1. Introduction
The androgen receptors play a central role in
prostate cancer development, their activation stimulates
proliferation and inhibits apoptosis of prostate cancer cells
(1). Androgen deprivation therapy
is thus the standard first line treatment for advanced prostate
cancer (2). When testicular
androgen production is suppressed and/or androgen receptor
signaling is prevented, clinical improvement is observed.
Unfortunately, these patients invariably relapse and develop
castration resistant prostate cancer (CRPC) within 18–24 months
(3). Relapse is due to
amplification of the expression (4), or to mutations of androgen receptors
allowing their activation by progesterone, estrogens (5) and androgen antagonists (6) or to androgen neo-synthesis in
prostate tumor or adrenals (7).
Furthermore, nuclear factor κB (NF-κB) and its target interleukin-8
(IL-8) contribute to androgen-independence of prostate cancer cells
by stimulating their proliferation (8,9).
It was shown that docetaxel increases significantly
the overall survival (OS) of CRPC patients with 16.5 months in the
prednisome plus mitoxantrone group and 18.9 in the prednisone plus
docetaxel group (10). The
analysis of 12 randomized trials provides evidence of clinical
benefit for docetaxel-based combination chemotherapy for CRPC
patients (11). Thus, the standard
treatment of CRPC patients remains docetaxel-based chemotherapy
(12). However, resistance to
docetaxel is a significant clinical problem given that about half
of patients do not show prostate specific antigen (PSA) response
(10,13). They are either spontaneously
resistant to docetaxel, or they become docetaxel resistant, but
finally and rapidly, the tumor progression goes or comes back in
all patients. To bypass this resistance, several new therapies have
been developed including cellular immunotherapy or hormonotherapy.
But to overcome docetaxel resistance it is critical to precise its
molecular mechanisms. Various mechanisms of taxane (docetaxel and
paclitaxel) resistance have been studied, but there is no consensus
to date regarding a definition of taxane resistance (14).
We reviewed docetaxel resistance in CRPC patients
(in vivo) and cell lines (in vitro). We start with a
description of mechanisms of action of docetaxel toxicity in CRPC
cells, mechanisms of resistance of CRPC to docetaxel, then we
provide insight into several biological markers of docetaxel
resistance and finally we present new prostate cancer therapies
proposed to overcome docetaxel resistance.
2. Mechanisms of action of docetaxel
toxicity in CRPC cells
Docetaxel is known to bind the β subunits of tubulin
in microtubules, which stabilizes them, inhibits their
depolymerization (15,16) blocks mitosis and finally induces
apoptosis (16,17). In CRPC cells, docetaxel induces
B-cell lymphoma 2 (Bcl-2) phosphorylation that prevents its
heterodimerization and leads to caspase activation and apoptosis
(18) in vivo and in
vitro (19). Apoptosis could
be induced via mitotic catastrophe (18) or could be independent (20). Moreover, docetaxel was able to
induce apoptosis by nuclear interaction of Smac-DIABLO with
survivin (an inhibitor of apoptosis protein, IAP, that regulates
the cell cycle) (21).
In addition, docetaxel reduces the expression of
androgen receptor on CRPC cells (22) and its mechanism of action seems to
involve androgen receptor (AR) nuclear localization and signalling
inhibition.
3. Resistance of CRPC to docetaxel
Prostate cancer cell population is heterogeneous,
including stem cells or neuroendocrine cells, and either uses
multiple mechanisms of resistance at the same time, leading to
selection of docetaxel resistant cells or is modified by docetaxel
to become resistant. To study docetaxel resistance, human prostate
cancer parental cells and their docetaxel-resistant derived cell
lines are currently used, as the androgen-dependent cell line
LNCaP, or the androgen-independent cell lines, PC3, DU145 and
22RV1.
Resistance of CRPC to docetaxel can be related to
endogenous cell mechanisms by production of secreted clusterin
(CLU) for example or to characteristics of the tumour
micro-environment, as hypoxia.
Endogenous CRPC cell mechanisms of
resistance to docetaxel
Some mechanisms of docetaxel resistance in prostate
cancer include constitutive production of molecules as multidrug
resistance proteins, p53, CLU. Some of these proteins may be
mutated (p53, Bax) or overexpressed [class III β-tubulin,
protease-activated receptor 1 (PAR1 or thrombin receptor)].
Furthermore, proteins constitutively expressed as CLU can be
overexpressed by stress signal and create a positive loop with
enhanced AKT signaling pathway. The ways of docetaxel resistance in
CRPC cells are thus numerous and cross overs exist (23). We spotlight below mechanisms
involving drug efflux, drug target, apoptosis, survival pathways,
inflammation mediators, as cytokines or chemokines and chaperone
molecules.
Drug efflux
Multidrug resistance proteins (MDRP) including P-gp
(P-glycoprotein), MRP1 (multidrug resistance-associated protein 1)
and BCRP (breast cancer resistance protein), act as pumps of drug
efflux with a wide range of substrates including docetaxel, and in
various tissues including prostate cancer cells. In CRPC patients
receiving docetaxel, MDR1 genetic variations are associated with
docetaxel resistance (24). P-gp
is not expressed in PC3 cells, while DU145 and 22RV1
docetaxel-resistance derived cell lines, overexpress P-gp by
comparison with parental cells, and inhibition of P-gp restores
their reactivity to docetaxel (25). In addition, docetaxel resistance
can be induced by phosphorylation of BCRP by serine/threonine
kinase PIM-1 in CRPC cells (26).
Drug target
By targeting β-tubulin, docetaxel can block CRPC
cell mitosis (see above). Moreover, class III β-tubulin isoform,
that reduced taxane binding, is highly upregulated in CRPC cells
(27). The overexpression of this
isoform confers docetaxel resistance to CRPC cells and inhibiting
class III β-tubulin expression increases their sensitivity to
docetaxel (28). Impaired tubulin
polymerization by docetaxel can also be evidenced in an LNCaP
derived docetaxel-resistant cell line and explained by an F270I
mutation in class I β-tubulin (29).
Blocking apoptosis
p53 protein by regulating the cell cycle and
apoptosis plays a key role in docetaxel resistance. It is
frequently overexpressed in prostate cancer, which is often
associated with mutations (30).
LNCaP cells (bearing wild-type p53) are more resistant to docetaxel
than DU145 (bearing mutant p53) and PC3 (lacking p53) cells.
Knocking down p53 sensitizes LNCaP cells to docetaxel, indicating
that p53 is involved in docetaxel resistance in prostate cancer
cells (31).
Bcl-2 mutation is an extremely rare event in
prostate cancer. However, it is shown in CRPC that missense
mutation affecting the BH3 domain of Bax is involved in the
formation of Bax-Bax and Bax-Bcl-2 dimers (32). It is, therefore, possible that the
pro-apoptotic activity of Bax-Bax dimer is lost. Modulation of the
expression of Bcl-xL, an anti-apoptotic member of the Bcl-2 family,
in PC3 cells renders cells more or less resistant to docetaxel,
showing that resistance of these cells to docetaxel is linked to
Bcl-xL level of expression (33).
In addition, activation of PAR1 decreases docetaxel
induced apoptosis through NF-κB activation and the upregulation of
Bcl-xL (34). PAR1 is
overexpressed in prostate cancer and its ligand thrombin is also
overexpressed in prostate cancer patients (34), thus, this signaling pathway could
be involved in vivo in the resistance to docetaxel.
Survival pathways
By itself, docetaxel can induce survival pathways.
Indeed, the binding of docetaxel to β-tubulin induces a stress
signal that in return activates different survival signaling
pathways such as c-Jun N-terminal kinase (JNK), leading to
transcription factor activation as signal transducers and activator
of transcription-1 (STAT-1), STAT-3, NF-κB. STAT proteins are a
family of transcription factors that regulates gene expression to
influence differentiation, proliferation, apoptosis and
angiogenesis. STAT-1 is involved when DU145 and PC3 CRPC cells are
made resistant to docetaxel and this is CLU-dependent (35). Furthermore, STAT-3 is also
implicated in the resistance to docetaxel but via the
serine-threonine kinase PIM1 that improves the survival of DU145
cells treated with docetaxel (36).
NF-κB in the cytoplasm is inhibited by IκB-α. Upon
stimulation of IκB kinases (IKK), IκB-α is degraded leaving NF-κB
free to translocate to the nucleus and activate a wide variety of
survival genes. NF-κB is activated constitutively through IKK
activation in both DU145 and PC3 cells but not in LNCaP cells
(37–39). The constitutive activation of NF-κB
prevents the cytotoxic activity of docetaxel, since its inhibition
increases docetaxel cytotoxicity in the three cell lines and that
docetaxel enhances NF-κB activation (31,39).
However, when PC3 cells are rendered docetaxel resistant,
inhibition of NF-κB activity before docetaxel treatment, increases
docetaxel cytotoxicity (31).
Cytokines and chemokines
Among the targets of NF-κB are the genes that encode
IL-6 and IL-8. These cytokines stimulate prostate cancer cell
growth in an autocrine and paracrine manner and are involved in the
development and progression of prostate cancer (40–43).
PC3 and DU145 cells, instead of LNCaP cells, show elevated IL-6 and
IL-8 production in conditioned media due to constitutive NF-κB
activity (40,42). Elevated IL-6 expression in CRPC
cells activates the JAK-STAT pathway. IL-6 and IL-8 productions
enhance proliferation and inhibit apoptosis in PC3 and DU145 cells
(42). By these pathways, IL-6 and
IL-8 are effectors of NF-κB in the resistance of CRPC cells to
docetaxel (39,42).
Another inflammatory molecule, the chemokine (C-C
motif) ligand 2 (CCL2), is involved in the resistance of CRPC cells
to docetaxel. CCL2 is expressed in prostate primary tumors
(44) and correlated with their
malignant potential (45). CCL2 is
also expressed in prostate cancer cell lines (44), its expression is increased in CRPC
cells by docetaxel (46). The
binding of docetaxel to β-tubulin is sensed by microtubule
interacting proteins which leads to activation of JNK signaling.
Docetaxel via the JNK pathways upregulates CCL2 expression in CRPC
cells (46). CCL2 acts in an
autocrine manner to stimulate the Erk/MAP kinase and PI3K/AKT
signaling pathways to promote tumor cell survival and resistance to
docetaxel (47,48).
Transforming growth factor-β1 (TGF-β1) is
upregulated by the transcriptional factors Twist1 and YB-1, and
participate to the docetaxel resistance of CRPC cells, as
demonstrated in PC3 cells (49).
By whole genome arrays, it was shown that TGF-β receptor III
(TGFBR3) is upregulated in both PC3 and DU145 docetaxel-resistant
cell lines and interestingly other TGF-β members appear deregulated
in the same network (TGFB2, TGF-β and LTBP2 for latent TGF-β
binding protein 2) (50).
The macrophage inhibitory cytokine-1 (MIC-1, also
named growth differenciation factor 15 GDF15) is a member of TGF-β
superfamily expressed in numerous cells including epithelial cells
and prostate cancer cells but its expression is higher in cancer
cells and increased with cancer progression, particularly in
prostate tumors and CRPC cells (51). Transcription of MIC-1 is suppressed
by Egr-1 and p53, and activated by castration (52), hypoxia (53), HIF-1α and NF-κB (reviewed in ref.
54). MIC-1 overexpression in CRPC
cells or their exposure to recombinant MIC-1 enhance their
resistance to docetaxel (55).
MIC-1 is upregulated in PC3 cells made resistant to docetaxel and
its downregulation by siRNA sensitizes to docetaxel (56). Enhanced level of secreted MIC-1 in
PC3 cells is associated with their acquisition of
epithelial-mesenchymal transition phenotype and docetaxel
resistance, the downregulation of MIC-1 improves the efficacy of
docetaxel cytotoxicity even in prostate cancer stem/progenitor
cells (57). Therefore, docetaxel
resistance and MIC-1 expression are directly linked in the total
prostate cancer cell mass.
Chaperone molecules
Chaperone proteins are key components of alternative
growth factor pathways upregulated in CRPC and are involved in
docetaxel resistance pathways.
Heat shock proteins, notably HSP27 and HSP90 are
overexpressed in CRPC cells and strategies to inhibit their
expression restore docetaxel sensitivity. Nevertheless, clinical
trials with HSP90 inhibitors have to date not succeed in improving
overall survival, but trials with HSP27, second generation
antisense drug (OGX-427) have shown a decrease in circulating tumor
cells (CTC) in mCRPC patients, validating this chaperone
therapeutic target. An ongoing study is now conducted with
co-treatment of CRPC patients with docetaxel and OGX-427 (58). Clinical studies in CRPC patients
targeting these chaperone molecules HSP90 and HSP27 and the
chaperone clusterin are reported in Table I.
 | Table IReported clinical studies targeting
chaperone molecules in castration resistant prostate cancer (CRPC)
patients. |
Table I
Reported clinical studies targeting
chaperone molecules in castration resistant prostate cancer (CRPC)
patients.
| Pathway | Inhibitors | Phase | Population | Intervention | Outcome | Patient Nb | Main ID |
|---|
| HSP90 | IPI504 | II | CRPC | IPI504 | No activity | 19 | NCT00564928 |
| 17-AAG | I | mCRPC + other
cancers | 17-AAG+DTX | Minor
responses | 80 | NCT00058253 |
| STA-9090 | II | mCRPC pretreated
DTX | STA-9090 | Suspended | 56 | NCT01270880 |
| HSP27 | OGX-427 | I | CRPC + other
cancers | OGX-427 + DTX | Ongoing study, not
recruiting | 54 | NCT00487786 |
| Clusterin | OGX-011 | I | CRPC + other
cancers | OGX-011 + DTX | Serum clusterin
decrease | 30 | NCT00471432 |
| OGX-011 | II | DTX resistant
CRPC | OGX-011 + DTX or
MTX | Low serum CLU
linked to superior survival | 70 | NCT00327340 |
| OGX-011 | II | mCRPC | DTX+/− OGX-011 | Improved survival
(23.8, 16.9%) | 82 | NCT00258388 |
| OGX-011 | III (synergy) | mCRPC chemotherapy
naive | DTX +/−
OGX-011 | Ongoing study, not
recruiting | 1000 | NTC01188187 |
| OGX-011 | III (tropic) | DTX-resistant
CRPC | OGX-011 +
Taxane | Ongoing study, not
recruiting | 292 | NCT01083615 |
| OGX-011 | III (affinity) | DTX-resistant
CRPC | Cabazitaxel +/−
OGX-011 | Recruiting | 630 | NCT01578655 |
CLU is a key protein in the resistance to cancer
chemotherapy, especially in the resistance of prostate cancer to
docetaxel. CLU exists in two forms: a truncated nuclear form (nCLU)
and a secreted form (sCLU). nCLU promotes CRPC cell death, however,
nCLU is not detected in prostate tumor cells (59,60)
and in contrast sCLU prevents cell death. nCLU and sCLU are not
produced at the same time, there is a shift from nCLU to sCLU
production or vice versa (61),
(reviewed in ref. 62). The level
of overexpression of sCLU in docetaxel PC3-resistant cells is
correlated to their level of docetaxel resistance (25). sCLU knockdown using antisense
oligonucleotide increases the sensitivity of resistant PC3 cells
in vitro (63) and in
vivo (64). sCLU expression is
induced by docetaxel via STAT-1 in PC3 and DU145 cells, it is
higher in cells made resistant to docetaxel than in parental cells
and inhibiting sCLU expression restores the reactivity to docetaxel
(35,63). It has been shown that AKT
inhibition, either by a pharmacologic agent or by overexpression of
dominant-negative AKT, suppresses CLU expression in the docetaxel
resistant CRPC cells and sensitizes them again to docetaxel
(59). Furthermore, these
experiments and the transfection of constitutive active AKT that
induces STAT-1 activation, show that CLU expression is dependent of
AKT. CLU exerts its anti-apoptotic effect by binding unfolded
proteins to prevent stress-induced protein aggregation. Notably,
its binding and stabilization of the Ku70-Bax complex is a key
factor preventing mitochondria-mediated apoptosis (60). Such CLU binding prevents the
release of Bax to the mitochondria to initiate cytochrome c
release and the resultant caspase-3-dependent apoptotic
pathway.
CLU is regulated by TGF-β1, via YB1 and contribute
to the epithelial-mesenchymal transition and metastasis of prostate
cancer cells (65). Moreover, the
early growth response-1 (Erg-1) regulates sCLU expression. An
inverse relationship between DOC-2/DAB2 interactive protein
(DAP2IP) and Erg-1 or sCLU shown in DAB2IP−/− mouse and
in CRPC patients, demonstrated that sCLU mediates the docetaxel
resistance of DAP2IP deficient prostate cancer cells, since DAP2IP
suppresses CLU expression by inhibiting Egr-1 gene transcription
(66). Thus, clusterin is a key
target to monitor and overcome docetaxel resistance in CRPC
patients and a second generation antisense drug, OGX-011 also named
custirsen, inhibiting the secretion of clusterin is now tested in 3
phase III clinical trials in CRPC patients, associated with taxane
chemotherapy, as reported in Table
I.
Microenvironment is involved in the
resistance of CRPC to docetaxel
Docetaxel resistance is due to endogenous cell
mechanisms but also to microenvironment. Microenvironment regulates
the interstitial fluid pressure that is increased in many solid
tumors (67). As a consequence,
transvascular gradient is close to zero, because microvascular
pressure is very close to the interstitial fluid pressure, reducing
the fluid filtration in solid tumors. As a result, the delivery of
drug including docetaxel in prostatic tissue is hindered (68).
Castration resistance is associated with increased
angiogenesis in experimental models of prostate cancer and in
patients (69–71). However, blood vessels of tumors,
including prostate tumors, have multiple structural and functional
abnormalities (72). Tumor vessels
are more leaky than normal vessels, that enhances the fluid
exchanges between the vascular and the interstitial space. Changes
in the transvascular gradient induce a rapid fluid redistribution
within the tumor tissue (73).
Enhancement in fluid extravasation improves the delivery of drugs
(68) including docetaxel in
CRPC.
On the other hand, docetaxel reduces tumor
angiogenesis (74). Docetaxel
decreases the number of small diameter blood vessels in PC3 tumors
induced in nude mice and acts also on endothelial cells in
vitro by inhibiting endothelial cell proliferation and
capillary development (75).
Moreover, docetaxel inhibits endothelial cell migration in
vitro and in vivo at low, not cytotoxic, doses (76). This is due to docetaxel inhibition
of the VEGF receptor activity (77). Thus, VEGF and bFGF which are
produced in PC3 and DU145 CRPC cells (78) and present in tumor microenvironment
(79) are involved in tumor
resistance to docetaxel. Consequently, during the course of the
treatment, docetaxel itself restricts its access and impairs its
delivery to tumor cells by reducing angiogenesis.
By increase of interstitial fluid pressure and
decrease of angiogenesis, oxygen diffusion also is prevented making
hypoxic areas in numerous solid tumors including prostate tumors
(80). Hypoxic conditions elicit
cellular responses designed to improve cell survival. The most
important protein regulating the molecular response to hypoxia is
the hypoxia-inducible factor-1 (HIF-1). HIF-1α expression activates
the transcription of genes that are involved in angiogenesis,
glucose metabolism, cell proliferation, apoptosis (decrease) and
invasion (increase). All these responses would oppose docetaxel
antitumor effect, thus, hypoxia is probably involved in docetaxel
resistance. However, the few reports dealing with this subject show
no modification of docetaxel cytotoxicity against CRPC cells
between normoxia or hypoxia (81)
or a decrease of apoptosis and an increase of necrosis under
hypoxia, but no changes in the overall cytotoxicity (82).
Moreover, the tumor microenvironment is
characterized by modified composition of extracellular matrix,
infiltration of immune cells and release of numerous molecules as
cytokines and chemokines. Stroma protects PC3 cells from docetaxel
cytotoxicity: inhibition of CXCR4 or CXCL12 with antagonist or
antibody, sensitized CRPC cells for docetaxel in vivo or
in vitro in the presence of bone marrow derived stromal
cells or of conditioned media from these cells (83). CXCR4 is the receptor of CXCL12, it
is expressed at the surface of most of tumor cells (84,85)
and is an independent prognostic factor for poor overall survival
in prostate cancer (86). Prostate
cancer cells home to the bone marrow toward a CXCL12 gradient
(87,88), CXCL12 binding to CXR4 protects CRPC
cells to docetaxel cytotoxicity (83).
The various intrinsic and extrinsic docetaxel
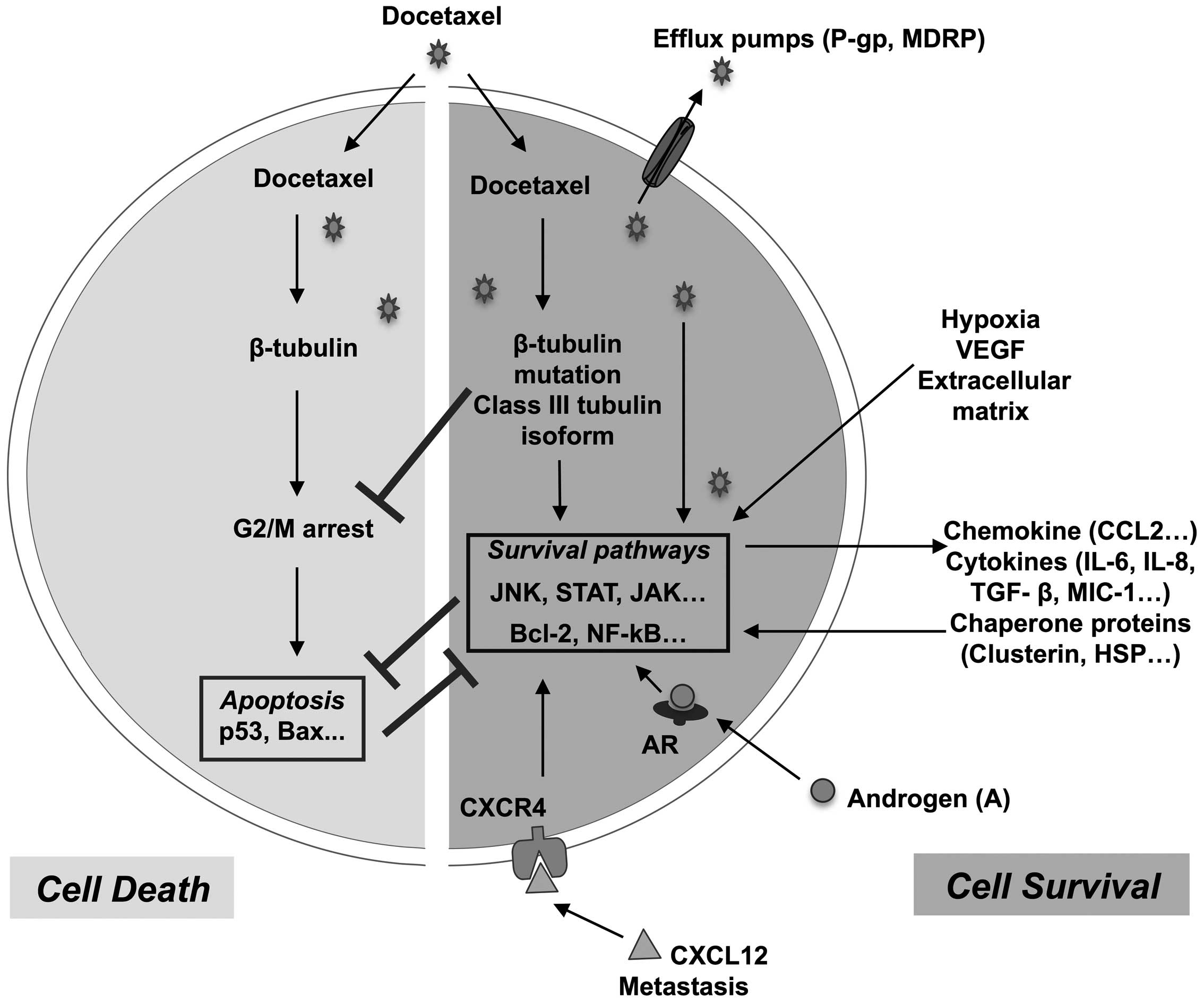
resistance mechanims are summerized in Fig. 1.
4. Biological markers of docetaxel
resistance
Genomic and proteomic comparisons of CRPC cells or
clinical samples from CRPC patients, treated or not with docetaxel,
reveal various proteins that are expressed when the resistance is
developing (50,89). It is noteworthy that a protein of
resistance is not necessarily a marker of resistance. There is an
implication of the protein of resistance in the docetaxel
resistance. While there is a correlation between the biological
marker of resistance, its expression in the tumor or elsewhere (in
the blood), and the resistance of tumor to docetaxel, that does not
mean an involvement of the biological marker in the resistance. On
the other hand, the correlation has to be evidenced in vitro
and in clinical trials, to be shown as general and reproducible.
Most, if not all, of the intracellular molecules that are candidate
cannot be specified as biological markers, mainly because of the
tumor heterogeneity that hinder this correlation and regardless to
the difficulty to monitor an intracellular biological marker of
resistance that would require tumor sampling during the treatment
(90). Thus, we will focus this
chapter on markers of resistance to docetaxel that are measured in
the blood (serum) of patients and easily monitored during their
treatment. There is consensus to accept some proteins as biological
markers of resistance, i.e. prostate specific antigen, and the
doubling time of its blood concentration, among the candidates are
CLU, MIC-1 and IL-6. Other molecules, such as CCL2, are associated
with the resistance of CRPC cells to docetaxel but are not
acknowledged as biological markers in patients.
Prostate specific antigen (PSA)
PSA is a serine protease which is routinely used as
a biological detection and diagnostic marker for prostate cancer
(91,92). PSA concentration is associated with
tumor volume and the velocity of its concentration doubling time in
the blood of patients is well correlated to cytotoxic treatment
effectiveness (93). This
parameter is used to monitoring treatment efficacy, for the
follow-up of prostate cancer patients and is as a classical outcome
measure in prostate cancer clinical trials (94,95).
Therefore, in a patient, increase in PSA blood concentration is a
biological marker of resistance to docetaxel of prostate tumors
(96,97).
PSA is, to date, the only relevant marker for
prostate cancer, nevertheless, it has a low predictive value.
Indeed, PSA does not discriminate between aggressive and
non-aggressive prostate tumors (98) and in this way PSA is not a
validated intermediate endpoint biomarker of survival in advanced
prostate cancer (99). This
highlights the importance for an effective prognostic and adapted
therapeutic follow-up, to develop new blood biological markers for
CRPC patients that would help to monitor the development of drug
resistance.
Among other blood biological measures that are now
evaluated as secondary outcomes in phase II or in observational
clinical trials on CRPC patients under taxane treatment, by
docetaxel alone or during the switch from docetaxel to cabazitaxel:
i) some are based on cells such as the circulating tumor cell (CTC)
enumeration, or the evaluation of the neutrophil lymphocyte ratio
(NLR), ii) some are based on RNA analysis by targeted RT-PCR for
gene expression profile evaluation, and iii) others are based on
protein quantification such as neuroendocrine markers (chromogranin
A, neuron specific enolase and progastrin-releasing peptide), bone
markers (N-telopeptide of type I collagen and bone-specific
alkaline phosphatase) to follow neuroendocrine and bone metastatic
tumor cell populations, respectively, as well as already identified
docetaxel resistance markers the chaperone clusterin, the cytokines
MIC-1 or IL-6. Examples of reported clinical studies recruiting
CRPC patients and monitoring their taxane response by different
blood biological measures is presented in Table II.
| Table IIReported clinical studies recruiting
CRPC patients and monitoring their taxane response by biomarker
analysis on blood samples. |
Table II
Reported clinical studies recruiting
CRPC patients and monitoring their taxane response by biomarker
analysis on blood samples.
| Main ID | Study type | Treatment | Blood analysis | Outcome | Year | Patient no. |
|---|
| NCT01160705 | Observational | DTX | RNA analysis,
RT-PCR, CTC, biomarker analysis | Recruiting | 2010 | 50 |
|
ACTRN12611000540910 | Phase II | DTX | MIC-1 | Recruiting | 2011 | 150 |
| NCT01718353 | Phase II | DTX->Cabazitaxel
switch | CTC | Recruiting | 2012 | 100 |
| DRKS00004797 | Observational | DTX or
Cabazitaxel | Neuroendocrine
markers CgA, NSE, ProGRP | Recruiting | 2013 | 75 |
Clusterin
The involvement of sCLU in docetaxel resistance is
now generally agreed, numerous data show that CLU is a protein of
resistance to docetaxel. sCLU expression is higher in prostate
tumors from patients treated with docetaxel than from control
patients (63). Inhibiting
clusterin expression in CRPC cells and CRPC patients prevents
docetaxel resistance (see last paragraph). The transcription of
sCLU is under the control of AP-1 (Jun and Fos), B-MYB, c-MYC,
Egr-1, NF-κB, HIF-1α, STAT-1 and p53, some of them can be
constitutively active in prostate cancer cells (100). Moreover, some of these
transcription factors are stimulated by constitutively activated
signaling pathways, by microenvironment (hypoxia), and the
expression of CLU can be induced in prostate cancer cells by
castration. As a consequence, most CRPC cells express and produce
sCLU and this expression can be enhanced by docetaxel. sCLU is
secreted out of CRPC cells and binds to a scavenger receptor
(megalin) at the surface or prostate cancer cells, the binding
activates the PI3K/AKT pathway that induces sCLU production. Thus,
sCLU constitutive expressed is increased by docetaxel and may be
able to auto-regulate its own overexpression preventing prostate
cancer apoptosis.
Only changes of concentration of the fully processed
sCLU identified in the medium of CRPC cells is associated with the
degree of cell line resistance to docetaxel (101). sCLU shows the characteristics of
biomarker of docetaxel resistance in CRPC. Its evolution is closely
linked with the resistance of CRPC cells to docetaxel. It is easily
measured in the blood of patients and can be used to monitor the
docetaxel effect. Biochemical recurrence-free survival in patients
with elevated clusterin density was significantly shorter than in
patients with normal density (101,102). CLU is also a protein of
resistance to cytotoxic cytokines TRAIL, TNF-α, in CRPC cells
(103,104) and a resistance protein to a
variety of drugs in different tumor types: such as taxol in human
mammary cancer cells (105) or in
human ovarian cancer cells (106)
or gemcitabine in pancreatic cancer cells (107). Therefore, sCLU is a protein of
resistance that could be useful as a therapeutic predictive
biological marker in various cancers. If the sCLU blood
concentration could be used to monitoring and predict docetaxel
effectiveness in CRPC patients, inhibition of the expression of
this resistance marker provide also an therapeutic approach to
overcome docetaxel resistance.
Macrophage inhibitory cytokine 1
(MIC-1)
MIC-1 inhibits tumor growth and induces apoptosis in
the early stages of cancer, while it promotes the proliferation,
migration and metastases formation in advanced stages of disease
(108), thereby increasing the
tumor cell resistance to docetaxel (reviewed in ref. 54). MIC-1 concentration in culture media
is higher in docetaxel resistant PC3 cells than with parental cells
(55), and is increased in the
presence of docetaxel (56).
Moreover, enhancement of the concentration of MIC-1 in blood from
CRPC patients after the first cycle of docetaxel is significantly
associated with cancer progression and shorter survival (56). This enhancement is due to tumor
cell secretion since after docetaxel treatment MIC-1 expression is
increased in tumor cells of prostate cancer tissue (55).
Furthermore, blood concentration of MIC-1 is an
independent marker of high grade prostate cancer (109) and elevated blood MIC-1
concentration correlates with bone metastases (110). Also in colon cancer, serum MIC-1
level rises with increasing adenoma numbers and high-risk
recurrence (111). Thus, MIC-1
could be a predictive biological marker for docetaxel effectiveness
in CRPC patients. MIC-1 could predict, after the first cycle of
docetaxel, the early resistance in CRPC patients and allow a
decision: to continue or to stop the treatment. The evolution of
this blood marker concentration will be followed in a clinical
trial recruting 150 CRPC patients on docetaxel treatment (Table II).
Interleukin 6 (IL-6)
In CRPC patients treated with docetaxel, elevated
baseline of IL-6 concentration in serum is inversely correlated
with time to progression and overall survival. Median overall
survival was 6.8 months in patients with high IL-6 (>14 ng/ml)
vs. 16.6 months in those with low IL-6 (≤14 ng/ml) (39). IL-6 appears to be involved in
docetaxel resistance, however, conflicting data exist in the
litterature (112). Recently,
pre-treatment IL-6 level was identified as independent prognostic
factor for overall survival in CRPC patients treated with docetaxel
(n=72), this pre-treatment IL-6 level correlating with nuclear
NF-κB/p65 tumor staining and response to docetaxel, but IL-6 level
changes under treatment and did not correlate with clinical outcome
(113). As for clusterin,
strategy to inhibit IL-6 synthesis and to overcome docetaxel
resistance are under evaluation. Serum C reactive protein (CRP)
production by the liver may reflect the activity of inflammatory
cytokines such as IL-6 that can be produce by docetaxel-resistant
prostate cancer cells. CRP functions adequately as a clinical
marker, therefore, a modified Glasgow prognostic score (mGPS) has
been proposed. CRP quantification is included as an independent
prognostic factor for overall survival (114).
5. New therapies intended to overcome
docetaxel resistance of CRPC
The aims of these therapies are either to bypass
docetaxel resistance using other antitumoral pathways or to inhibit
the docetaxel resistance mechanisms.
Among the other antitumoral approaches,
immunotherapy is challenging. Sipuleucel-T in 2010 was the first
antitumoral cellular vaccine approved by FDA. In patients with
metastatic CRPC, this cell vaccine resulted in 4.1 months
improvement in the median overall survival (25.8 months in the
sipuleucel-T group vs. 21.7 months in the placebo group) (115). New generation of
hormonotherapeutics have also been approved, as the androgen
biosynthesis inhibitor abiraterone (CYP17 inhibitor) or the AR
antagonist enzalutamide (MDV 3100). In addition is the
radiopharmaceutical alpharadin (radium-223 chloride).
The number of new therapies intended to overcome
docetaxel resistance development in CRPC are far less numerous.
The therapies are based on different mechanisms. For
example, new drugs have been developed, that keep the same
molecular target but with limiting drug efflux. This is the case of
cabazitaxel, a second-generation taxane that has low affinity for
MDR proteins, unlike docetaxel, which is a known P-gp substrate.
Cabazitaxel, was recently reported to have an OS benefit in
patients with prostate cancer who have progressed on docetaxel
(116) and is now approved by
FDA.
Moreover, an inhibitor of the NF-κB survival pathway
implied in the endogenous docetaxel cell resistance pathway have
also been approved for the treatment of CRPC. This is the case of
denosumab an antibody against the receptor activator of nuclear
factor κB ligand (RANK-L).
Thus, among the treatments which have been approved
by FDA for docetaxel resistant CRPC patients, only cabazitaxel and
denosumab can be attributed more or less directly to interfere with
the pathways of endogenous docetaxel cell resistance (117).
New therapies for CRPC resistant to docetaxel are
currently tested in phase II and III trials, and many new ones are
currently on the horizon.
Target therapy to stimulate apoptosis or inhibit
survival pathways with monoclonal antibodies or small inhibitors
(as IKK inhibitors PS1145, BAY 11-7082) are under evaluation.
Concerning the targeting of chaperone molecules, this is the case
of custirsen that prevents the production of CLU induced by
docetaxel. CLU knockdown using antisense oligonucleotides or siRNA,
decreases sCLU expression in CRPC cells and reduces their
resistance to docetaxel in vitro and in vivo in nude
mice (63). A phase II trial using
a CLU inhibitor (custirsen OGX-011 an antisense inhibitor of CLU)
in association with docetaxel is ongoing. Serum CLU concentration
was negatively correlated with survival, better PSA response rates
is seen in the docetaxel arm (40 vs. 27%) and overall survival
confirms this (14.7 vs. 11.4 months) (118). Three phase III studies looking at
the activity of custirsen both in combination with taxanes
(docetaxel or cabazitaxel) and after docetaxel are currently
underway (Table I).
Cytokines are also potential targets to overcome
docetaxel resistance. A phase I study of a chimeric antibody
against IL-6 (Siltuximab) given combined with docetaxel
demonstrates the safety of this approach and shows preliminary
efficacy in patients with metastatic CRPC (119). As expected, CRP concentration
were suppressed during this treatment.
Moreover, targeting the tumor microenvironment is
also challenging to control docetaxel resistance development.
Inhibition of angiogenesis theoretically would not overcome
docetaxel resistance since docetaxel itself reduces CRPC
angiogenesis. However, inhibition of VEGF activity is more than the
inhibition of angiogenesis, it is to inhibit VEGF signaling.
Combination of bevacizumab, anti-VEGF antibody, with docetaxel in
CRPC docetaxel resistant patients in phase II trial showed
interesting findings. Major PSA responses were observed in previous
responders to docetaxel alone, evidencing that bevacizumab could
return activity to docetaxel (120). However, a phase III study with
docetaxel plus prednisone with or without bevacizumab failed to
obtain OS advantage and serious morbidity and mortality rates were
seen (121). Another combination
of docetaxel in first line and VEGF-trap (aflibercept) under
investigation in phase III with CRPC patients has completed accrual
and results are awaited (122).
Other clinical trials are currently investigating
the combination of tyrosine kinase inhibitors or anti-apoptotic
protein inhibitors with docetaxel in CRPC (reviewed in ref.
123) but, with anti-VEGF, they
are not truly inhibitors of resistance mechanisms induced by
docetaxel, but inhibitors of general mechanisms induced by
oncogenesis. Intermittent therapy to avoid docetaxel resistance has
not be sucessful (124). Since
many therapeutic options are under evaluation to overome docetaxel
resistance, more investigations should be performed on the optimal
combination or the sequence of treatments needed in order to
maximize the clinical benefits for the patients.
6. Conclusion
There is no standard second line treatment for CRPC
patients resistant to docetaxel. Since 2010 Food and Drug
Administration (FDA) and European Medicines Agency (EMA) have
approved six new molecules: cabazitaxel, abiraterone, enzalutamide,
Sipuleucel T, denosumab and Alpharadin. Cabazitaxel is a
second-generation taxane, abiratrone is an inhibitor of cytochrome
P450 that inhibits de novo biosynthesis of androgens,
enzalutamide is an androgen receptor antagonist, Sipuleucel T is an
immunotherapy, denosumab is an antibody against RANK ligand and
Alpharadin is a radiopharmaceutical. Most of these new molecules
are not intended to directly overcome docetaxel resistance.
Although to bypass docetaxel resistance using molecules that go by
other pathways is intellectually, a priori, more satisfactory, to
inhibit the mechanisms of docetaxel resistance could be perhaps
more secure. We know that docetaxel is effective, it would be
perhaps easier and more profitable to overcome its mechanisms of
resistance than to find new effective pathways. Indeed, as we
report in the present review many data support the investigation of
targeted therapies to enhance the antitumor activity of docetaxel,
and custirsen may give the answer.
References
|
1
|
Feldman BJ and Feldman D: The development
of androgen-independent prostate cancer. Nat Rev Cancer. 1:34–45.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yap TA, Zivi A, Omlin A and de Bono JS:
The changing therapeutic landscape of castration-resistant prostate
cancer. Nat Rev Clin Oncol. 8:597–610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eisenberger MA and Walsh PC: Early
androgen deprivation for prostate cancer? N Engl J Med.
341:1837–1838. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Visakorpi T, Hyytinen E, Koivisto P, et
al: In vivo amplification of the androgen receptor gene and
progression of human prostate cancer. Nat Genet. 9:401–406. 1995.
View Article : Google Scholar
|
|
5
|
Taplin ME, Bubley GJ, Shuster TD, et al:
Mutation of the androgen-receptor gene in metastatic
androgen-independent prostate cancer. N Engl J Med. 332:1393–1398.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Taplin ME, Bubley GJ, Ko YJ, et al:
Selection for androgen receptor mutations in prostate cancers
treated with androgen antagonist. Cancer Res. 59:2511–2515.
1999.PubMed/NCBI
|
|
7
|
Yamaoka M, Hara T and Kusaka M: Overcoming
persistent dependency on androgen signaling after progression to
castration-resistant prostate cancer. Clin Cancer Res.
16:4319–4324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Araki S, Omori Y, Lyn D, et al:
Interleukin-8 is a molecular determinant of androgen independence
and progression in prostate cancer. Cancer Res. 67:6854–6862. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu Y, Josson S, Fang F, et al: RelB
enhances prostate cancer growth: implications for the role of the
nuclear factor-κB alternative pathway in tumorigenicity. Cancer
Res. 69:3267–3271. 2009.PubMed/NCBI
|
|
10
|
Tannock IF, de Wit R, Berry WR, et al:
Docetaxel plus prednisone or mitoxantrone plus prednisone for
advanced prostate cancer. N Engl J Med. 351:1502–1512. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Serpa Neto A, Tobias-Machado M, Kaliks R,
Wroclawski ML, Pompeo AC and Del Giglio A: Ten years of
docetaxel-based therapies in prostate adenocarcinoma: a systematic
review and meta-analysis of 2244 patients in 12 randomized clinical
trials. Clin Genitourin Cancer. 9:115–123. 2011.PubMed/NCBI
|
|
12
|
Marech I, Vacca A, Ranieri G, Gnoni A and
Dammacco F: Novel strategies in the treatment of
castration-resistant prostate cancer (Review). Int J Oncol.
40:1313–1320. 2012.PubMed/NCBI
|
|
13
|
Seruga B and Tannock IF:
Chemotherapy-based treatment for castration-resistant prostate
cancer. J Clin Oncol. 29:3686–3694. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
El-Amm J and Aragon-Ching JB: The changing
landscape in the treatment of metastatic castration-resistant
prostate cancer. Ther Adv Med Oncol. 5:25–40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shelanski ML, Gaskin F and Cantor CR:
Microtubule assembly in the absence of added nucleotides. Proc Natl
Acad Sci USA. 70:765–768. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McGrogan BT, Gilmartin B, Carney DN and
McCann A: Taxanes, microtubules and chemoresistant breast cancer.
Biochim Biophys Acta. 1785:96–132. 2008.PubMed/NCBI
|
|
17
|
Zhu J, Beattie EC, Yang Y, Wang HJ, Seo JY
and Yang LX: Centrosome impairments and consequent cytokinesis
defects are possible mechanisms of taxane drugs. Anticancer Res.
25:1919–1925. 2005.PubMed/NCBI
|
|
18
|
Fabbri F, Amadori D, Carloni S, et al:
Mitotic catastrophe and apoptosis induced by docetaxel in
hormone-refractory prostate cancer cells. J Cell Physiol.
217:494–501. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kramer G, Schwarz S, Hägg M, Havelka AM
and Linder S: Docetaxel induces apoptosis in hormone refractory
prostate carcinomas during multiple treatment cycles. Br J Cancer.
94:1592–1598. 2006.
|
|
20
|
Mediavilla-Varela M, Pacheco FJ, Almaguel
F, et al: Docetaxel-induced prostate cancer cell death involves
concomitant activation of caspase and lysosomal pathways and is
attenuated by LEDGF/p75. Mol Cancer. 8:682009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim JY, Chung JY, Lee SG, et al: Nuclear
interaction of Smac/DIABLO with Survivin at G2/M arrest prompts
docetaxel-induced apoptosis in DU145 prostate cancer cells. Biochem
Biophys Res Commun. 350:949–954. 2006. View Article : Google Scholar
|
|
22
|
Kuroda K, Liu H, Kim S, Guo M, Navarro V
and Bander NH: Docetaxel down-regulates the expression of androgen
receptor and prostate-specific antigen but not prostate-specific
membrane antigen in prostate cancer cell lines: implications for
PSA surrogacy. Prostate. 69:1579–1585. 2009. View Article : Google Scholar
|
|
23
|
Seruga B, Ocana A and Tannock IF: Drug
resistance in metastatic castration-resistant prostate cancer. Nat
Rev Clin Oncol. 8:12–23. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sissung TM, Baum CE, Deeken J, et al:
ABCB1 genetic variation influences the toxicity and clinical
outcome of patients with androgen-independent prostate cancer
treated with docetaxel. Clin Cancer Res. 14:4543–4549. 2008.
View Article : Google Scholar
|
|
25
|
O’Neill AJ, Prencipe M, Dowling C, et al:
Characterisation and manipulation of docetaxel resistant prostate
cancer cell lines. Mol Cancer. 10:1262011.PubMed/NCBI
|
|
26
|
Xie Y, Xu K, Linn DE, et al: The 44-kDa
Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its
multimerization and drug-resistant activity in human prostate
cancer cells. J Biol Chem. 283:3349–3356. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Terry S, Ploussard G, Allory Y, et al:
Increased expression of class III β-tubulin in castration-resistant
human prostate cancer. Br J Cancer. 101:951–956. 2009.
|
|
28
|
Ploussard G, Terry S, Maille P, et al:
Class III β-tubulin expression predicts prostate tumor
aggressiveness and patient response to docetaxel-based
chemotherapy. Cancer Res. 70:9253–9264. 2010.
|
|
29
|
Hara T, Ushio K, Nishiwaki M, et al: A
mutation in β-tubulin and a sustained dependence on androgen
receptor signalling in a newly established docetaxel-resistant
prostate cancer cell line. Cell Biol Int. 34:177–184. 2010.
|
|
30
|
Heidenberg HB, Bauer JJ, McLeod DG, Moul
JW and Srivastava S: The role of the p53 tumor suppressor gene in
prostate cancer: a possible biomarker? Urology. 48:971–979. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gan L, Wang J, Xu H and Yang X: Resistance
to docetaxel-induced apoptosis in prostate cancer cells by
p38/p53/p21 signaling. Prostate. 71:1158–1166. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yoshino T, Shiina H, Urakami S, et al:
Bcl-2 expression as a predictive marker of hormone-refractory
prostate cancer treated with taxane-based chemotherapy. Clin Cancer
Res. 12:6116–6124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lebedeva I, Rando R, Ojwang J, Cossum P
and Stein CA: Bcl-xL in prostate cancer cells: effects of
overexpression and down-regulation on chemosensitivity. Cancer Res.
60:6052–6060. 2000.PubMed/NCBI
|
|
34
|
Tantivejkul K, Loberg RD, Mawocha SC, et
al: PAR1-mediated NF-κB activation promotes survival of prostate
cancer cells through a Bcl-xL-dependent mechanism. J Cell Biochem.
96:641–652. 2005.PubMed/NCBI
|
|
35
|
Patterson SG, Wei S, Chen X, et al: Novel
role of Stat1 in the development of docetaxel resistance in
prostate tumor cells. Oncogene. 25:6113–6122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zemskova M, Sahakian E, Bashkirova S and
Lilly M: The PIM1 kinase is a critical component of a survival
pathway activated by docetaxel and promotes survival of
docetaxel-treated prostate cancer cells. J Biol Chem.
283:20635–20644. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shimada K, Nakamura M, Ishida E, Kishi M,
Yonehara S and Konishi N: Contributions of mitogen-activated
protein kinase and nuclear factor kappa B to
N-(4-hydroxyphenyl)retinamide-induced apoptosis in prostate
cancer cells. Mol Carcinog. 35:127–137. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Palayoor ST, Youmell MY, Calderwood SK,
Coleman CN and Price BD: Constitutive activation of IκB kinase α
and NF-κB in prostate cancer cells is inhibited by ibuprofen.
Oncogene. 18:7389–7394. 1999.
|
|
39
|
Domingo-Domenech J, Oliva C, Rovira A, et
al: Interleukin 6, a nuclear factor-κB target, predicts resistance
to docetaxel in hormone-independent prostate cancer and nuclear
factor-κB inhibition by PS-1145 enhances docetaxel antitumor
activity. Clin Cancer Res. 12:5578–5586. 2006.
|
|
40
|
Zerbini LF, Wang Y, Cho JY and Libermann
TA: Constitutive activation of nuclear factor κB p50/p65 and Fra-1
and JunD is essential for deregulated interleukin 6 expression in
prostate cancer. Cancer Res. 63:2206–2215. 2003.
|
|
41
|
Michalaki V, Syrigos K, Charles P and
Waxman J: Serum levels of IL-6 and TNF-α correlate with
clinicopathological features and patient survival in patients with
prostate cancer. Br J Cancer. 90:2312–2316. 2004.
|
|
42
|
Singh RK and Lokeshwar BL: Depletion of
intrinsic expression of Interleukin-8 in prostate cancer cells
causes cell cycle arrest, spontaneous apoptosis and increases the
efficacy of chemotherapeutic drugs. Mol Cancer. 8:572009.
View Article : Google Scholar
|
|
43
|
Inoue K, Slaton JW, Eve BY, et al:
Interleukin 8 expression regulates tumorigenicity and metastases in
androgen-independent prostate cancer. Clin Cancer Res. 6:2104–2119.
2000.PubMed/NCBI
|
|
44
|
Lu Y, Cai Z, Galson DL, et al: Monocyte
chemotactic protein-1 (MCP-1) acts as a paracrine and autocrine
factor for prostate cancer growth and invasion. Prostate.
66:1311–1318. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shirotake S, Miyajima A, Kosaka T, et al:
Regulation of monocyte chemoattractant protein-1 through
angiotensin II type 1 receptor in prostate cancer. Am J Pathol.
180:1008–1016. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qian DZ, Rademacher BL, Pittsenbarger J,
et al: CCL2 is induced by chemotherapy and protects prostate cancer
cells from docetaxel-induced cytotoxicity. Prostate. 70:433–442.
2010.
|
|
47
|
Roca H, Varsos ZS and Pienta KJ: CCL2 is a
negative regulator of AMP-activated protein kinase to sustain mTOR
complex-1 activation, survivin expression, and cell survival in
human prostate cancer PC3 cells. Neoplasia. 11:1309–1317. 2009.
|
|
48
|
Loberg RD, Day LL, Harwood J, et al: CCL2
is a potent regulator of prostate cancer cell migration and
proliferation. Neoplasia. 8:578–586. 2006. View Article : Google Scholar
|
|
49
|
Shiota M, Kashiwagi E, Yokomizo A, et al:
Interaction between docetaxel resistance and castration resistance
in prostate cancer: implications of Twist1, YB-1, and androgen
receptor. Prostate. 73:1336–1344. 2013. View Article : Google Scholar
|
|
50
|
Marin-Aguilera M, Codony-Servat J, Kalko
SG, et al: Identification of docetaxel resistance genes in
castration-resistant prostate cancer. Mol Cancer Ther. 11:329–339.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Patrikainen L, Porvari K, Kurkela R,
Hirvikoski P, Soini Y and Vihko P: Expression profiling of PC3 cell
line variants and comparison of MIC-1 transcript levels in benign
and malignant prostate. Eur J Clin Invest. 37:126–133. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Karan D, Kelly DL, Rizzino A, Lin MF and
Batra SK: Expression profile of differentially-regulated genes
during progression of androgen-independent growth in human prostate
cancer cells. Carcinogenesis. 23:967–975. 2002. View Article : Google Scholar
|
|
53
|
Kelly JA, Lucia MS and Lambert JR: p53
controls prostate-derived factor/macrophage inhibitory
cytokine/NSAID-activated gene expression in response to cell
density, DNA damage and hypoxia through diverse mechanisms. Cancer
Lett. 277:38–47. 2009. View Article : Google Scholar
|
|
54
|
Mimeault M and Batra SK: Divergent
molecular mechanisms underlying the pleiotropic functions of
macrophage inhibitory cytokine-1 in cancer. J Cell Physiol.
224:626–635. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Huang CY, Beer TM, Higano CS, et al:
Molecular alterations in prostate carcinomas that associate with
in vivo exposure to chemotherapy: identification of a
cytoprotective mechanism involving growth differentiation factor
15. Clin Cancer Res. 13:5825–5833. 2007.PubMed/NCBI
|
|
56
|
Zhao L, Lee BY, Brown DA, et al:
Identification of candidate biomarkers of therapeutic response to
docetaxel by proteomic profiling. Cancer Res. 69:7696–7703. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mimeault M, Johansson SL and Batra SK:
Marked improvement of cytotoxic effects induced by docetaxel on
highly metastatic and androgen-independent prostate cancer cells by
downregulating macrophage inhibitory cytokine-1. Br J Cancer.
108:1079–1091. 2013. View Article : Google Scholar
|
|
58
|
Shiota M, Bishop JL, Nip KM, et al: Hsp27
regulates epithelial mesenchymal transition, metastasis, and
circulating tumor cells in prostate cancer. Cancer Res.
73:3109–3119. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhong B, Sallman DA, Gilvary DL, et al:
Induction of clusterin by AKT - role in cytoprotection against
docetaxel in prostate tumor cells. Mol Cancer Ther. 9:1831–1841.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang H, Kim JK, Edwards CA, Xu Z,
Taichman R and Wang CY: Clusterin inhibits apoptosis by interacting
with activated Bax. Nat Cell Biol. 7:909–915. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Pucci S, Bonanno E, Pichiorri F, Angeloni
C and Spagnoli LG: Modulation of different clusterin isoforms in
human colon tumorigenesis. Oncogene. 23:2298–2304. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shannan B, Seifert M, Leskov K, et al:
Challenge and promise: roles for clusterin in pathogenesis,
progression and therapy of cancer. Cell Death Differ. 13:12–19.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sowery RD, Hadaschik BA, So AI, et al:
Clusterin knockdown using the antisense oligonucleotide OGX-011
re-sensitizes docetaxel-refractory prostate cancer PC3 cells to
chemotherapy. BJU Int. 102:389–397. 2008. View Article : Google Scholar
|
|
64
|
Springate CM, Jackson JK, Gleave ME and
Burt HM: Efficacy of an intratumoral controlled release formulation
of clusterin antisense oligonucleotide complexed with chitosan
containing paclitaxel or docetaxel in prostate cancer xenograft
models. Cancer Chemother Pharmacol. 56:239–247. 2005. View Article : Google Scholar
|
|
65
|
Shiota M, Zardan A, Takeuchi A, et al:
Clusterin mediates TGF-β-induced epithelial-mesenchymal transition
and metastasis via Twist1 in prostate cancer cells. Cancer Res.
72:5261–5272. 2012.PubMed/NCBI
|
|
66
|
Wu K, Xie D, Zou Y, et al: The mechanism
of DAB2IP in chemoresistance of prostate cancer cells. Clin Cancer
Res. 19:4740–4749. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Heldin CH, Rubin K, Pietras K and Ostman
A: High interstitial fluid pressure - an obstacle in cancer
therapy. Nat Rev Cancer. 4:806–813. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Netti PA, Hamberg LM, Babich JW, et al:
Enhancement of fluid filtration across tumor vessels: implication
for delivery of macromolecules. Proc Natl Acad Sci USA.
96:3137–3142. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tomic TT, Gustavsson H, Wang W, Jennbacken
K, Welen K and Damber JE: Castration resistant prostate cancer is
associated with increased blood vessel stabilization and elevated
levels of VEGF and Ang-2. Prostate. 72:705–712. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gustavsson H, Welen K and Damber JE:
Transition of an androgen-dependent human prostate cancer cell line
into an androgen-independent subline is associated with increased
angiogenesis. Prostate. 62:364–373. 2005. View Article : Google Scholar
|
|
71
|
Gustavsson H, Wang W, Jennbacken K, Welen
K and Damber JE: ADAMTS1, a putative anti-angiogenic factor, is
decreased in human prostate cancer. BJU Int. 104:1786–1790. 2009.
View Article : Google Scholar
|
|
72
|
Eberhard A, Kahlert S, Goede V, Hemmerlein
B, Plate KH and Augustin HG: Heterogeneity of angiogenesis and
blood vessel maturation in human tumors: implications for
antiangiogenic tumor therapies. Cancer Res. 60:1388–1393. 2000.
|
|
73
|
Netti PA, Baxter LT, Boucher Y, Skalak R
and Jain RK: Time-dependent behavior of interstitial fluid pressure
in solid tumors: implications for drug delivery. Cancer Res.
55:5451–5458. 1995.PubMed/NCBI
|
|
74
|
Wilson C, Scullin P, Worthington J, et al:
Dexamethasone potentiates the antiangiogenic activity of docetaxel
in castration-resistant prostate cancer. Br J Cancer. 99:2054–2064.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sweeney CJ, Miller KD, Sissons SE, et al:
The antiangiogenic property of docetaxel is synergistic with a
recombinant humanized monoclonal antibody against vascular
endothelial growth factor or 2-methoxyestradiol but antagonized by
endothelial growth factors. Cancer Res. 61:3369–3372. 2001.
|
|
76
|
Hotchkiss KA, Ashton AW, Mahmood R,
Russell RG, Sparano JA and Schwartz EL: Inhibition of endothelial
cell function in vitro and angiogenesis in vivo by
docetaxel (taxotere): association with impaired repositioning of
the microtubule organizing center. Mol Cancer Ther. 1:1191–1200.
2002.PubMed/NCBI
|
|
77
|
Murtagh J, Lu H and Schwartz EL:
Taxotere-induced inhibition of human endothelial cell migration is
a result of heat shock protein 90 degradation. Cancer Res.
66:8192–8199. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Erten C, Karaca B, Kucukzeybek Y, et al:
Regulation of growth factors in hormone- and drug-resistant
prostate cancer cells by synergistic combination of docetaxel and
octreotide. BJU Int. 104:107–114. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bok RA, Halabi S, Fei DT, et al: Vascular
endothelial growth factor and basic fibroblast growth factor urine
levels as predictors of outcome in hormone-refractory prostate
cancer patients: a cancer and leukemia group B study. Cancer Res.
61:2533–2536. 2001.
|
|
80
|
Tatum JL, Kelloff GJ, Gillies RJ, et al:
Hypoxia: importance in tumor biology, noninvasive measurement by
imaging, and value of its measurement in the management of cancer
therapy. Int J Radiat Biol. 82:699–757. 2006. View Article : Google Scholar
|
|
81
|
Forde JC, Perry AS, Brennan K, et al:
Docetaxel maintains its cytotoxic activity under hypoxic conditions
in prostate cancer cells. Urol Oncol. 30:912–919. 2012. View Article : Google Scholar
|
|
82
|
Thews O, Gassner B, Kelleher DK, Schwerdt
G and Gekle M: Impact of hypoxic and acidic extracellular
conditions on cytotoxicity of chemotherapeutic drugs. Adv Exp Med
Biol. 599:155–161. 2007. View Article : Google Scholar
|
|
83
|
Domanska UM, Timmer-Bosscha H, Nagengast
WB, et al: CXCR4 inhibition with AMD3100 sensitizes prostate cancer
to docetaxel chemotherapy. Neoplasia. 14:709–718. 2012.PubMed/NCBI
|
|
84
|
Sun YX, Wang J, Shelburne CE, et al:
Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers
(PCa) in vivo. J Cell Biochem. 89:462–473. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Engl T, Relja B, Marian D, et al: CXCR4
chemokine receptor mediates prostate tumor cell adhesion through
α5 and β3 integrins. Neoplasia. 8:290–301.
2006.
|
|
86
|
Akashi T, Koizumi K, Tsuneyama K, Saiki I,
Takano Y and Fuse H: Chemokine receptor CXCR4 expression and
prognosis in patients with metastatic prostate cancer. Cancer Sci.
99:539–542. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Taichman RS, Cooper C, Keller ET, Pienta
KJ, Taichman NS and McCauley LK: Use of the stromal cell-derived
factor-1/CXCR4 pathway in prostate cancer metastasis to bone.
Cancer Res. 62:1832–1837. 2002.PubMed/NCBI
|
|
88
|
Shiozawa Y, Pedersen EA, Havens AM, et al:
Human prostate cancer metastases target the hematopoietic stem cell
niche to establish footholds in mouse bone marrow. J Clin Invest.
121:1298–1312. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Desarnaud F, Geck P, Parkin C, Carpinito G
and Makarovskiy AN: Gene expression profiling of the androgen
independent prostate cancer cells demonstrates complex mechanisms
mediating resistance to docetaxel. Cancer Biol Ther. 11:204–212.
2011. View Article : Google Scholar
|
|
90
|
Bjartell A, Montironi R, Berney DM and
Egevad L: Tumour markers in prostate cancer II: diagnostic and
prognostic cellular biomarkers. Acta Oncol. 50(Suppl 1): 76–84.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Stamey TA, Yang N, Hay AR, McNeal JE,
Freiha FS and Redwine E: Prostate-specific antigen as a serum
marker for adenocarcinoma of the prostate. N Engl J Med.
317:909–916. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Catalona WJ, Smith DS, Ratliff TL, et al:
Measurement of prostate-specific antigen in serum as a screening
test for prostate cancer. N Engl J Med. 324:1156–1161. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Smith DC, Dunn RL, Strawderman MS and
Pienta KJ: Change in serum prostate-specific antigen as a marker of
response to cytotoxic therapy for hormone-refractory prostate
cancer. J Clin Oncol. 16:1835–1843. 1998.PubMed/NCBI
|
|
94
|
Petrylak DP, Ankerst DP, Jiang CS, et al:
Evaluation of prostate-specific antigen declines for surrogacy in
patients treated on SWOG 99–16. J Natl Cancer Inst. 98:516–521.
2006.PubMed/NCBI
|
|
95
|
Colloca G: Prostate-specific antigen
kinetics as a surrogate endpoint in clinical trials of metastatic
castration-resistant prostate cancer: a review. Cancer Treat Rev.
38:1020–1026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Berry W and Eisenberger M: Achieving
treatment goals for hormone-refractory prostate cancer with
chemotherapy. Oncologist. 10(Suppl 3): 30–39. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ross RW, Galsky MD, Febbo P, et al: Phase
2 study of neoadjuvant docetaxel plus bevacizumab in patients with
high-risk localized prostate cancer: a Prostate Cancer Clinical
Trials Consortium trial. Cancer. 118:4777–4784. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Sokoll LJ, Sanda MG, Feng Z, et al: A
prospective, multicenter, National Cancer Institute Early Detection
Research Network study of [-2]proPSA: improving prostate cancer
detection and correlating with cancer aggressiveness. Cancer
Epidemiol Biomarkers Prev. 19:1193–1200. 2010.
|
|
99
|
Yap TA, Swanton C and de Bono JS:
Personalization of prostate cancer prevention and therapy: are
clinically qualified biomarkers in the horizon? EPMA J. 3:32012.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Sala A, Bettuzzi S, Pucci S, Chayka O,
Dews M and Thomas-Tikhonenko A: Regulation of CLU gene expression
by oncogenes and epigenetic factors implications for tumorigenesis.
Adv Cancer Res. 105:115–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Girard FP, Byrne J, Downes M, et al:
Detecting soluble clusterin in in-vitro and in-vivo models of
prostate cancer. Neoplasma. 57:488–493. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Miyake H, Muramaki M, Furukawa J,
Kurahashi T and Fujisawa M: Serum level of clusterin and its
density in men with prostate cancer as novel biomarkers reflecting
disease extension. Urology. 75:454–459. 2010. View Article : Google Scholar
|
|
103
|
Sensibar JA, Sutkowski DM, Raffo A, et al:
Prevention of cell death induced by tumor necrosis factor α in
LNCaP cells by overexpression of sulfated glycoprotein-2
(clusterin). Cancer Res. 55:2431–2437. 1995.
|
|
104
|
Sallman DA, Chen X, Zhong B, et al:
Clusterin mediates TRAIL resistance in prostate tumor cells. Mol
Cancer Ther. 6:2938–2947. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
So A, Sinnemann S, Huntsman D, Fazli L and
Gleave M: Knockdown of the cytoprotective chaperone, clusterin,
chemosensitizes human breast cancer cells both in vitro and
in vivo. Mol Cancer Ther. 4:1837–1849. 2005. View Article : Google Scholar
|
|
106
|
Park DC, Yeo SG, Wilson MR, et al:
Clusterin interacts with paclitaxel and confer paclitaxel
resistance in ovarian cancer. Neoplasia. 10:964–972.
2008.PubMed/NCBI
|
|
107
|
Tang Y, Liu F, Zheng C, Sun S and Jiang Y:
Knockdown of clusterin sensitizes pancreatic cancer cells to
gemcitabine chemotherapy by ERK1/2 inactivation. J Exp Clin Cancer
Res. 31:732012. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Breit SN, Johnen H, Cook AD, et al: The
TGF-β superfamily cytokine, MIC-1/GDF15: a pleotrophic cytokine
with roles in inflammation, cancer and metabolism. Growth Factors.
29:187–195. 2011.
|
|
109
|
Brown DA, Stephan C, Ward RL, et al:
Measurement of serum levels of macrophage inhibitory cytokine 1
combined with prostate-specific antigen improves prostate cancer
diagnosis. Clin Cancer Res. 12:89–96. 2006. View Article : Google Scholar
|
|
110
|
Selander KS, Brown DA, Sequeiros GB, et
al: Serum macrophage inhibitory cytokine-1 concentrations correlate
with the presence of prostate cancer bone metastases. Cancer
Epidemiol Biomarkers Prev. 16:532–537. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Brown DA, Hance KW, Rogers CJ, et al:
Serum macrophage inhibitory cytokine-1 (MIC-1/GDF15): a potential
screening tool for the prevention of colon cancer? Cancer Epidemiol
Biomarkers Prev. 21:337–346. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ignatoski KM, Friedman J, Escara-Wilke J,
et al: Change in markers of bone metabolism with chemotherapy for
advanced prostate cancer: interleukin-6 response is a potential
early indicator of response to therapy. J Interferon Cytokine Res.
29:105–112. 2009. View Article : Google Scholar
|
|
113
|
Codony-Servat J, Marin-Aguilera M, Visa L,
et al: Nuclear factor-kappa B and interleukin-6 related docetaxel
resistance in castration-resistant prostate cancer. Prostate.
73:512–521. 2013. View Article : Google Scholar
|
|
114
|
Ito M, Saito K, Yasuda Y, et al:
Prognostic impact of C-reactive protein for determining overall
survival of patients with castration-resistant prostate cancer
treated with docetaxel. Urology. 78:1131–1135. 2011. View Article : Google Scholar
|
|
115
|
Kantoff PW, Higano CS, Shore ND, et al:
Sipuleucel-T immunotherapy for castration-resistant prostate
cancer. N Engl J Med. 363:411–422. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
de Bono JS, Oudard S, Ozguroglu M, et al:
Prednisone plus cabazitaxel or mitoxantrone for metastatic
castration-resistant prostate cancer progressing after docetaxel
treatment: a randomised open-label trial. Lancet. 376:1147–1154.
2010.
|
|
117
|
Yin L, Hu Q and Hartmann RW: Recent
progress in pharmaceutical therapies for castration-resistant
prostate cancer. Int J Mol Sci. 14:13958–13978. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Saad F, Hotte S, North S, et al:
Randomized phase II trial of custirsen (OGX-011) in combination
with docetaxel or mitoxantrone as second-line therapy in patients
with metastatic castrate-resistant prostate cancer progressing
after first-line docetaxel: CUOG trial P-06c. Clin Cancer Res.
17:5765–5773. 2011. View Article : Google Scholar
|
|
119
|
Hudes G, Tagawa ST, Whang YE, et al: A
phase 1 study of a chimeric monoclonal antibody against
interleukin-6, siltuximab, combined with docetaxel in patients with
metastatic castration-resistant prostate cancer. Invest New Drugs.
31:669–676. 2013. View Article : Google Scholar
|
|
120
|
Di Lorenzo G, Figg WD, Fossa SD, et al:
Combination of bevacizumab and docetaxel in docetaxel-pretreated
hormone-refractory prostate cancer: a phase 2 study. Eur Urol.
54:1089–1094. 2008.
|
|
121
|
Kelly WK, Halabi S, Carducci M, et al:
Randomized, double-blind, placebo-controlled phase III trial
comparing docetaxel and prednisone with or without bevacizumab in
men with metastatic castration-resistant prostate cancer: CALGB
90401. J Clin Oncol. 30:1534–1540. 2012. View Article : Google Scholar
|
|
122
|
Adamo V, Noto L, Franchina T, et al:
Emerging targeted therapies for castration-resistant prostate
cancer. Front Endocrinol (Lausanne). 3:732012.PubMed/NCBI
|
|
123
|
Galsky MD and Vogelzang NJ:
Docetaxel-based combination therapy for castration-resistant
prostate cancer. Ann Oncol. 21:2135–2144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Beer TM, Ryan CW, Venner PM, et al:
Intermittent chemotherapy in patients with metastatic
androgen-independent prostate cancer: results from ASCENT, a
double-blinded, randomized comparison of high-dose calcitriol plus
docetaxel with placebo plus docetaxel. Cancer. 112:326–330. 2008.
View Article : Google Scholar
|