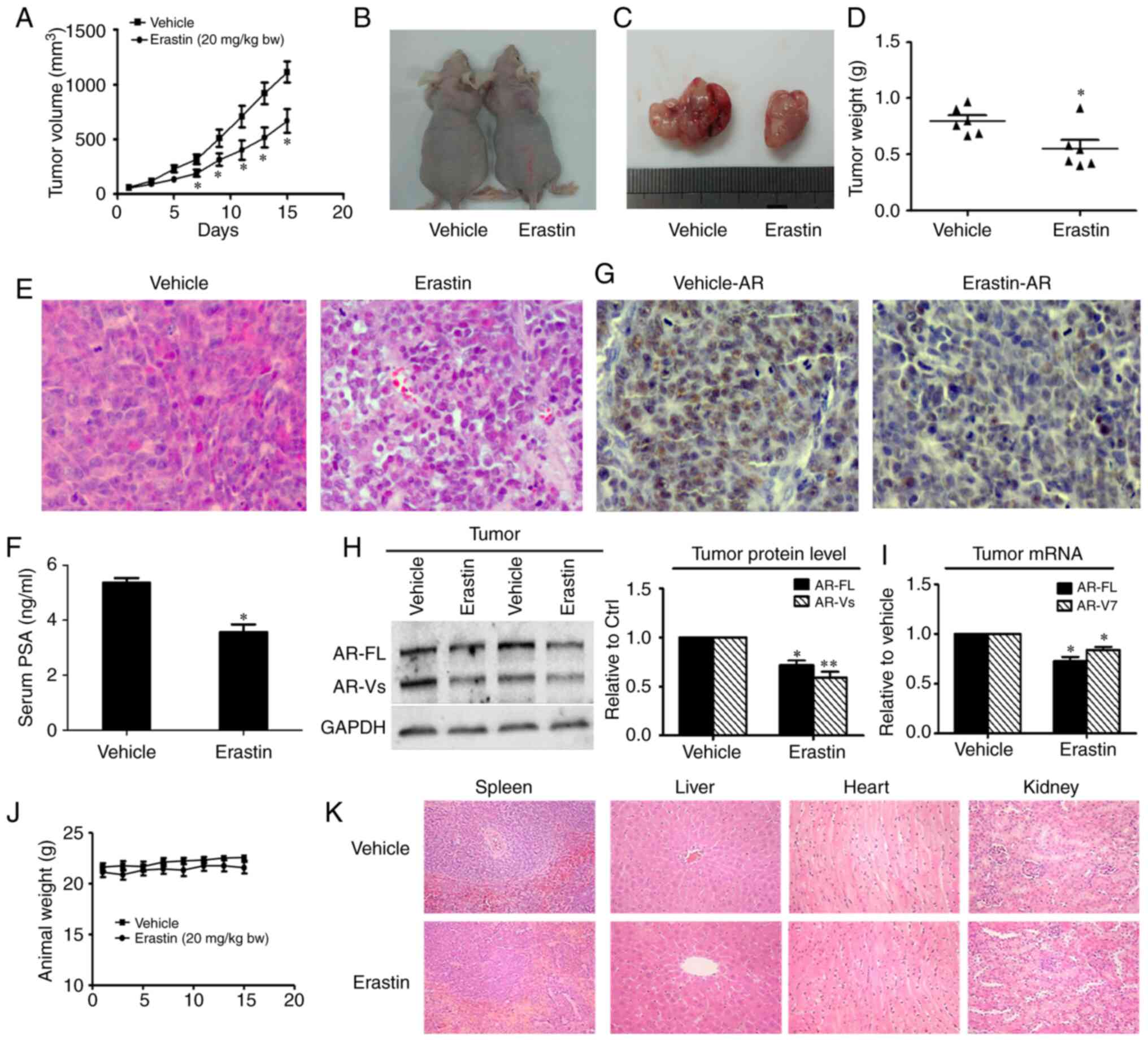
Erastin inhibits the 22Rv1 ×enograft
tumor growth rate
To investigate whether erastin inhibits the tumor
growth in vivo, 22Rv1 cells were implanted into the
subcutaneous space of immune-deficient nude mice. The tumor growth
curve results are shown in Fig. 6A
and revealed that erastin inhibited the growth of 22Rv1 tumors,
with significance differences found on day 7 of the treatment. At
the end of the experiments, the average tumor weight in the control
group was 0.80±0.11 g, while that in the erastin-treated group was
0.55±0.17 g (Fig. 6B-D). The results
of H&E staining of the tumor tissues showed that the
nuclear/cytoplasmic ratio of tumor cells was notably decreased in
the erastin group (Fig. 6E). Given
that serum PSA concentration is one of most important clinical
indexes for the diagnosis of prostate cancer, serum PSA levels were
measured by ELISA, and a significant reduction in PSA serum levels
in response to erastin treatment was observed (Fig. 6F). Then, AR protein levels were
measured using IHC in tumor tissues. In erastin-treated tumor
specimens, AR was obviously downregulated (Fig. 6G). In addition, the protein and mRNA
levels of AR-FL and AR-V in the tumor tissues were measured by
western blotting and RT-qPCR. Both the protein levels and mRNA
levels of AR-FL and AR-V7 in the tumor tissues were decreased
(Fig. 6H and I), which was consistent
with the results obtained in vitro.
Evaluation of erastin safety in
mice
To evaluate the toxicity of erastin in vivo,
the body weights of mice were measured and mouse organs were
collected, including the heart, liver, spleen and kidney, to
observe changes in morphology by H&E staining. The mice
appeared to tolerate erastin well, and neither a significant
difference in body weight (Fig. 6J)
nor noticeable organ damage (Fig. 6K)
was detected between the treatment group and the control group.
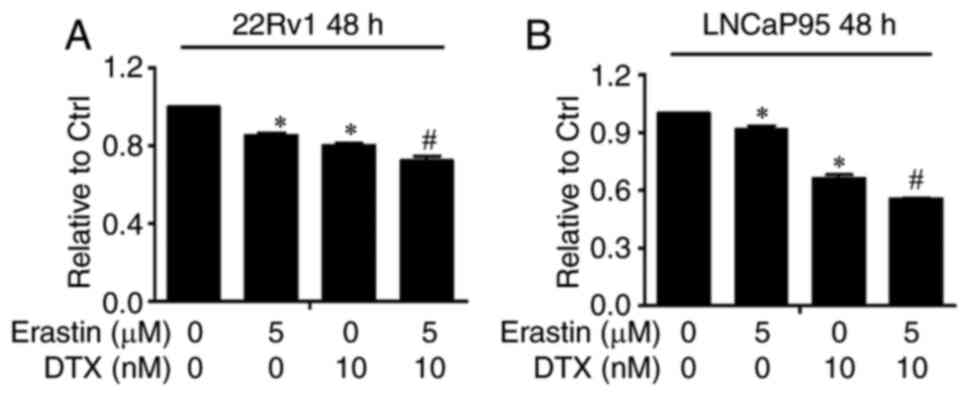
Erastin synergistically enhances the
growth inhibitory efficacy of docetaxel
Docetaxel, the standard therapy for CRPC, represents
the only class of chemotherapy drugs that prolongs the survival of
patients with CRPC. Previous studies have reported that docetaxel
induces microtubule stabilization and abrogates AR nuclear
translocation and transcriptional activity (29–31).
However, microtubule stabilization has been found to have no effect
on AR-Vs, especially AR-V7, with neither subcellular localization
nor nuclear activity affected, indicating one of the mechanisms of
docetaxel resistance (29,32). Given that erastin has the ability to
downregulate AR-V expression and activity and that the underlying
mechanism by which docetaxel inhibits AR-FL is different from that
of erastin, it was hypothesized in the present study that erastin
can enhance the efficacy of docetaxel in CRPC. To test this
hypothesis, the growth of 22Rv1 and LNCaP95 cells was measured
after treatment with erastin and docetaxel, and the combined index
values were calculated. All the combinations produced a CI value
<1, suggesting synergy between erastin and docetaxel in
inhibiting cell growth (Tables I and
II). When 5 µM erastin and 10 nM
docetaxel were added to 22Rv1 and LNCaP95 cells alone or in
combination, as shown in Fig. 7A and
B, combination therapy inhibited tumor cell growth to a
significantly greater extent than monotherapy. These data provided
preliminary support for using erastin to enhance docetaxel efficacy
in CRPC.
 | Table I.Combination index values of DTX and
erastin treatment in 22Rv1 cells. |
Table I.
Combination index values of DTX and
erastin treatment in 22Rv1 cells.
|
| Erastin, µM |
|---|
|
|
|
|---|
| DTX, nM | 2.5 | 5 | 10 |
|---|
| 5 | 0.313 | 0.717 | 0.989 |
| 10 | 0.550 | 0.615 | 0.252 |
| 20 | 0.480 | 0.706 | 0.821 |
| Table II.Combination index values of DTX and
erastin treatment in LNCaP95 cells. |
Table II.
Combination index values of DTX and
erastin treatment in LNCaP95 cells.
|
| Erastin, µM |
|---|
|
|
|
|---|
| DTX, nM | 2.5 | 5 | 10 |
|---|
| 5 | 0.765 | 0.837 | 0.961 |
| 10 | 0.545 | 0.617 | 0.868 |
Discussion
As the first ferroptosis inducer discovered, erastin
has been found to have a significant antitumor effect in multiple
types of tumors through different mechanisms (33). However, there has been relatively
little research on elastin in prostate cancer. In the present
study, erastin inhibited the proliferation of 22RV1 and LNCaP95
cells in a dose-dependent manner. Although the mechanism of
erastin-induced ferroptosis in prostate cancer remains unknown, the
ferroptosis marker GPX4 protein was downregulated in both cell
lines after treatment with erastin, indicating the inherent
ferroptosis. In addition, it was confirmed that erastin can inhibit
the expression of AR-FL and AR-V proteins by reducing the
transcriptional activity of AR-FL and AR-Vs and downregulating the
transcription level of the AR gene. In addition, in vivo
experiments confirmed that erastin inhibited the growth rate of
tumors and downregulated the levels of AR protein and mRNA in
tumors. There was no evident damage induced by erastin, as the
weight of the body and organs, such as the heart, liver, spleen and
kidney, was unaffected in the treated in mice.
While, previous studies have confirmed that erastin
exerts an antitumor effect in multiple types of cancer, such as
colorectal, breast and cervical cancer (33–35), the
results of the present study revealed a novel underlying mechanism
in prostate cancer in which erastin reduces AR-FL and AR-V protein
expression by downregulating their mRNA levels. The increased
expression of the full-length and splice variants of AR has been
indicated as an important mechanism of resistance to traditional
androgen deprivation therapy and newly developed androgen
deprivation drugs, such as abiraterone and enzalutamide (5,36,37). However, none of the anti-androgens
currently used in clinics can target AR-Vs directly to reduce their
availability. In addition to erastin, the selective AR degradants
UT-69, UT-155 and (R)-UT-155 bind to the AR transcriptional
activation domain AF-1 in the amino terminus, and UT-69 and UT-155
can also bind to the carboxy terminal LBD, significantly reducing
the activity of wild-type and splice mutants even in the presence
of small amounts of AR (38). ASC-J9,
an AR degradation enhancer, can degrade both AR-FL and AR-V7 in
22Rv1 cells and in C4-2 and C81 cells upon the addition of AR-V7
(39). These compounds may serve as
effective antidotes for overcoming resistance to androgen
deprivation therapy for the treatment of CRPC in the future.
Erastin can also enhance the efficacy of docetaxel
chemotherapy in prostate cancer (Fig. 7A
and B). It is important for AR-FL to translocate from the
cytoplasm to the nucleus to form dimers, which have transcriptional
activity (29,30,40,41).
Docetaxel has been reported to attenuate the nuclear input of AR-FL
by stabilizing microtubules, which play important roles in the
process of AR translocation (29).
However, the nuclear localization of AR-Vs, especially AR-V7, is
not dependent on microtubules, and AR-V expression is proposed to
be one of the mechanisms of docetaxel resistance. The results of
the present study showed that erastin enhanced the growth
inhibitory effect of docetaxel in CRPC cells, thus it is necessary
to determine the optimal ratio for the two drugs in combination
therapy in future studies.
One of the chemotherapeutic mechanisms of docetaxel
is the promotion of the mitochondrial release of cytochrome C,
interrupting the mitochondrial electron transport chain, and
leading to the production of a large amount of ROS, which cause
lipid peroxidation, DNA oxidation modification, protein oxidation
and inactivation of various enzymes, ultimately causing apoptosis
and necrosis (42,43). However, when CRPC cells become
resistant to docetaxel, the levels of ROS in cells are quite low
(44,45). In the process of erastin-induced
ferroptosis, the xCT light chain of the cystine/glutamate
transporter is blocked, thus depleting GSH and reducing GPX4
activity, and as a result ROS cannot be catalyzed by GPX4.
Ultimately, ROS accumulate, which can reverse the decrease in ROS
levels caused by the docetaxel resistance of CRPC cells.
In summary, the present study demonstrated that
erastin can significantly downregulate the expression and
activities of AR-FL and AR-Vs in prostate cancer in vitro
and in vivo, and enhance the growth inhibitory efficacy of
docetaxel in CRPC cells. These results indicated that erastin may
be a promising therapeutic strategy for the treatment of human
prostate cancer in the future, although further studies will be
needed.
Acknowledgements
We would like to thank Dr Alan Meeker at the Johns
Hopkins University (Baltimore, Maryland) for providing LNCaP95
cells, and to Dr Robert Matusik at Vanderbilt School of Medicine
(Nashville, Tennessee) for providing the ARR3-luc construct.
Funding
This work was supported by the National Natural
Science Foundation of China Project (grant nos. 81302206 and
81602228), Jilin Scientific and Technological Development Program
(grant no. 20160101237JC), and Project of Jilin Provincial
Department of Education (grant no. JJKH20170831KJ).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ, YY and TL designed and conducted the study. CH,
HX, WeL, JC and SW performed the experiments. YY, TL and YJ
performed the data analysis and interpretation. YX and WaL were
responsible for data collection, validation and visualization. YY
and TL drafted the manuscript with critical revision from LZ. LZ
and YY confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Ethics
Committee of Basic Medical College of Jilin University (approval
no. 2016045).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferraldeschi R, Welti J, Luo J, Attard G
and de Bono JS: Targeting the androgen receptor pathway in
castration-resistant prostate cancer: Progresses and prospects.
Oncogene. 34:1745–1757. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sun S, Sprenger CC, Vessella RL, Haugk K,
Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, et
al: Castration resistance in human prostate cancer is conferred by
a frequently occurring androgen receptor splice variant. J Clin
Invest. 120:2715–2730. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Noble RL: The development of prostatic
adenocarcinoma in Nb rats following prolonged sex hormone
administration. Cancer Res. 37:1929–1933. 1977.PubMed/NCBI
|
|
5
|
Egan A, Dong Y, Zhang H, Qi Y, Balk SP and
Sartor O: Castration-resistant prostate cancer: Adaptive responses
in the androgen axis. Cancer Treat Rev. 40:426–433. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Centenera MM, Harris JM, Tilley WD and
Butler LM: The contribution of different androgen receptor domains
to receptor dimerization and signaling. Mol Endocrinol.
22:2373–2382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chandrasekar T, Yang JC, Gao AC and Evans
CP: Mechanisms of resistance in castration-resistant prostate
cancer (CRPC). Transl Androl Urol. 4:365–380. 2015.PubMed/NCBI
|
|
8
|
Hu R, Dunn TA, Wei S, Isharwal S, Veltri
RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al:
Ligand-independent androgen receptor variants derived from splicing
of cryptic exons signify hormone-refractory prostate cancer. Cancer
Res. 69:16–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dolma S, Lessnick SL, Hahn WC and
Stockwell BR: Identification of genotype-selective antitumor agents
using synthetic lethal chemical screening in engineered human tumor
cells. Cancer Cell. 3:285–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yagoda N, von Rechenberg M, Zaganjor E,
Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM,
Boniface JJ, et al: RAS-RAF-MEK-dependent oxidative cell death
involving voltage-dependent anion channels. Nature. 447:864–868.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e025232014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sato M, Kusumi R, Hamashima S, Kobayashi
S, Sasaki S, Komiyama Y, Izumikawa T, Conrad M, Bannai S and Sato
H: The ferroptosis inducer erastin irreversibly inhibits system xc-
and synergizes with cisplatin to increase cisplatin's cytotoxicity
in cancer cells. Sci Rep. 8:9682018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze
MT, Zeh HJ, Kang R and Tang D: The ferroptosis inducer erastin
enhances sensitivity of acute myeloid leukemia cells to
chemotherapeutic agents. Mol Cell Oncol. 2:e10545492015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kwon MY, Park E, Lee SJ and Chung SW: Heme
oxygenase-1 accelerates erastin-induced ferroptotic cell death.
Oncotarget. 6:24393–24403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hasegawa M, Takahashi H, Rajabi H, Alam M,
Suzuki Y, Yin L, Tagde A, Maeda T, Hiraki M, Sukhatme VP and Kufe
D: Functional interactions of the cystine/glutamate antiporter,
CD44v and MUC1-C oncoprotein in triple-negative breast cancer
cells. Oncotarget. 7:11756–11769. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roh JL, Kim EH, Jang HJ, Park JY and Shin
D: Induction of ferroptotic cell death for overcoming cisplatin
resistance of head and neck cancer. Cancer Lett. 381:96–103. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dong Y, Zhang H, Hawthorn L, Ganther HE
and Ip C: Delineation of the molecular basis for selenium-induced
growth arrest in human prostate cancer cells by oligonucleotide
array. Cancer Res. 63:52–59. 2003.PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gordon CA, Gulzar ZG and Brooks JD: NUSAP1
expression is upregulated by loss of RB1 in prostate cancer cells.
Prostate. 75:517–526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu S, Zhao F, Zhao J, Li H, Chen J, Xia Y,
Wang J, Zhao B, Zhao S and Li N: Dioscin improves postmenopausal
osteoporosis through inducing bone formation and inhibiting
apoptosis in ovariectomized rats. Biosci Trends. 13:394–401. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng J, Zhao S, Yu X, Huang S and Liu HY:
Simultaneous targeting of CD44 and EpCAM with a bispecific aptamer
effectively inhibits intraperitoneal ovarian cancer growth.
Theranostics. 7:1373–1388. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dehm SM, Schmidt LJ, Heemers HV, Vessella
RL and Tindall DJ: Splicing of a novel androgen receptor exon
generates a constitutively active androgen receptor that mediates
prostate cancer therapy resistance. Cancer Res. 68:5469–5477. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kita K, Shiota M, Tanaka M, Otsuka A,
Matsumoto M, Kato M, Tamada S, Iwao H, Miura K, Nakatani T and
Tomita S: Heat shock protein 70 inhibitors suppress androgen
receptor expression in LNCaP95 prostate cancer cells. Cancer Sci.
108:1820–1827. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao N, Peacock SO, Lo CH, Heidman LM,
Rice MA, Fahrenholtz CD, Greene AM, Magani F, Copello VA, Martinez
MJ, et al: Arginine vasopressin receptor 1a is a therapeutic target
for castration-resistant prostate cancer. Sci Transl Med.
11:eaaw46362019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W,
Gambhir SS, Lee P, Sartor O, Flemington EK, et al: Androgen
receptor splice variants dimerize to transactivate target genes.
Cancer Res. 75:3663–3671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Paschalis A, Sharp A, Welti JC, Neeb A,
Raj GV, Luo J, Plymate SR and de Bono JS: Alternative splicing in
prostate cancer. Nat Rev Clin Oncol. 15:663–675. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thadani-Mulero M, Portella L, Sun S, Sung
M, Matov A, Vessella RL, Corey E, Nanus DM, Plymate SR and
Giannakakou P: Androgen receptor splice variants determine taxane
sensitivity in prostate cancer. Cancer Res. 74:2270–2282. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Darshan MS, Loftus MS, Thadani-Mulero M,
Levy BP, Escuin D, Zhou XK, Gjyrezi A, Chanel-Vos C, Shen R, Tagawa
ST, et al: Taxane-induced blockade to nuclear accumulation of the
androgen receptor predicts clinical responses in metastatic
prostate cancer. Cancer Res. 71:6019–6029. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu ML, Horbinski CM, Garzotto M, Qian DZ,
Beer TM and Kyprianou N: Tubulin-targeting chemotherapy impairs
androgen receptor activity in prostate cancer. Cancer Res.
70:7992–8002. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bai S, Zhang BY and Dong Y: Impact of
taxanes on androgen receptor signaling. Asian J Androl. 21:249–252.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao Y, Li Y, Zhang R, Wang F, Wang T and
Jiao Y: The role of erastin in ferroptosis and its prospects in
cancer therapy. Onco Targets Ther. 13:5429–5441. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huo H, Zhou Z, Qin J, Liu W, Wang B and Gu
Y: Erastin disrupts mitochondrial permeability transition pore
(mPTP) and induces apoptotic death of colorectal cancer cells. PLoS
One. 11:e01546052016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu M, Gai C, Li Z, Ding D, Zheng J, Zhang
W, Lv S and Li W: Targeted exosome-encapsulated erastin induced
ferroptosis in triple negative breast cancer cells. Cancer Sci.
110:3173–3182. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Antonarakis ES, Lu C, Wang H, Luber B,
Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et
al: AR-V7 and resistance to enzalutamide and abiraterone in
prostate cancer. N Engl J Med. 371:1028–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li Y, Chan SC, Brand LJ, Hwang TH,
Silverstein KA and Dehm SM: Androgen receptor splice variants
mediate enzalutamide resistance in castration-resistant prostate
cancer cell lines. Cancer Res. 73:483–489. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ponnusamy S, Coss CC, Thiyagarajan T,
Watts K, Hwang DJ, He Y, Selth LA, McEwan IJ, Duke CB, Pagadala J,
et al: Novel selective agents for the degradation of androgen
receptor variants to treat castration-resistant prostate cancer.
Cancer Res. 77:6282–6298. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yamashita S, Lai KP, Chuang KL, Xu D,
Miyamoto H, Tochigi T, Pang ST, Li L, Arai Y, Kung HJ, et al:
ASC-J9 suppresses castration-resistant prostate cancer growth
through degradation of full-length and splice variant androgen
receptors. Neoplasia. 14:74–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang G, Liu X, Li J, Ledet E, Alvarez X,
Qi Y, Fu X, Sartor O, Dong Y and Zhang H: Androgen receptor splice
variants circumvent AR blockade by microtubule-targeting agents.
Oncotarget. 6:23358–23371. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shan X, Danet-Desnoyers G, Aird F, Kandela
I, Tsui R, Perfito N and Iorns E: Replication study: Androgen
receptor splice variants determine taxane sensitivity in prostate
cancer. PeerJ. 6:e46612018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cristofani R, Montagnani Marelli M,
Cicardi ME, Fontana F, Marzagalli M, Limonta P, Poletti A and
Moretti RM: Dual role of autophagy on docetaxel-sensitivity in
prostate cancer cells. Cell Death Dis. 9:8892018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang L, Chen H, Liu F, Madigan MC, Power
CA, Hao J, Patterson KI, Pourgholami MH, O'Brien PM, Perkins AC and
Li Y: Monoclonal antibody targeting MUC1 and increasing sensitivity
to docetaxel as a novel strategy in treating human epithelial
ovarian cancer. Cancer Lett. 300:122–133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang D, Cui Y, Niu L, Xu X, Tian K, Young
CYF, Lou H and Yuan H: Regulation of SOD2 and β-arrestin1 by
interleukin-6 contributes to the increase of IGF-1R expression in
docetaxel resistant prostate cancer cells. Eur J Cell Biol.
93:289–298. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shan W, Zhong W, Zhao R and Oberley TD:
Thioredoxin 1 as a subcellular biomarker of redox imbalance in
human prostate cancer progression. Free Radic Biol Med.
49:2078–2087. 2010. View Article : Google Scholar : PubMed/NCBI
|