Introduction
Retinoblastoma is the most common primary malignant
intra-ocular tumor in infants and children. In the United States,
it affects 12 per million children aged 0–4 years, representing
6.1% of all childhood cancers under the age of 5 years (1). Slightly more than half of the
patients have the sporadic or non-inherited form of the disease,
which results from the spontaneous inactivation of the
retinoblastoma gene (RB1). Despite progress in the treatment of
retinoblastoma, significant problems remain unsolved and metastatic
disease is all too often fatal (2).
Although several treatment modalities are available
for retinoblastoma, including local control of small to
intermediate size tumors with laser and/or cryotherapy sometimes in
combination with radiation and/or chemotherapy, or enucleation with
or without systemic chemotherapy to control metastatic disease,
each of them has major drawbacks, especially in pediatric patients.
For example, conventional external beam radiation, which is used to
control large tumors, has many complications, including an
increased appearance of secondary malignancies, such as
osteosarcoma. This complication occurs more frequently in patients
with the hereditary-form of retinoblastoma. The 30-year cumulative
incidence of second malignancies is >35% for patients who
received external beam therapy vs. 6% for those patients without
radiation (3). Intra-arterial
chemiotherapy is currently novel treatment option for
retinoblastoma, however, variables that affect blood flow can
greatly affect drug delivery and therapy success (4–6).
Also retinal and choroidal vasculopathy may occur in 10 to 20% of
patients (7,8). Studies show that direct intravitreal
injection of melphalan may be effective in controlling active
vitreous seeds, however, major concern is the potential for tumor
dissemination (6,9–12).
Systemic chemotherapy used as a first line treatment for
intraocular retinoblastoma with subsequent consolidation with
photocoagulation, cryotherapy or radiotherapy has a recurrence rate
of 24% by 5 years (13). This
increases to 50% for patients with vitreous seeds (14). Recent analyses by several research
groups (15–18) show success for local control
approaching 90–100% for group A–C, but in less than 50% for group D
(new international classification). In addition, significant
morbidity with the chemotherapy has been described previously
(19). One of the drugs used for
chemotherapy (etoposide) is thought to be associated with increased
incidence of acute myeloblastic leukemia although the actual number
of cases implicated so far has been low with just ~20 cases
reported in the literature (20).
For these reasons, there is a pressing need for alternative
treatment modalities for retinoblastoma with better safety and
efficacy profiles.
Metformin is a biguanide drug that is widely used
for the treatment of type II diabetes (4,21–23).
A significant body of preclinical studies have shown that metformin
decreases cancer cell viability and tumor growth in xenograft
models (6,11,24–28).
However, other studies have shown that metformin in vivo may
accelerate tumor growth. For example, BRAF-mutant melanoma cells
that are resistant to metformin in vitro show accelerated
growth in vivo when treated with metformin (29). Likewise, metformin/AMPK activation
promoted an angiogenic phenotype in the ERα negative MDA-MB-435
breast cancer model (30).
Some of the effects of metformin have been linked to
activation of AMP-activated protein kinase (AMPK) in muscle,
adipose and liver tissue (22,31).
AMPK is activated by cellular stress resulting in the restoration
of energy levels through regulation of metabolism and growth
(32–34). Insufficient AMPK activity allows
uncontrolled cell growth despite the conditions of cellular stress
(such as those occurring during tumorigenesis). Furthermore,
metformin has been shown to inhibit the mTOR pathway and S6K1
phosphorylation implicated in protein synthesis (4,6). Of
note, these effects have been observed only at millimolar doses of
metformin and recent studies indicate that metformin may exert its
action through AMPK-independent mechanisms (6,11,24,28,35–41).
Thus the effects of metformin on the proliferation
of cancer cells appear to be cell type dependent and not fully
elucidated. For this reason, we investigated the effects of
metformin on human retinoblastoma cancer cell lines in vitro
and in vivo.
Materials and methods
Reagents
Metformin, MTT (3-(4,5-dimethylthiazol-
2-yl)-2,5-diphenyltetrazolium bromide) and ribonuclease-A were
purchased from Sigma-Aldrich (St. Louis, MO, USA). Propidium
iodide, calcein and DAPI were purchased from Invitrogen (Carlsbad,
CA, USA). The following primary antibodies were purchased from Cell
Signaling Technology (Danvers, MA, USA): phospho-ACC (Ser79),
phospho-AMPK (Thr172), phospho-S6 ribosomal protein (Ser235/236),
phospho-4E-BP1 (Thr37/46), p21 Waf1/Cip1, p27Kip1, LC3B,
phospho-p38 MAPK (Thr180/Tyr182), phospho-Akt (S473),
phospho-p44/42 MAPK (Erk1/2), β-tubulin, GAPDH. The following
antibodies were purchased from Epitomics (Burlingame, CA, USA)
cyclin E1, E2, D1, D3, A2, CDK4 and CDK2. Anti-Ki67 was purchased
from Dako (Carpinteria, CA, USA), anti-CD31 and anti-CD11b from BD
Bioscience (Franklin Lakes, NJ, USA).
Cell culture
The human retinoblastoma cells WERI and Y79 (ATCC,
Manassas, VA, USA) were grown in RPMI-1640 medium (Invitrogen,
Grand Island, NY, USA) supplemented with 15% fetal bovine serum
(ATCC), penicillin and streptomycin (both at 100 μg/ml;
Invitrogen), 2 mM L-glutamine (Invitrogen) and 10 mM HEPES
(Invitrogen). Cells were incubated at 37°C in a humidified
atmosphere of 95% air and 5% CO2 and split when the
cells reached approximately 80% confluence.
Trypan blue exclusion test, growth curve
and doubling time
Retinoblastoma cells were seeded in 6-well plates at
a concentration of 4.5×105 cells per well. On days 3, 6
and 9 cell number and viability was determined by trypan blue
(0.4%) dye exclusion and growth-inhibition curves were drawn.
Experiments were performed in triplicate with 2 wells per
condition.
Measurement of cell viability by the MTT
assay
Cell viability was assessed by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. MTT assay is used to measure the reduction of a tetrazolium
compound by the cellular mitochondria, producing an optically
active soluble formazan.
Cells were cultured in 48-well plates at density
60,000 cells per well in 300 μl growth medium. After 1 and 3 days
of treatment with metformin, MTT (5 mg/ml in PBS) was added to each
well at a 1/10 volume. Cells were incubated for 1 h at 37°C and
resuspended in DMSO. The absorbance at 595 nm was measured using a
microplate reader. Data are displayed as percentage of control.
Flow cytometry assessment of cell
viability
Live and dead cells were quantified using the
fluorescent probes calcein AM and DAPI. Cells were cultured in
6-well plates at 500,000 cells per 2 ml growth medium and were
treated with 5 mM metformin for 48 h. The calcein was added at
final concentration 0.1 μM and DAPI at 3 μM. The samples were read
on Becton Dickinson FACScan. Results were analyzed with Summit 4.3
software.
Flow cytometry assessment of the cell
cycle
Cellular DNA content was assessed by flow cytometry.
Cells were seeded in 6-well plates at density 500,000 cells per 2
ml growth medium and were treated with 5 mM metformin for 48 h.
After overnight fixation in 75% ethanol, cells were suspended in
PBS with DNase-free RNase A at final concentration 0.3 mg/ml and
propidium iodide at final concentration 1 mg/ml. DNA content
assessed on Becton Dickinson LSRII flow cytometer. Results were
analyzed with Modfit LF software.
Protein extraction and western blot
analysis
For in vitro experiments, cells were
incubated for 48 h in the presence or absence of metformin at
various concentrations (12 μM to 10 mM). For in vivo
experiments, tumor pieces were cut. The samples lysed in M-PER
Mammalian Protein Extraction Reagent (Thermo-Scientific, Pierce
Protein Research Products) with protease (according to
manufacturer’s suggestions; Roche Applied Science) and phosphatase
inhibitor cocktails (dilution 1:50; Thermo-Scientific, Pierce
Protein Research Products). Total amount of protein (10 μg) was
loaded onto a 4–12% Bis-Tris Gel (NuPAGE; Invitrogen). The
electrophoresis was done using NuPAGE MOPS Running Buffer
(Invitrogen) and then samples were transferred onto a PVDF membrane
(Millipore, Billerica, MA, USA). The membranes were blocked for 45
min at room temperature in 5% wt/vol BSA, 1× TBS 0.1% Tween-20. The
primary antibodies were diluted in 5% wt/vol BSA 1× TBS, 0.1%
Tween-20 1:1,000 for all except CCNE1, E2, D1, D3, A2, CDK4 and
CDK2 which were used at concentrations 1:5,000. After overnight
incubation at 4°C, the membranes were washed three times 1× TBS
0.1% Tween-20 and incubated for 45 min at room temperature with the
horseradish peroxidase-labeled secondary anti-rabbit antibody at
1:50,000 (Jackson Immuno Research, West Grove, PA, USA). The
immunoreactive bands were visualized with ECL exposured to Fuji RX
film (Fujifilm, Tokyo, Japan). The results were quantified using
ImageJ software.
Animals
All animal experiments complied with guidelines
established by the Association for Research in Vision and
Ophthalmology for the use of animals in ophthalmic and vision
research, and were approved by the Animal Care and Use Committee of
the Massachusetts Eye and Ear Infirmary (Boston, MA, USA). Four to
five-weeks-old BALB/c (nu/nu) female mice were purchased from
Charles River Laboratories (MA) and maintained in a facility under
specific pathogen-free conditions in a climate controlled room with
a 12 h light/dark cycle.
Xenograft tumor growth assay
Xenograft tumors were established bilaterally in
nu/nu mice by means of a single subcutaneous injection in each
flank consisting of 4 million Y79 retinoblastoma cells suspended in
0.3 ml of a 1:1 mixture of ice-cold matrigel basement membrane
matrix (BD Bioscience, MA, USA) and RPMI-1640 medium. Once a tumor
mass became visible (within the week from injection of the cells),
mice were randomly assigned to receive either daily peritoneal
injections of metformin (250 mg/kg) or normal saline for 31 days.
Two independent experiments were performed with five mice assigned
to each group. The dose was based on the LD50 of metformin (420
mg/kg), as well as on human therapeutic and maximum prescribed
doses for human patients (2,000–2,500 mg/day) (6,11).
The tumor volume was monitored by external measurement in two
dimensions with calipers every week and determined according to the
equation: volume (mm3) = 4/3 × phi × (length/2) ×
(width/2)2 (9). Mice
were weighted once a week.
Immunohistochemistry assay and
pathological evaluation
Five tumors from each group were frozen, cut into 10
μm sections and analyzed for retinoblastoma cell proliferation,
vessel area and macrophage infiltration. Cryosections were also
used for immunohistochemistry, first being fixed in 4%
paraformaldehyde, blocked with 5% goat serum, and permeabilized
with 0.1% Triton X-100. The sections were incubated in a humid
chamber with primary antibodies, including anti-Ki67 (1:100),
anti-CD31 (1:100) and anti-CD11b (1:100). A fluorophore-conjugated
secondary antibody (Molecular Probes, Carlsbad, CA, USA) was used
to detect fluorescence using a confocal microscope (Leica
Microsystems, Wetzler, Germany). Nuclei were stained with DAPI.
Cryostat sections were examined at random fields at ×20
magnification and the percentage of fluorescent-positive
cells/DAPI-positive cells in each field was measured. Tumor vessel
area was calculated as the number of image pixels that stained
positive for CD31 per high-power field.
TUNEL assay in tissue sections
Frozen 10 μm sections were prepared from tumors as
above and stained with TUNEL cell death detection kit (Roche
Diagnostics Corp., Indianapolis, IN, USA) according to the
manufacturer’s recommendations. Sections were counter stained with
DAPI and examined under an epifluorescent microscope (Leica
Microsystems, Wetzler, Germany). Cryostat sections were examined at
random fields at ×20 magnification and the percentage of
TUNEL-positive cells/DAPI-positive cells in each field was
measured.
Serum levels of metformin, insulin-like
growth factor-1 (IGF-1) and insulin-like growth factor binding
protein 3 (IGFBP-3)
Retro-orbital blood was collected 3 and 15 h after
metformin injection for ELISA testing and metformin levels
assessment from all mice after euthanization. The samples were
mixed with 4 mM EDTA and left at 4°C for 2 h, then centrifuged for
15 min at 180 × g. Serum levels of IGF-1 and IGFBP-3 were measured
using a Mouse/Rat IGF-I and IGFBP-3 ELISA kit (R&D Systems,
Minneapolis, MN, USA). Metformin levels were assayed (3 and 15 h
after i.p. metformin injection), using high-performance liquid
chromatography (NMS Lab, Willow Grove, PA, USA).
Statistical analysis
The data are expressed as mean ± standard error of
the mean (SEM). Statistical significance was evaluated using the
one-way ANOVA test with Dunnett’s modification for multiple means
comparison or t-test for two means. *p<0.05 was
considered statistically significant. Two-tailed tests were used
for all comparisons.
Results
Metformin inhibits the growth and
increases doubling time of human retinoblastoma cells in vitro at
mM, but not μM levels
In order to determine whether metformin affects
human retinoblastoma cell viability and proliferation, we analyzed
the effect of the drug on two human retinoblastoma cell lines: WERI
and Y79. Cells were treated with various concentrations of
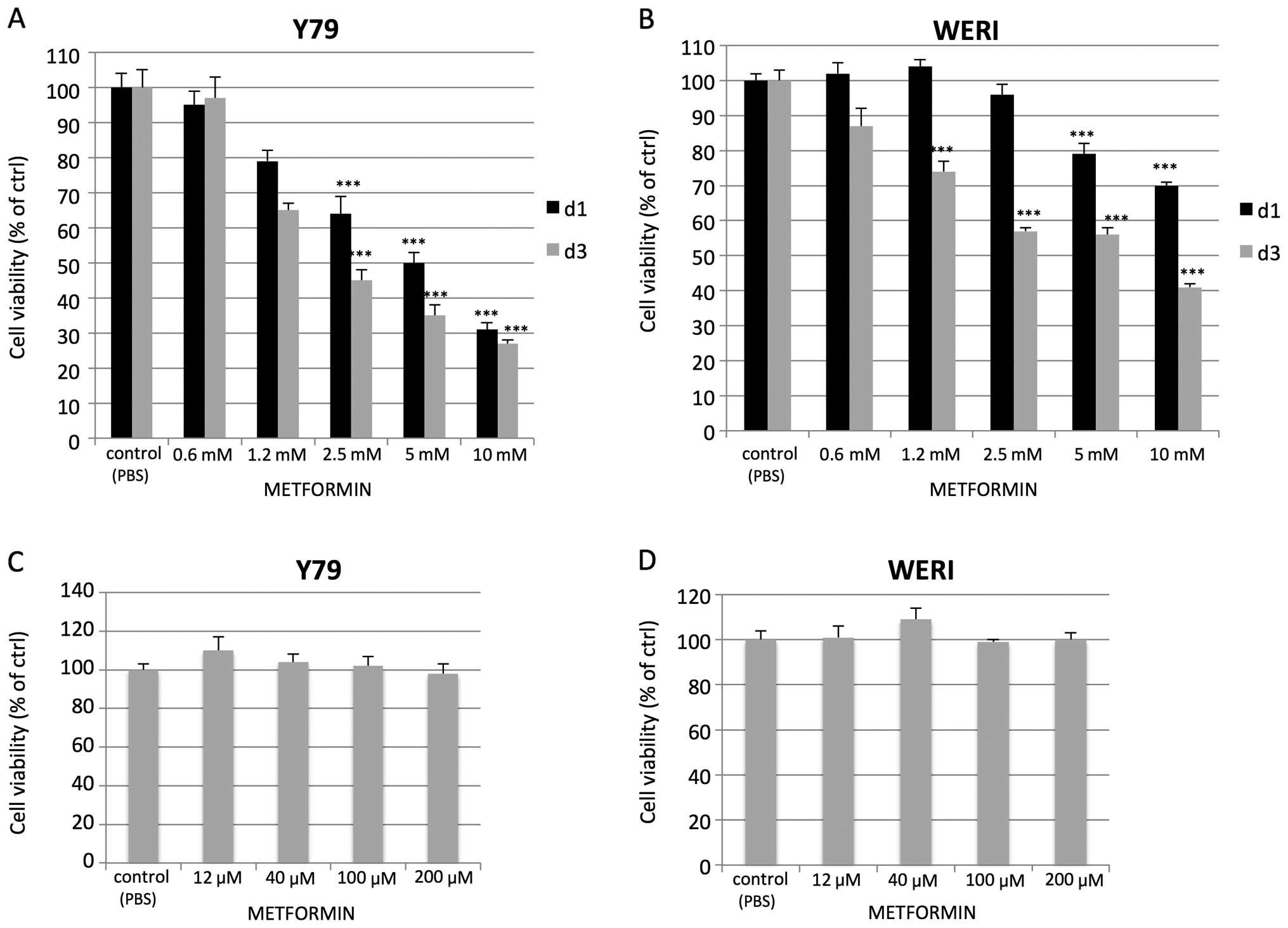
metformin (12 μM up to 10 mM) and the viability was assed by the
MTT assay. Increasing doses of metformin led to a corresponding
reduction in cell viability but at doses in the mM range of
concentrations (Fig. 1A and B).
Reduced viability was not observed at μM concentrations (Fig. 1C and D). Assessment of cell growth
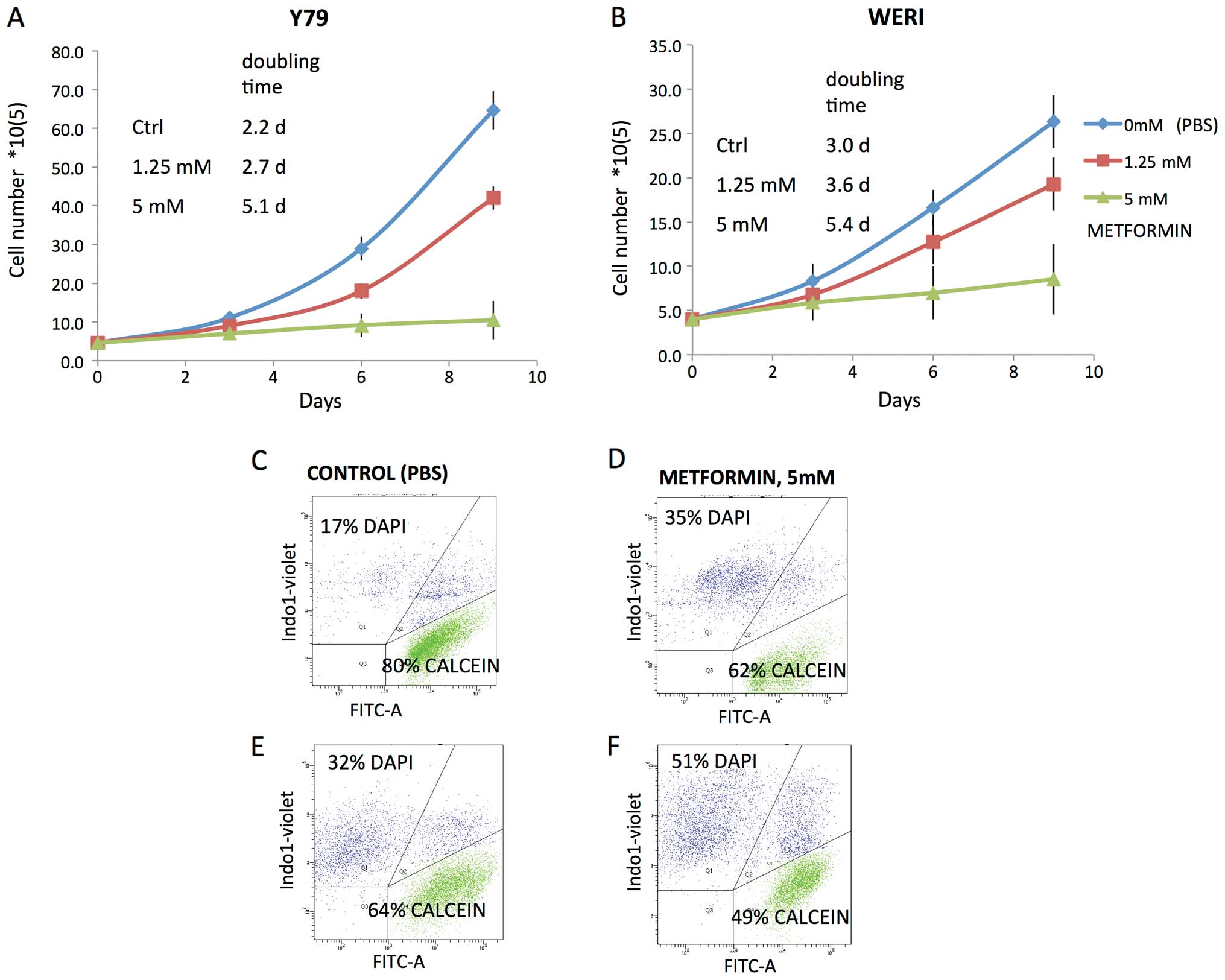
and doubling time by trypan blue exclusion showed decreased growth
rates in the presence of mM levels of metformin. Doubling time
increased from 2.2 to 5.1 days for the Y79 cell line and from 3 to
5.4 days for the WERI cell line (Fig.
2A and B). Metformin treatment at 5 mM also increased the
proportion of non-viable cells and decreased the proportion of
viable cells (Fig. 2D and F) as
judged by calcein AM/DAPI flow cytometry when compared to control
(Fig. 2C and E).
Metformin at higher mM levels leads to
variable cell cycle changes in human retinoblastoma cells and to a
global reduction in cell cycle regulators
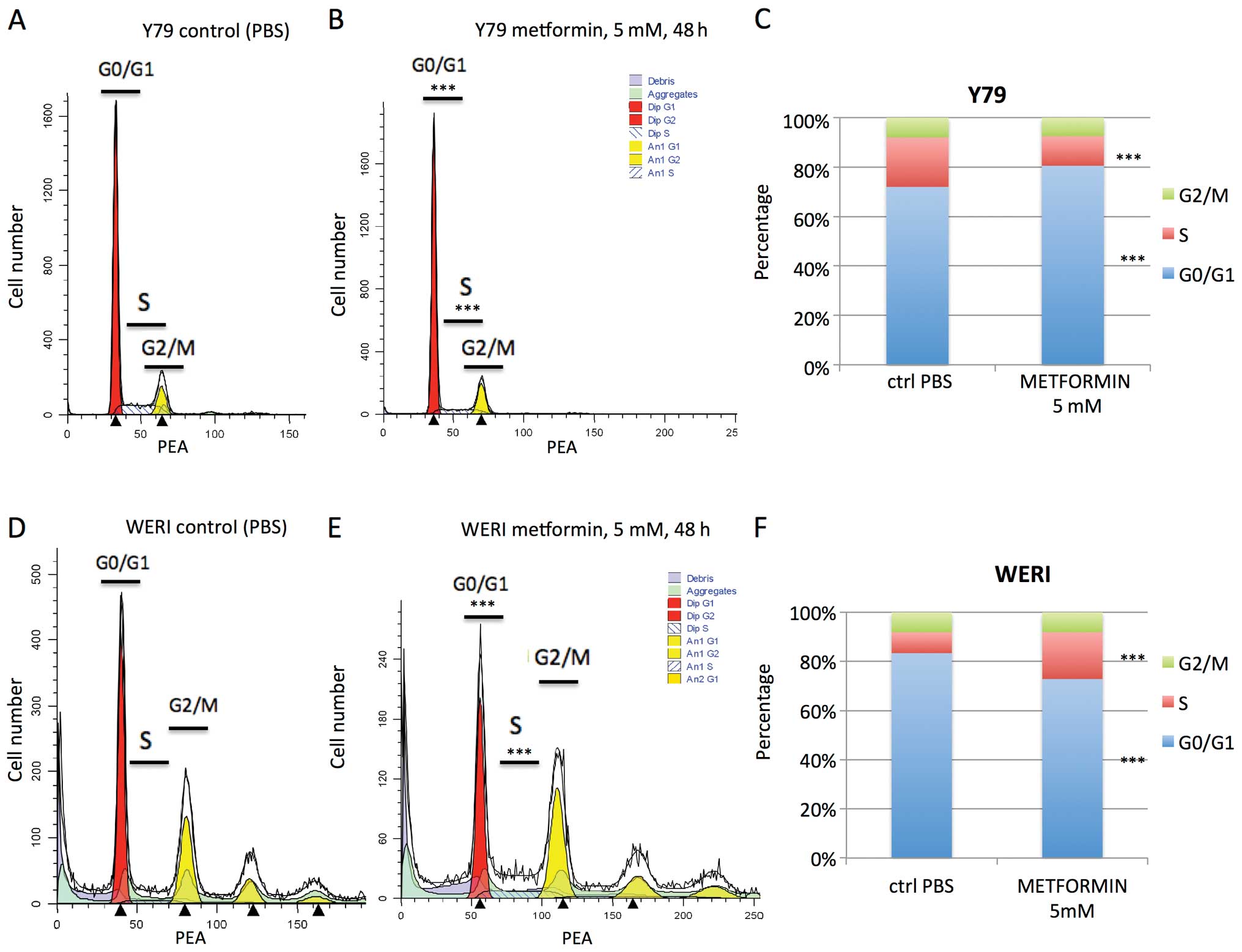
Previous reports have shown arrest in G0/G1 or S
phase by mM levels of metformin (32). In our study, cell cycle analysis
revealed that metformin treatment (5 mM for 48 h) of Y79 cells led
to a statistically significant increase in cells in G0/G1 phase (72
to 81%, p<0.001), and a decrease in S phase (20 to 12%,
p<0.001) (Fig. 3A–C). In
contrast the reverse was seen when WERI were treated with
metformin. There was a decrease in G0/G1 phase (83 to 73%,
p<0.001) and an increase in cells in S phase (9 to 19%,
p<0.001) (Fig. 3D–F). These
cell cycle effects were not associated with specific cyclin and CDK
changes but rather they were associated with non-specific global
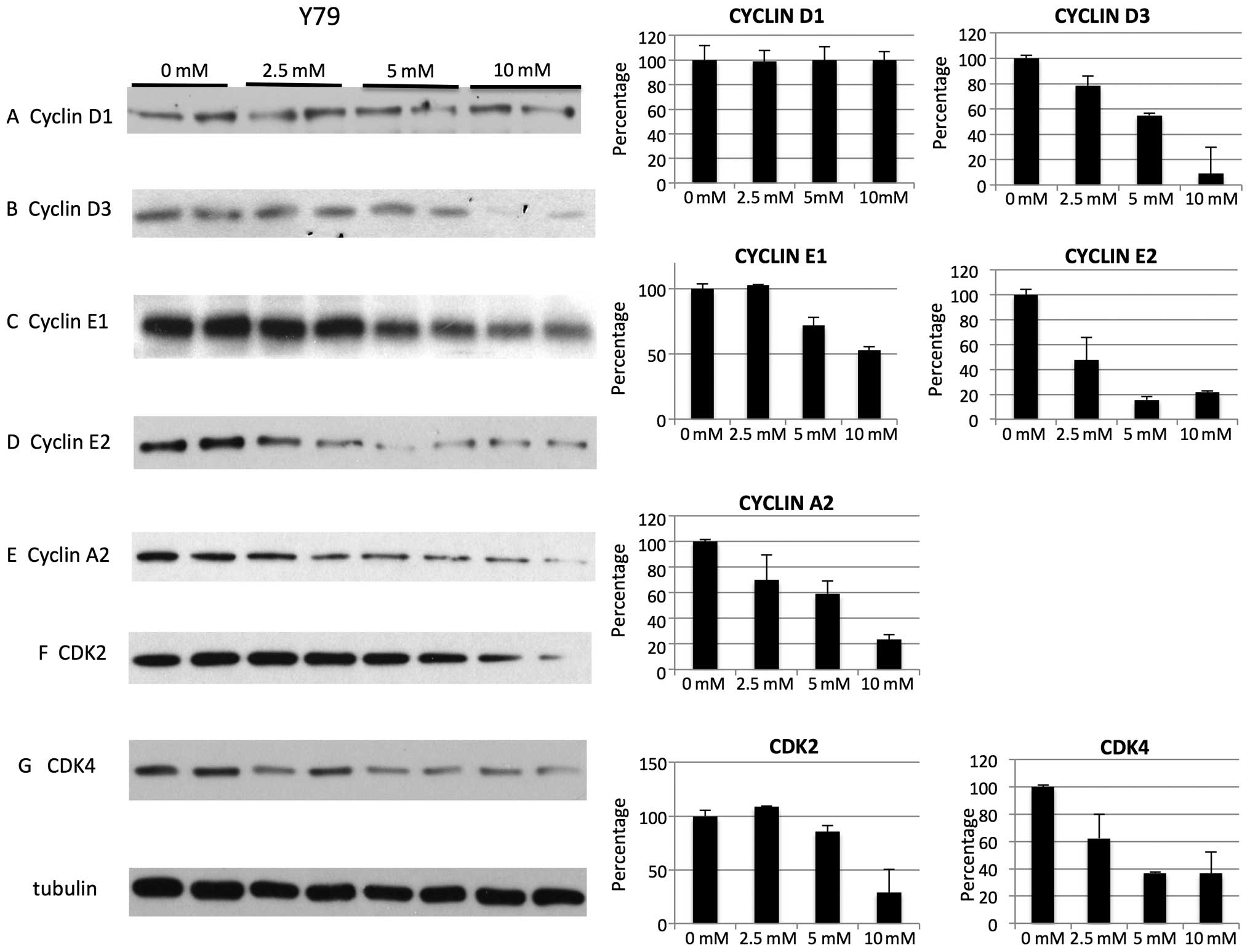
reduction in cyclins. For Y79 cell line on treatment with metformin
we noted decrease of cyclin D3, E1, E2, A2 (Fig. 4B–E), cyclin dependent kinases CDK2
and CDK4 (Fig. 4F and G). Levels
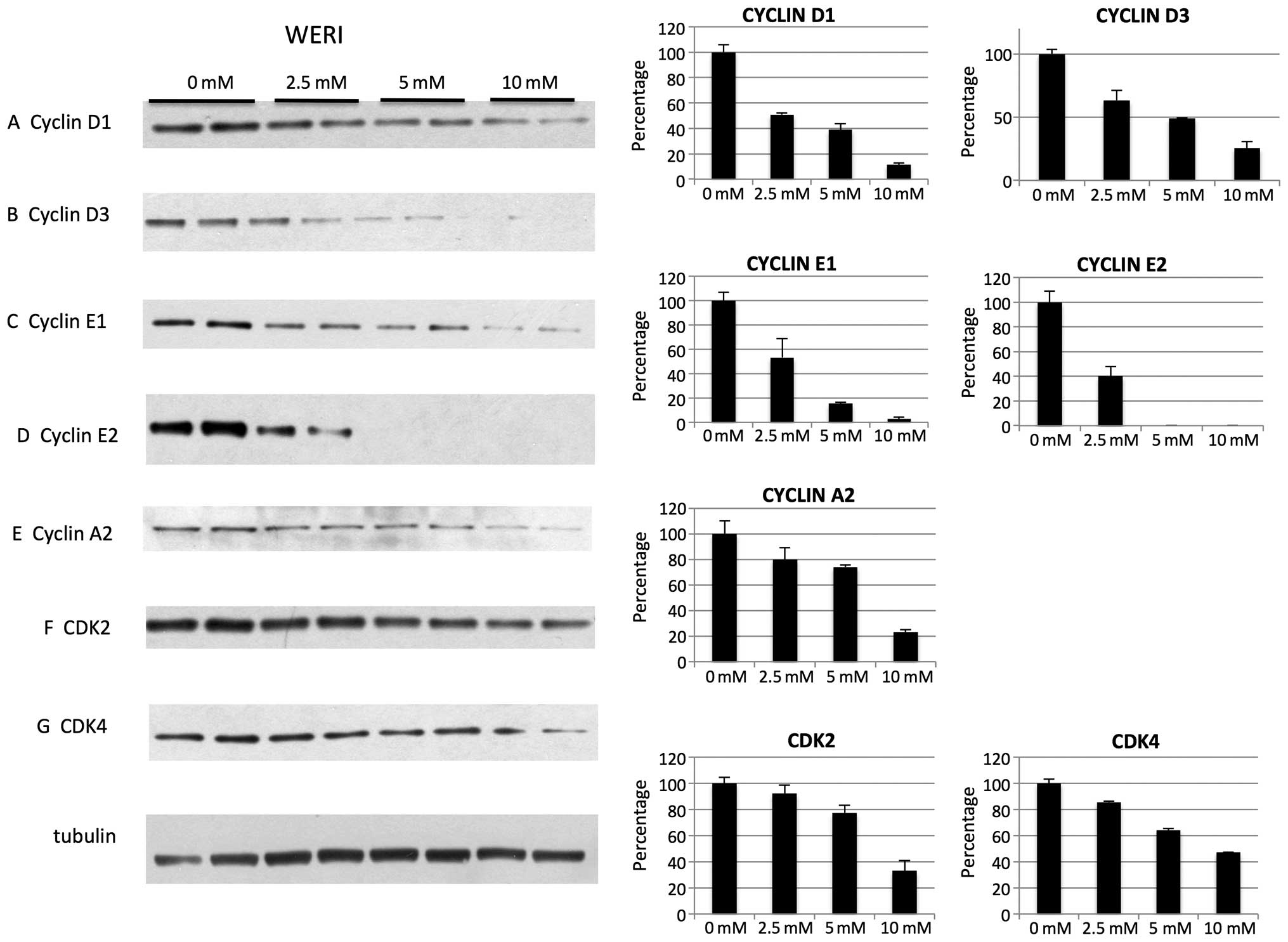
of cyclin D1 were not decreased for Y79 (Fig. 1A). For WERI cell line on treatment
with metformin we noted decrease of cyclin D1, D3, E1, E2, A2
(Fig. 5A–E) as well as CDK2 and
CDK4 (Fig. 5F and G). In addition,
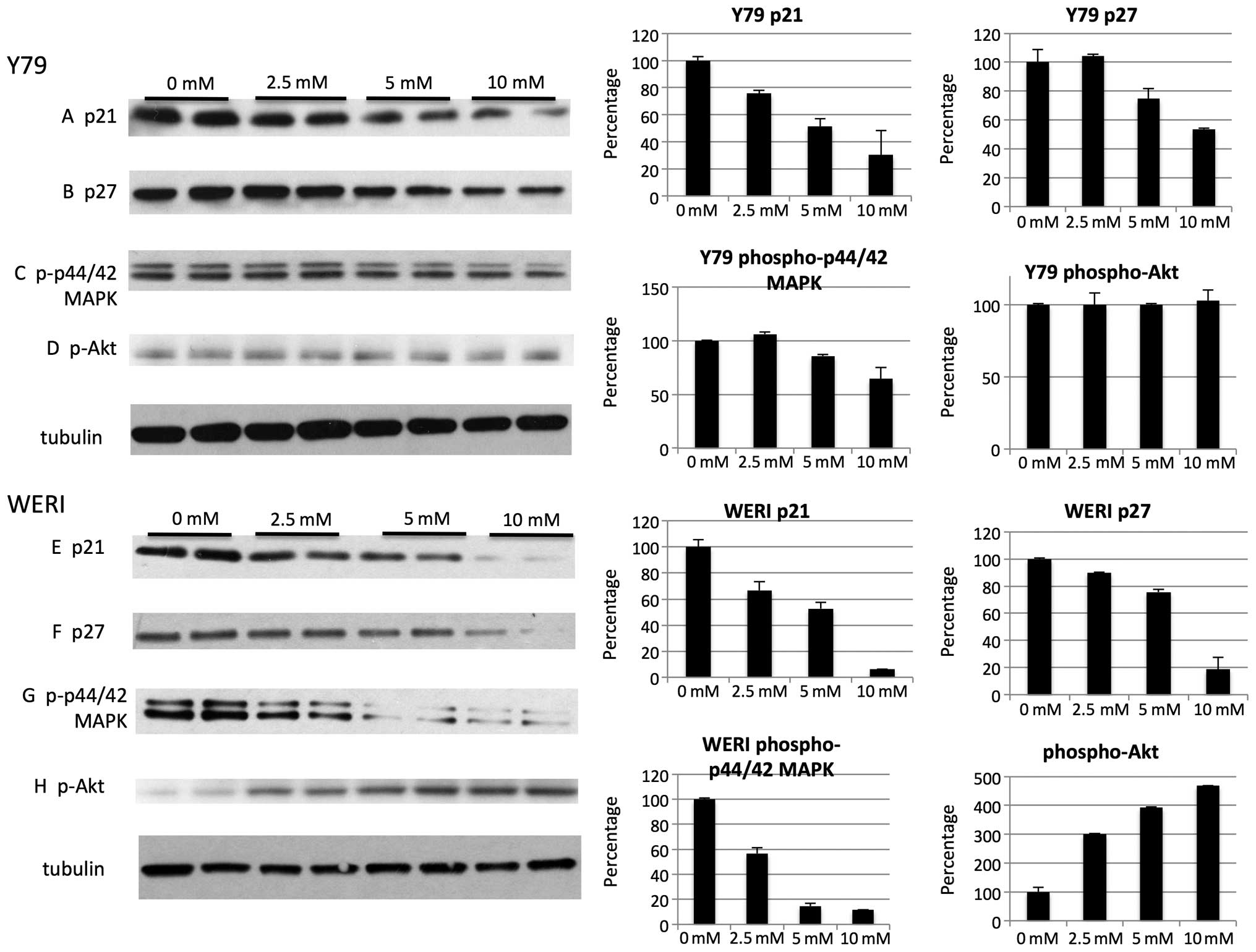
metformin treatment reduced CDK inhibitors p21 (Fig. 6A and E) and p27 (Fig. 6B and F) in both cell lines.
Metformin reduced levels of positive cell growth regulators, such
as phospho-p44/42 MAPK in Y79 (Fig.
6C) and WERI (Fig. 6G). Other
cell proliferation and survival factors, such as phospho-Akt were
unchanged in the Y79 cell line (Fig.
6D) but were found to be activated in the WERI cell line
(Fig. 6H), suggesting that some of
the effects of metformin on cell cycle may be non-specific.
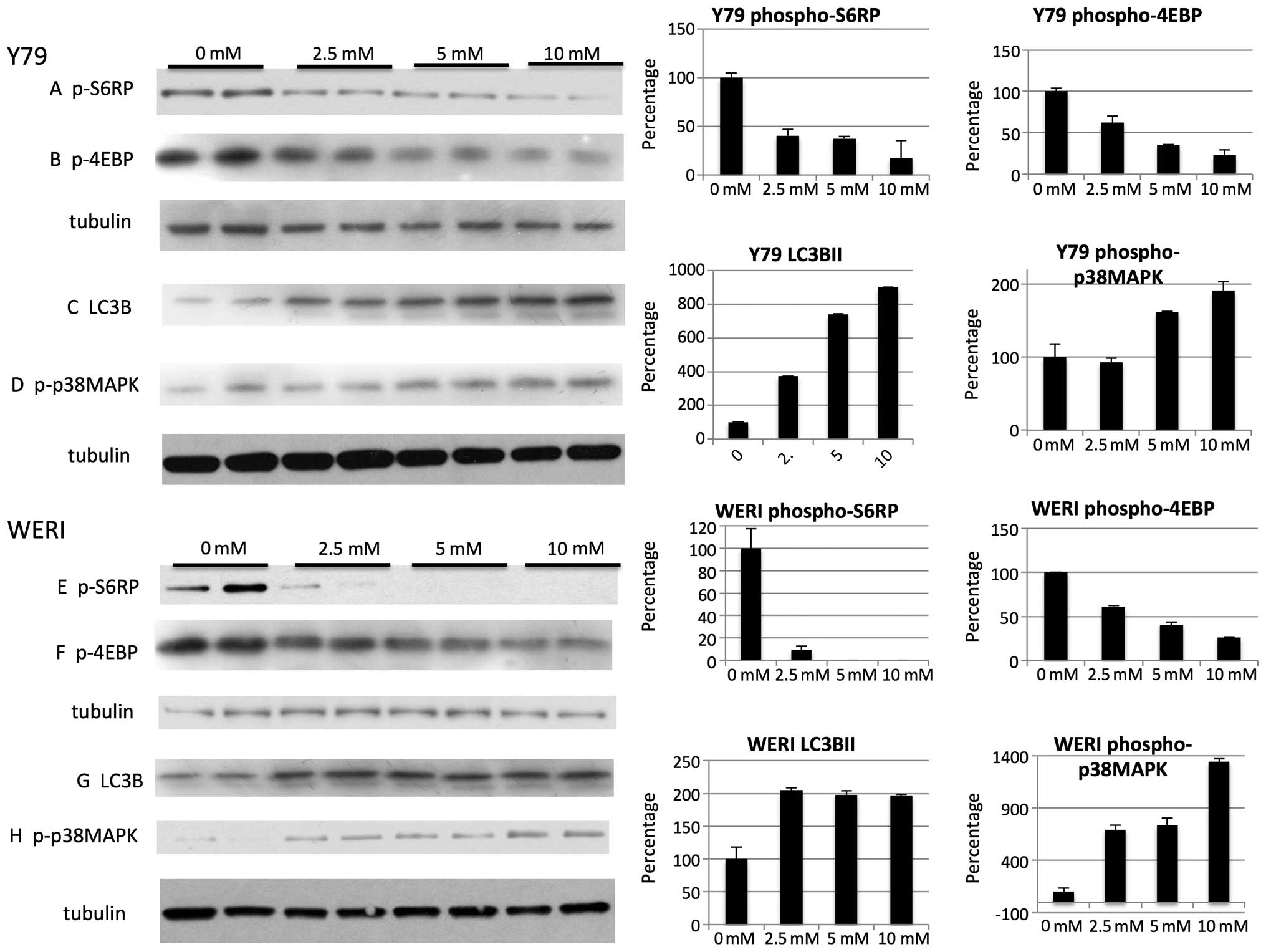
Metformin at higher mM levels inhibits
the mTOR pathway, upregulates phospho-p38MAPK, autophagy marker
LC3B and activates AMPK
Autophagy is usually activated under conditions of
cell stress and is inhibited by the mTOR pathway, an intracellular
signaling pathway important in apoptosis. Indeed mM levels of
metformin decreased the mTOR pathway as judged by phosphorylation
of S6RP (Fig. 7A and E) and 4E-BP1
(Fig. 7B and F) and led to
variable increases in LC3B-I and LC3B-II protein levels (Fig. 7C and G). Similar to some (35,37,38)
but not other studies (39,40)
the induction of LC3 was associated with increases in p38 MAPK
(Fig. 7D and H). Similar to other
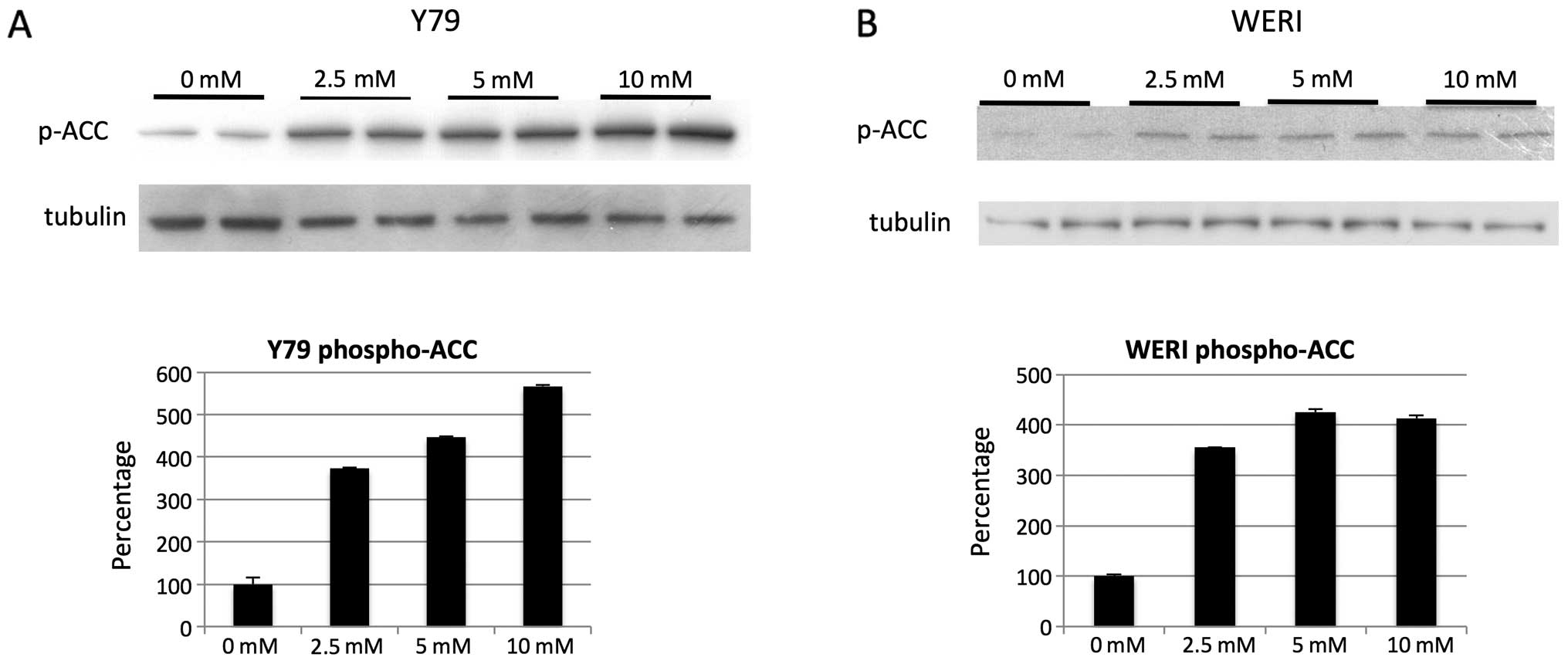
investigators (21) we found AMPK
to be activated in retinoblastoma cells at the mM level as
determined by phospho-ACC (Fig. 8A and
B).
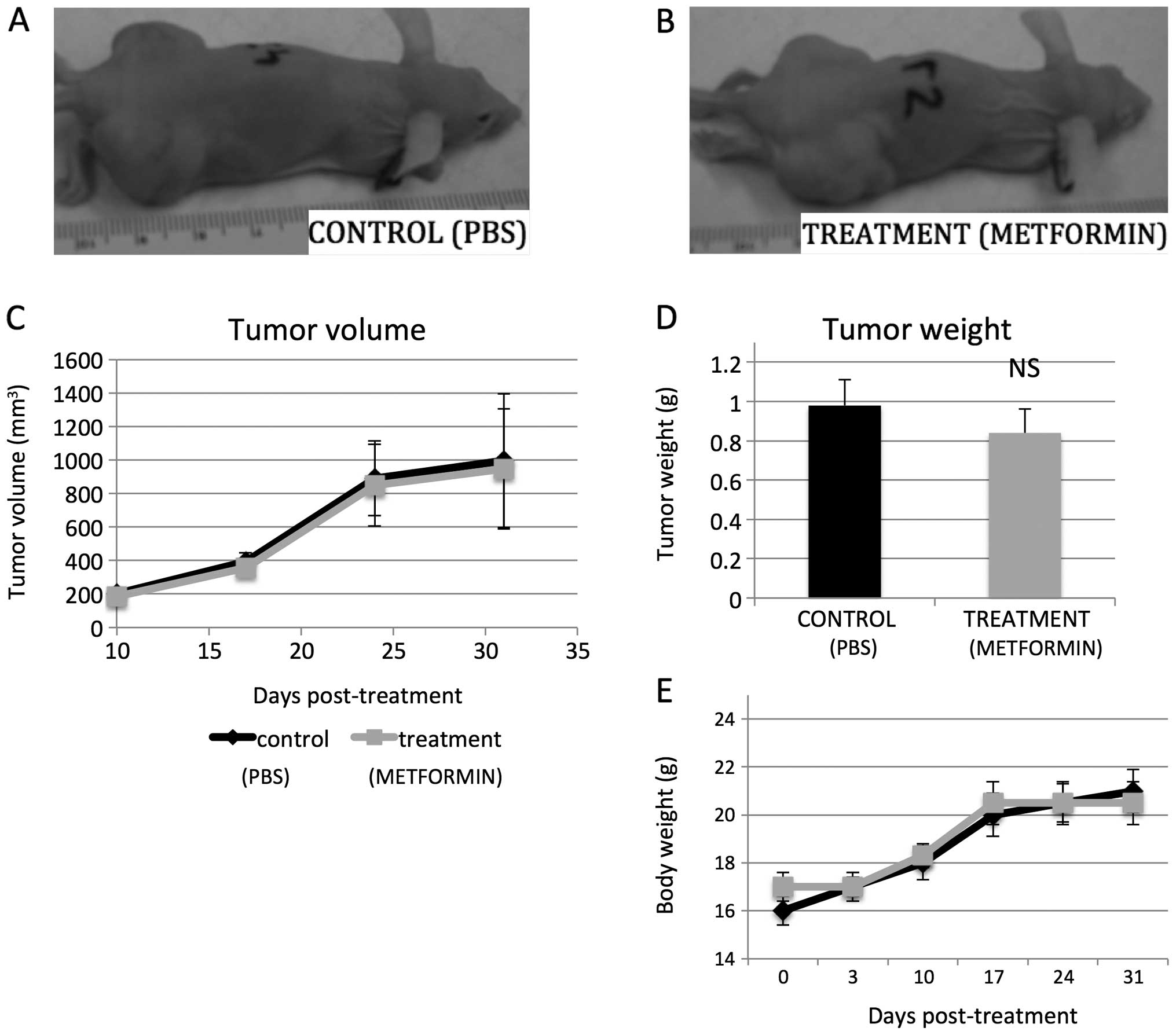
Metformin at pharmacologic levels fails
to suppress growth of human retinoblastoma xenografts in vivo
In order to evaluate the in vivo effect of
metformin on retinoblastoma growth, heterotopic tumor xenografts of
human Y79 retinoblastoma cells were established and mice were
treated with metformin (250 mg/kg every 24 h) or equal volume of
normal saline delivered i.p. (intraperitoneally). The dose of
metformin was based on previous studies (11,28)
and the LD50 for mice (420 mg/kg), as well as on the typical
therapeutic and maximally prescribed human doses (2,000–2,500
mg/day) (6,11).
In our in vivo experiments, metformin levels
in mouse sera were on average 2.13 and 0.66 μg/ml for peak and
trough, respectively (measured via high-performance liquid
chromatography). The level of 2.13 μg/ml metformin equals about 12
μM. For comparison human peak levels are 1.03 (±0.33), 1.60
(±0.38), 2.01 (±0.42) for 500 mg p.o. (orally) daily, 850 mg p.o.
daily or 850 mg p.o. taken three times per day, respectively
(42). Despite achieving
equivalent pharmacologic levels of metformin in mice, tumor growth
was not significantly different than in the vehicle treated animals
(Fig. 9A–C). The mean tumor
weight, determined at necropsy, in the control mice was 0.98 g, as
compared to 0.82 g in the metformin-treated mice (p=0.89, n=10, two
independent experiments; Fig. 9D).
The body weight of the tumor-injected mice was not found to differ
significantly from controls (Fig.
9E).
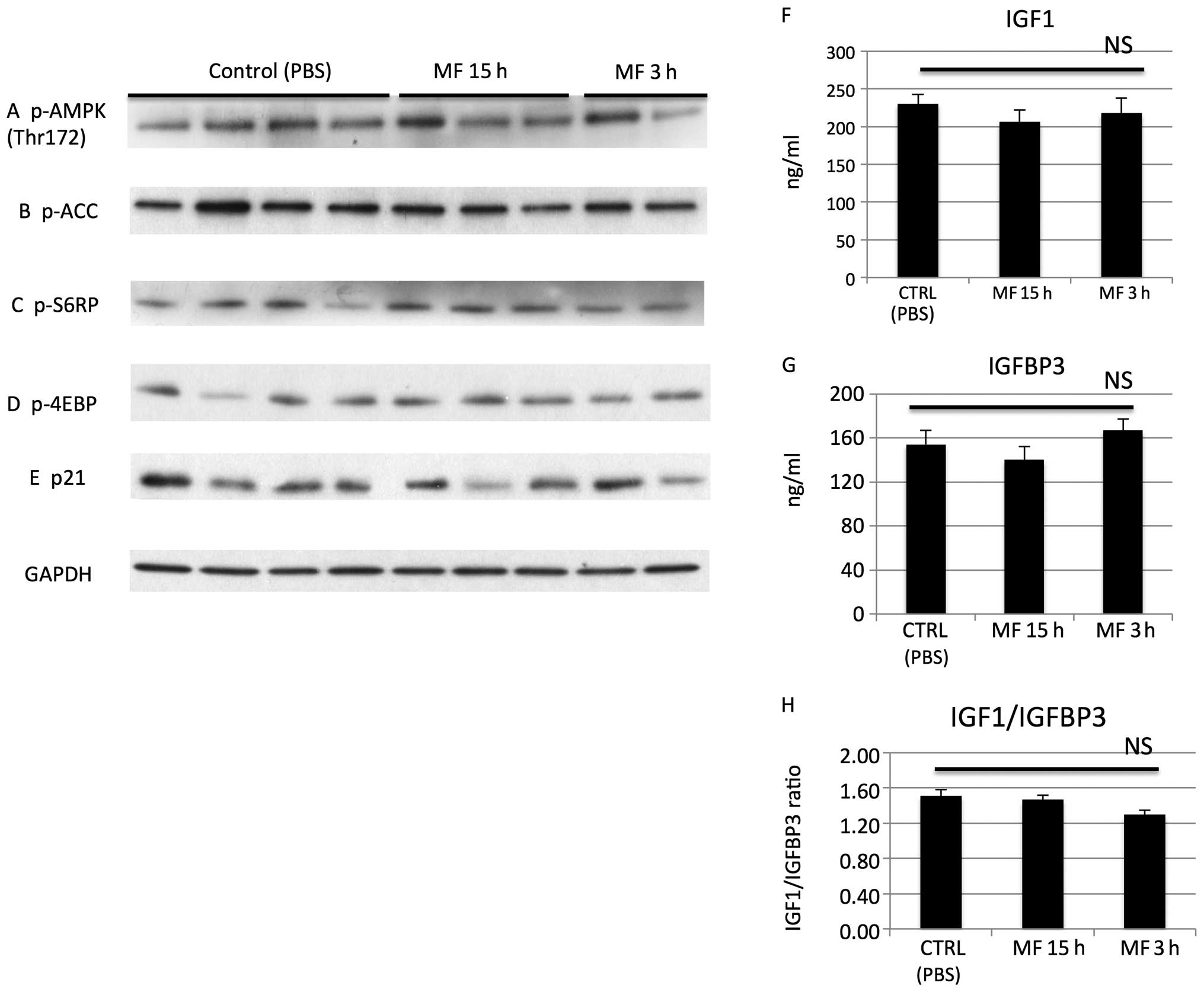
We observed that metformin 3 and 15 h after i.p.
administration did not affect proteins/pathways in vivo
thought to be affected by metformin at mM levels in vitro
such as AMPK, phospho-ACC, mTOR, p21 (Fig. 10A–E). Also the drug did not
significantly affect the IGF1, IGFBP3 or the IGF1/IGFBP3 ratio in
our experiments (Fig. 10F–H).
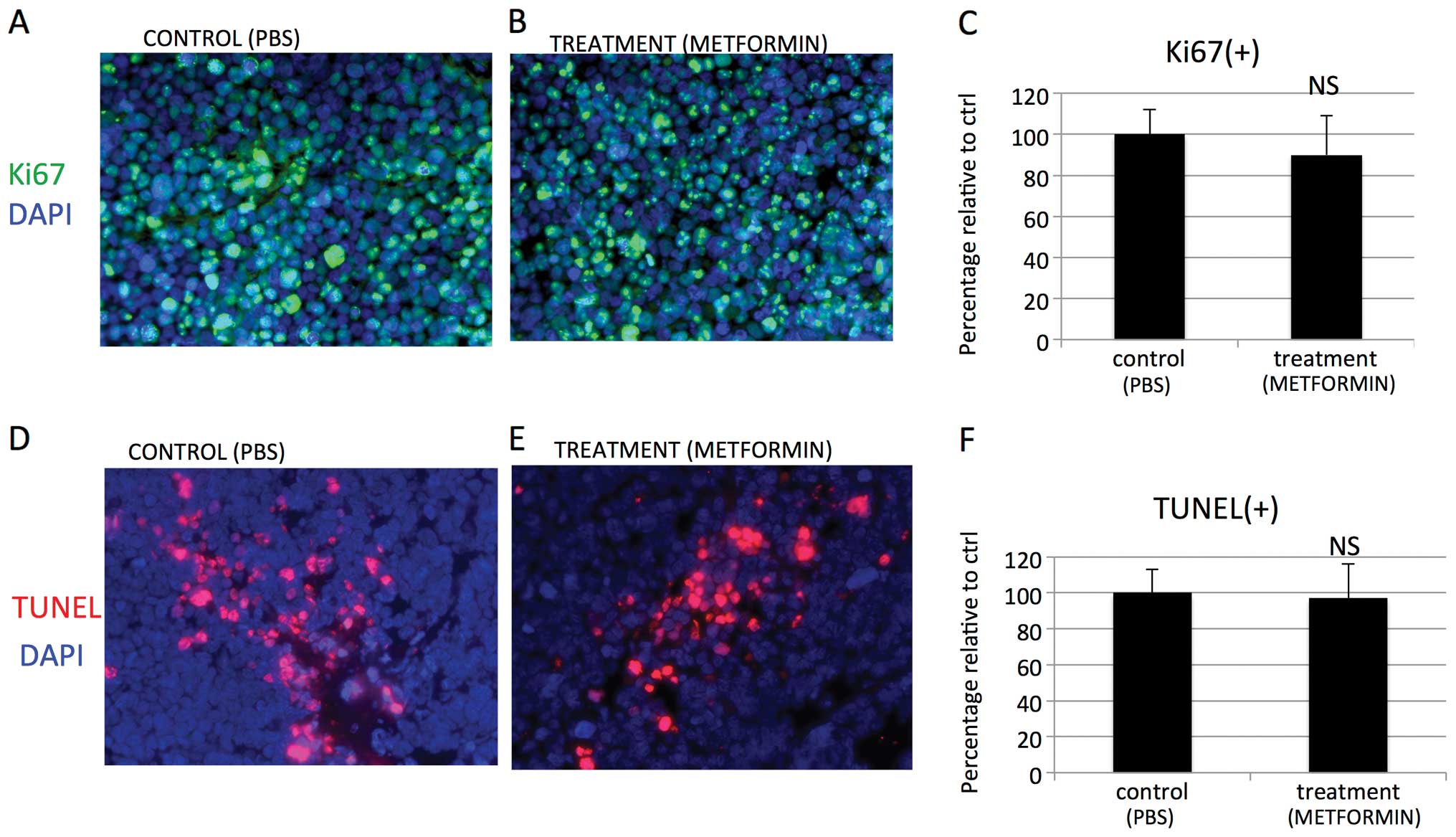
When tumors were examined histologically significant changes of
Ki-67 proliferative index [Ki67(+) cells/DAPI(+) cells; Fig. 11A–C] were not observed. Apoptosis
labeling was similar in both groups [TUNEL(+) cells/DAPI(+) cells;
Fig. 11D–F]. On treatment with
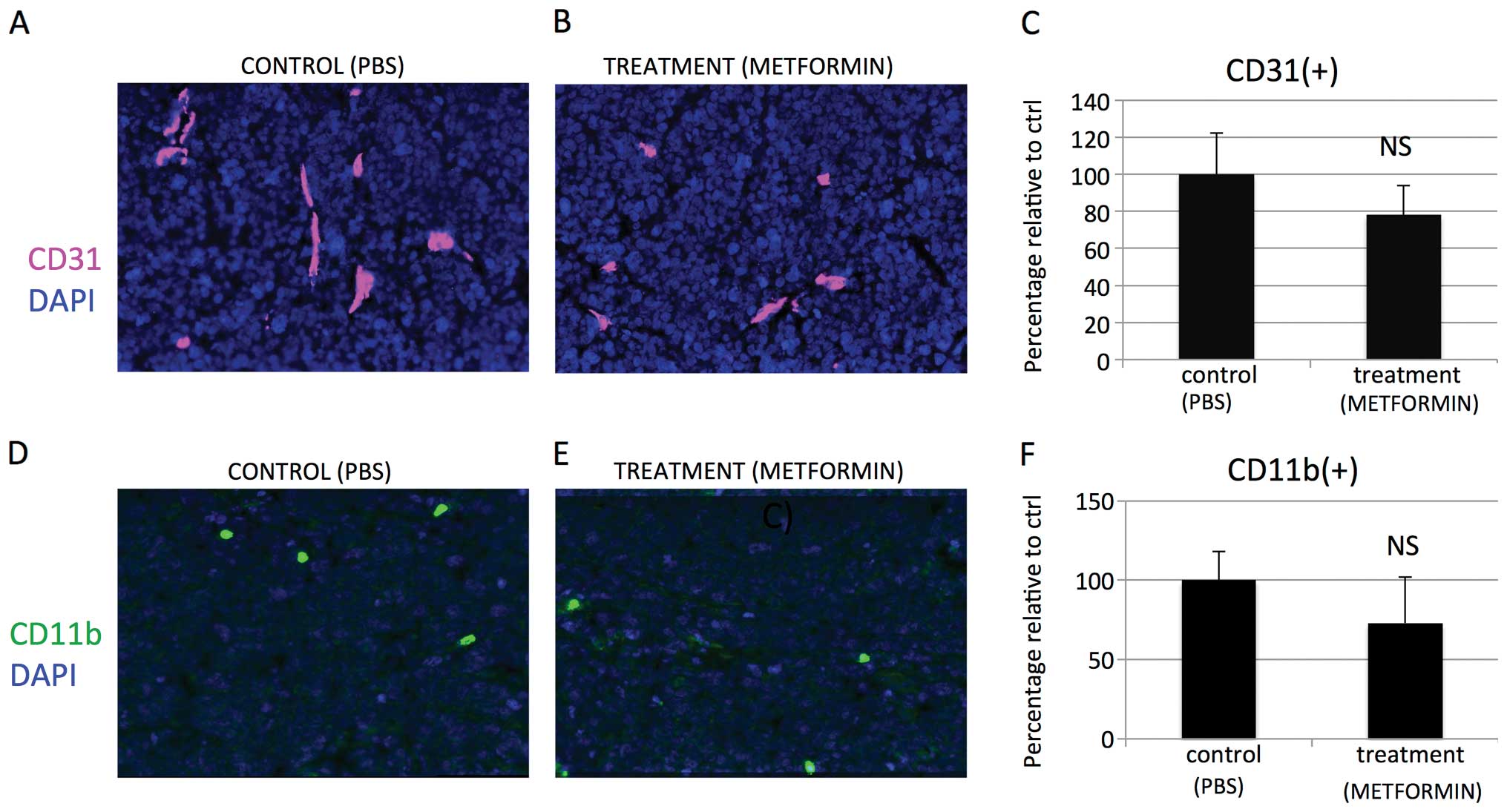
metformin we observed a small, nonsignificant decrease in tumor
vascularity (vessel area: μm2/hpf; Fig. 12A–C) and a small, nonsignificant
decrease of infiltration by CD11b cells [CD11b(+) cells/DAPI(+)
cells Fig. 12D–F].
Discussion
Metformin, a drug used primarily for the treatment
of type II diabetes, has been reported in epidemiological studies
to reduce the incidence of certain cancers among diabetic patients
(43). Initial studies examining
the anti-proliferative effects of metformin have focused on tissues
involved in insulin signaling and glucose/fatty acid metabolism,
such as muscle and liver (4,21,23).
However, the effects of metformin on other tissues or cells in
culture have not been well characterized. Of the studies available
(6,11,28),
the anticancer effects of metformin are reportedly seen at mM
levels, levels that are 100–1,000 times in excess of doses capable
of being achieved by pharmacotherapy with metformin in humans. In
addition, some conflicting data have arisen in in vitro and
in vivo studies, with most indicating that the drug may have
the potential to directly suppress tumor growth (11,24,28,32,44),
while other reports indicate that metformin may not halt the growth
of tumors (25,29,45,46).
Increased tissue accumulation of metformin relative to blood levels
have been hypothesized to explain metformin anticancer activity
in vivo, although the concentration seen in most tissues
still remains at the low 100 μM level (47). Other explanations proposed relate
to metformin’s well-known effects on cholesterol, leptin, insulin
levels and adiponectin, suggesting that some metabolic changes
account for a reduction in tumor growth.
Some retrospective epidemiologic studies have
revealed a decrease in the incidence of certain cancers in patients
treated with metformin (48–50).
The most recent meta-analysis suggests that metformin reduces the
risk for colorectal cancer and hepatocellular cancer, but not for
pancreatic, breast, gastric, prostate, bladder or lung cancer
(50). Other case-control trials
indicate that taking metformin is not associated with altered risk
for esophagus cancer (51),
endometrial cancer (52), lung
cancer (53), colorectal cancer
(54), prostate cancer recurrence
and related mortality (55). Some
trails suggest that metformin is associated with a decreased risk
of pancreatic cancer but in women only (56). Others indicate that although
metformin decreases risk of lung cancer, diabetics who develop lung
cancer while receiving metformin may have a more aggressive cancer
phenotype (57). Some trials show
that only long-term use of metformin is associated with a tendency
towards a decreased risk of ovarian cancer (58). Similarly long-term use of metformin
(>5 years) but not short-term use was associated with lower risk
for developing breast cancer compared with no use of metformin
(59).
In this study, we examined the effects of metformin
on human retinoblastoma growth in vivo, as well as in
vitro, ranging from mM down to μM concentrations. We show that
metformin inhibition of retinoblastoma cells in vitro, like
all other cancer-related studies involving metformin, is seen at mM
levels. Furthermore, levels similar to therapeutic levels achieved
in humans (μM) do not have an impact on retinoblastoma growth
either in vitro or in vivo.
High dose metformin has been shown to increase the
activity of AMPK in various cell lines at mM levels similar to
those used in our study (6,24,27,31,32,60)
and activation of AMPK has been shown to be involved in cell
proliferation (61,62). AMPK activation leads to inhibition
of the mTOR pathway through tuberous sclerosis complex 2 (TSC2)
(63) or directly without
involvement of TSC2 after stimulation with pharmacological agent or
with nutrient deprivation/stress (64). Indeed, in our study metformin
activated the AMPK pathway in retinoblastoma cell lines at mM
levels, as indicated by ACC phosphorylation and decreased
phosphorylation of ribosomal protein S6 (a downstream effector of
mTOR) and 4E-BP1 (a downstream effector of S6K). However, those
effects were not observed in vivo. Energy deprivation and
inhibition of the mTOR pathway (65) regulate autophagy (66), a process that maintains cellular
homeostasis. Indeed, mM dose metformin inhibited the mTOR pathway
associated with increased LC3-II expression (an autophagic marker).
This is in agreement with some studies which showed that high dose
metformin induces autophagy in cancer cells (25), however, others have not observed
induction of autophagy (27).
Although use of metformin has been extensively used
to study the AMPK pathway, like many pharmacological tools, it may
have other unknown functions that are independent of its initially
characterized action. Indeed, biguanides do not directly activate
AMPK in cell free assays (67),
and some studies have suggested that metformin mediates its effects
completely independently of AMPK (24,36,68).
Thus, to determine which proteins mediate the intracellular effect
of metformin, further studies are warranted.
When the in vitro effects of metformin on the
cell cycle are examined, it has been demonstrated that cells arrest
either in the G1 phase (24,32,69),
S phase (69), and/ or increase
the proportion of cells in the sub-G0/G1 population (69) depending on the cell type. In our
study, cell cycle analysis revealed that metformin treatment led to
a significant increase of cells in G0/G1 phase and a decrease in S
phase in Y79 cells, but the reverse was seen when WERI were treated
with metformin (Fig. 3). Similarly
to some researchers (24), we
observed decrease of cyclin D1 at mM levels in WERI cell line, but
in contrast to those reports, not in Y79 cells despite arrest in
G0/G1 (Figs. 3A, 4A and 5A). Additionally, the different cell
cycle changes observed in these two cell lines were not associated
with any specific cyclin and CDK change, but a rather non-specific
global reduction in cyclins (E1, E2, D3 and A2), cyclin-dependent
kinase (CDK2 and CDK4) as well as the CDK inhibitors p27 and p21
(Figs. 4, 5 and 6).
The downregulation of p27 at mM doses of metformin in our study is
in contrast to research that showed upregulation of p27 in prostate
cancer and ovarian caner cells (24,70),
or no effect in breast cancer cells (69). Several studies have also indicated
metformin may be involved in regulating the positive cell growth
regulator phospho-Akt (69,71–73).
In our study, the high (mM) dose of metformin in vitro
resulted in variable effects on the two retinoblastoma cell lines.
No effect was seen in the Y79 cell line, while in the WERI cell
line metformin lead to increased phospho-Akt (Fig. 6D and H). The data on cell cycle,
cyclins, and Akt taken together suggest that the in vitro
high dose metformin effects on cell cycle of retinoblastoma cells
may be non-specific.
Most in vitro experiments have shown effects
in various cancer cell lines but they typically use concentrations
in 2–50 mM, which are much higher than the plasma and tissue
concentrations measured in individuals who receive recommended
therapeutic doses (6,11,27,28).
Studies with μM levels of metformin usually have little effect on
cancer cell proliferation, as shown by our study and others
(74,75). Yet several epidemiological studies
have suggested that patients on metformin may have reduced cancer
risk (76,77) and some animal studies have shown
effects with μM levels (still almost 10-fold higher than the levels
seen in patients on metformin) (78,79).
In these studies (78,79) metformin was used with combination
chemotherapy and was shown to have a preferential effect on
tumor-forming, self-renewing cancer stem cells, which are resistant
to mainstream chemotherapy, yet were found to be sensitive to
metformin. Other additional hypothesis claim that metformin exerts
its antitumor effects in vivo via its effects on insulin,
IGF1 or IGFBP3 (reviewed in ref. 80), however, in our experiments the
levels of IGF1, IGFBP3 or IGF1/IGFBP3 ratio remained unchanged
(Fig. 10F–H).
Importantly, in our in vivo study, metformin
administration lead to levels of the drug equivalent to those seen
in patients on metformin, yet we did not detect statistically
significant effect on tumor growth, apoptosis, proliferation,
vascularity or infiltration by CD11b cells. It is possible that the
effects of metformin may be cancer cell specific and/or may involve
other pathways in the presence of concurrent chemotherapy. We can
not exclude that long-term treatment with metformin may have cancer
preventive effects for some cancer types which would be in
agreement with some but not all clinical trials (4,21,23,42,43,58,59).
In conclusion, we found that while mM concentration
of metformin inhibit growth of human retinoblastoma cell lines
in vitro, μM levels comparable to those achieved in
vivo do not. Furthermore, achieving therapeutic levels of
metformin in plasma (μM levels) did not affect tumor growth in
xenogratfs in Balb/c nude mice. Analysis of molecular signal
changes suggests that the effects seen in vitro at mM
metformin concentrations are possibly non-specific and due to the
very high drug dose causing toxicity. Any potential beneficial
effects of metformin seen in some, but not other, epidemiological
studies of cancer require extensive further investigation with
careful attention to the tumor type, as well as other indirect
effects and mechanisms.
Acknowledgements
This study was supported by National Eye Institute
grant EY014104 (MEEI Core Grant), an unrestricted grant to the
institution by the Research to Prevent Blindness Foundation (RPB),
a Physician Scientist Award by RPB to D.G.V. and by the
Massachusetts Lions Eye Research Fund.
References
|
1
|
Broaddus E, Topham A and Singh AD:
Incidence of retinoblastoma in the USA: 1975–2004. Br J Ophthalmol.
93:21–23. 2009.
|
|
2
|
Rodriguez-Galindo CC, Wilson MWM, Haik
BGB, et al: Treatment of metastatic retinoblastoma. Ophthalmology.
110:1237–1240. 2003. View Article : Google Scholar
|
|
3
|
Roarty JD, McLean IW and Zimmerman LE:
Incidence of second neoplasms in patients with bilateral
retinoblastoma. Ophthalmology. 95:1583–1587. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shaw RJ, Lamia KA, Vasquez D, et al: The
kinase LKB1 mediates glucose homeostasis in liver and therapeutic
effects of metformin. Science. 310:1642–1646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marr BP, Hung C, Gobin YP, Dunkel IJ,
Brodie SE and Abramson DH: Success of intra-arterial chemotherapy
(chemo-surgery) for retinoblastoma: effect of orbitovascular
anatomy. Arch Ophthalmol. 130:180–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu J, Li M, Song B, et al: Metformin
inhibits renal cell carcinoma in vitro and in vivo xenograft. Urol
Oncol. 31:264–270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bianciotto C, Shields CL, Iturralde JC,
Sarici A, Jabbour P and Shields JA: Fluorescein angiographic
findings after intra-arterial chemotherapy for retinoblastoma.
Ophthalmology. 119:843–849. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Muen WJ, Kingston JE, Robertson F, Brew S,
Sagoo MS and Reddy MA: Efficacy and complications of
super-selective intra-ophthalmic artery melphalan for the treatment
of refractory retinoblastoma. Ophthalmology. 119:611–616. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyano-Kurosaki N, Kurosaki K, Hayashi M,
et al: 2-amino-phenoxazine-3-one suppresses the growth of mouse
malignant melanoma B16 cells transplanted into C57BL/6Cr Slc mice.
Biol Pharm Bull. 29:2197–2201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ghassemi F and Shields CL: Intravitreal
melphalan for refractory or recurrent vitreous seeding from
retinoblastoma. Arch Ophthalmol. 130:1268–1271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qu Z, Zhang Y, Liao M, Chen Y, Zhao J and
Pan Y: In vitro and in vivo antitumoral action of metformin on
hepatocellular carcinoma. Hepatol Res. 42:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Munier FL, Gaillard M-C, Balmer A, et al:
Intravitreal chemotherapy for vitreous disease in retinoblastoma
revisited: from prohibition to conditional indications. Br J
Ophthalmol. 96:1078–1083. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shields CLC, Honavar SGS, Shields JAJ,
Demirci HH, Meadows ATA and Naduvilath TJT: Factors predictive of
recurrence of retinal tumors, vitreous seeds, and subretinal seeds
following chemoreduction for retinoblastoma. Arch Ophthalmol.
120:460–464. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shields CL, Shelil A, Cater J, Meadows AT
and Shields JA: Development of new retinoblastomas after 6 cycles
of chemo-reduction for retinoblastoma in 162 eyes of 106
consecutive patients. Arch Ophthalmol. 121:1571–1576. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sussman DA, Escalona-Benz E, Benz MS, et
al: Comparison of retinoblastoma reduction for chemotherapy vs
external beam radiotherapy. Arch Ophthalmol. 121:979–984. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schefler AC, Cicciarelli N, Feuer W,
Toledano S and Murray TG: Macular retinoblastoma: evaluation of
tumor control, local complications, and visual outcomes for eyes
treated with chemotherapy and repetitive foveal laser ablation.
Opthalmology. 114:162–169. 2007. View Article : Google Scholar
|
|
17
|
Shields CL: Forget-me-nots in the care of
children with retinoblastoma. Semin Ophthalmol. 23:324–334. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shields CL, Palamar M, Sharma P, et al:
Retinoblastoma regression patterns following chemoreduction and
adjuvant therapy in 557 tumors. Arch Ophthalmol. 127:282–290. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Benz MS, Scott IU, Murray TG, Kramer D and
Toledano S: Complications of systemic chemotherapy as treatment of
retinoblastoma. Arch Ophthalmol. 118:577–578. 2000.PubMed/NCBI
|
|
20
|
Nishimura S, Sato T, Ueda H and Ueda K:
Acute myeloblastic leukemia as a second malignancy in a patient
with hereditary retinoblastoma. J Clin Oncol. 19:4182–4183.
2001.PubMed/NCBI
|
|
21
|
Stumvoll M, Nurjhan N, Perriello G, Dailey
G and Gerich JE: Metabolic effects of metformin in
non-insulin-dependent diabetes mellitus. N Engl J Med. 333:550–554.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou G, Myers R, Li Y, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hundal HS, Ramlal T, Reyes R, Leiter LA
and Klip A: Cellular mechanism of metformin action involves glucose
transporter translocation from an intracellular pool to the plasma
membrane in L6 muscle cells. Endocrinology. 131:1165–1173.
1992.
|
|
24
|
Sahra IB, Laurent K, Loubat A, et al: The
antidiabetic drug metformin exerts an antitumoral effect in vitro
and in vivo through a decrease of cyclin D1 level. Oncogene.
27:3576–3586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Buzzai M, Jones RG, Amaravadi RK, et al:
Systemic treatment with the antidiabetic drug metformin selectively
impairs p53-deficient tumor cell growth. Cancer Res. 67:6745–6752.
2007. View Article : Google Scholar
|
|
26
|
Cufi S, Corominas-Faja B, Vazquez-Martin
A, et al: Metformin-induced preferential killing of breast cancer
initiating CD44+CD24−/low cells is sufficient
to overcome primary resistance to trastuzumab in HER2+
human breast cancer xenografts. Oncotarget. 3:395–398.
2012.PubMed/NCBI
|
|
27
|
Tomic T, Botton T, Cerezo M, et al:
Metformin inhibits melanoma development through autophagy and
apoptosis mechanisms. Cell Death Dis. 2:e1992011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kato K, Gong J, Iwama H, et al: The
antidiabetic drug metformin inhibits gastric cancer cell
proliferation in vitro and in vivo. Mol Cancer Ther. 11:549–560.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Martin MJ, Hayward R, Viros A and Marais
R: Metformin accelerates the growth of BRAF V600E-driven melanoma
by upregulating VEGF-A. Cancer Discov. 2:344–355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Phoenix KN, Vumbaca F and Claffey KP:
Therapeutic metformin/AMPK activation promotes the angiogenic
phenotype in the ERalpha negative MDA-MB-435 breast cancer model.
Breast Cancer Res Treat. 113:101–111. 2009. View Article : Google Scholar
|
|
31
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhuang Y and Miskimins WK: Cell cycle
arrest in metformin treated breast cancer cells involves activation
of AMPK, down-regulation of cyclin D1, and requires p27Kip1 or
p21Cip1. J Mol Signal. 3:182008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Towler MC and Hardie DG: AMP-activated
protein kinase in metabolic control and insulin signaling. Circ
Res. 100:328–341. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vijg J and Campisi J: Puzzles, promises
and a cure for ageing. Nature. 454:1065–1071. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Moruno-Manchón JF, Pérez-Jiménez E and
Knecht E: Glucose induces autophagy under starvation conditions by
a p38 MAPK-dependent pathway. Biochem J. 449:497–506.
2013.PubMed/NCBI
|
|
36
|
Hardie DG: The LKB1-AMPK pathway-friend or
foe in cancer? Cancer Cell. 23:131–142. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Doyle A, Zhang G, Abdel Fattah EA, Eissa
NT and Li YP: Toll-like receptor 4 mediates
lipopolysaccharide-induced muscle catabolism via coordinate
activation of ubiquitin-proteasome and autophagy-lysosome pathways.
FASEB J. 25:99–110. 2011. View Article : Google Scholar
|
|
38
|
Matsuzawa T, Kim B-H, Shenoy AR, Kamitani
S, Miyake M and Macmicking JD: IFN-γ elicits macrophage autophagy
via the p38 MAPK signaling pathway. J Immunol. 189:813–818.
2012.
|
|
39
|
Webber JL and Tooze SA: Coordinated
regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP.
EMBO J. 29:27–40. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thyagarajan A, Jedinak A, Nguyen H, et al:
Triterpenes from Ganoderma Lucidum induce autophagy in colon cancer
through the inhibition of p38 mitogen-activated kinase (p38 MAPK).
Nutr Cancer. 62:630–640. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Osborne CK, Bolan G, Monaco ME and Lippman
ME: Hormone responsive human breast cancer in long-term tissue
culture: effect of insulin. Proc Natl Acad Sci USA. 73:4536–4540.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Metformin insert 1–6, 2009. Distributed
by: Bristol-Myers Squibb Company; Princeton, NJ 08543, USA:
http://packagein-serts.bms.com/pi/pi_glucophage_xr.pdf.
|
|
43
|
Quinn BJ, Kitagawa H, Memmott RM, Gills JJ
and Dennis PA: Repositioning metformin for cancer prevention and
treatment. Trends Endocrinol Metabol. 24:469–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Janjetovic K, Harhaji-Trajkovic L,
Misirkic-Marjanovic M, et al: In vitro and in vivo anti-melanoma
action of metformin. Eur J Pharmacol. 668:373–382. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hadad SM, Appleyard V and Thompson AM:
Therapeutic metformin/AMPK activation promotes the angiogenic
phenotype in the ERα negative MDA-MB-435 breast cancer model.
Breast Cancer Res Treat. 114:391–399. 2009.
|
|
46
|
Dool CJ, Mashhedi H, Zakikhani M, et al:
IGF1/insulin receptor kinase inhibition by BMS-536924 is better
tolerated than alloxan-induced hypoinsulinemia and more effective
than metformin in the treatment of experimental insulin-responsive
breast cancer. Endocr Relat Cancer. 18:699–709. 2011. View Article : Google Scholar
|
|
47
|
Wilcock C and Bailey CJ: Accumulation of
metformin by tissues of the normal and diabetic mouse. Xenobiotica.
24:49–57. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cazzaniga M, Bonanni B, Guerrieri-Gonzaga
A and Decensi A: Is it time to test metformin in breast cancer
clinical trials? Cancer Epidemiol Biomarkers Prev. 18:701–705.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Noto H, Goto A, Tsujimoto T and Noda M:
Cancer risk in diabetic patients treated with metformin: a
systematic review and meta-analysis. PLoS One. 7:e334112012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Becker C, Meier CR, Jick SS and Bodmer M:
Case-control analysis on metformin and cancer of the esophagus.
Cancer Causes Control. 24:1763–1770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Becker C, Jick SS, Meier CR and Bodmer M:
Metformin and the risk of endometrial cancer: a case-control
analysis. Gynecol Oncol. 129:565–569. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bodmer M, Becker C, Jick SS and Meier CR:
Metformin does not alter the risk of lung cancer: a case-control
analysis. Lung Cancer. 78:133–137. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bodmer M, Becker C, Meier C, Jick SS and
Meier CR: Use of metformin is not associated with a decreased risk
of colorectal cancer: a case-control analysis. Cancer Epidemiol
Biomarkers Prev. 21:280–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kaushik D, Karnes RJ, Eisenberg MS, Rangel
LJ, Carlson RE and Bergstralh EJ: Effect of metformin on prostate
cancer outcomes after radical prostatectomy. Urol Oncol.
32:43.e1–7. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bodmer M, Becker C, Meier C, Jick SS and
Meier CR: Use of antidiabetic agents and the risk of pancreatic
cancer: a case-control analysis. Am J Gastroenterol. 107:620–626.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mazzone PJ, Rai H, Beukemann M, Xu M, Jain
A and Sasidhar M: The effect of metformin and thiazolidinedione use
on lung cancer in diabetics. BMC Cancer. 12:4102012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bodmer M, Becker C, Meier C, Jick SS and
Meier CR: Use of metformin and the risk of ovarian cancer: a
case-control analysis. Gynecol Oncol. 123:200–204. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bodmer M, Meier C, Krähenbühl S, Jick SS
and Meier CR: Long-term metformin use is associated with decreased
risk of breast cancer. Diabetes Care. 33:1304–1308. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tomimoto A, Endo H, Sugiyama M, et al:
Metformin suppresses intestinal polyp growth in ApcMin/+
mice. Cancer Science. 99:2136–2141. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Theodoropoulou S, Kolovou PE, Morizane Y,
et al: Retinoblastoma cells are inhibited by aminoimidazole
carboxamide ribonucleotide (AICAR) partially through activation of
AMP-dependent kinase. FASEB J. 24:2620–2630. 2010. View Article : Google Scholar
|
|
62
|
Rattan R, Giri S, Singh AK and Singh I:
5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits
cancer cell proliferation in vitro and in vivo via AMP-activated
protein kinase. J Biol Chem. 280:39582–39593. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Inoki KK, Zhu TT and Guan K-LK: TSC2
mediates cellular energy response to control cell growth and
survival. Cell. 115:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cheng SWY, Fryer LGD, Carling D and
Shepherd PR: Thr2446 is a novel mammalian target of rapamycin
(mTOR) phosphorylation site regulated by nutrient status. J Biol
Chem. 279:15719–15722. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sarbassov DD, Ali SM and Sabatini DM:
Growing roles for the mTOR pathway. Curr Opin Cell Biol.
17:596–603. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hawley SA, Gadalla AE, Olsen GS and Hardie
DG: The antidiabetic drug metformin activates the AMP-activated
protein kinase cascade via an adenine nucleotide-independent
mechanism. Diabetes. 51:2420–2425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kalender A, Selvaraj A, Kim SY, et al:
Metformin, independent of AMPK, inhibits mTORC1 in a rag
GTPase-dependent manner. Cell Metab. 11:390–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu B, Fan Z, Edgerton SM, et al:
Metformin induces unique biological and molecular responses in
triple negative breast cancer cells. Cell Cycle. 8:2031–2040. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Li C, Liu VWS, Chan DW, Yao KM and Ngan
HYS: LY294002 and metformin cooperatively enhance the inhibition of
growth and the induction of apoptosis of ovarian cancer cells. Int
J Gynecol Cancer. 22:15–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Capano M and Crompton M: Bax translocates
to mitochondria of heart cells during simulated ischaemia:
involvement of AMP-activated and p38 mitogen-activated protein
kinases. Biochem J. 395:57–64. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Xi X, Han J and Zhang JZ: Stimulation of
glucose transport by AMP-activated protein kinase via activation of
p38 mitogen-activated protein kinase. J Biol Chem. 276:41029–41034.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Würth R, Pattarozzi A, Gatti M, et al:
Metformin selectively affects human glioblastoma tumor-initiating
cell viability: A role for metformin-induced inhibition of Akt.
Cell Cycle. 12:145–156. 2013.PubMed/NCBI
|
|
74
|
Bao B, Wang Z, Ali S, et al: Metformin
inhibits cell proliferation, migration and invasion by attenuating
CSC function mediated by deregulating miRNAs in pancreatic cancer
cells. Cancer Prevent Res. 5:355–364. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ben Sahra I, Regazzetti C, Robert G, et
al: Metformin, independent of AMPK, induces mTOR inhibition and
cell-cycle arrest through REDD1. Cancer Res. 71:4366–4372.
2011.PubMed/NCBI
|
|
76
|
Soranna D, Scotti L, Zambon A, et al:
Cancer risk associated with use of metformin and sulfonylurea in
type 2 diabetes: a meta-analysis. Oncologist. 17:813–822. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bost F, Sahra IB, Le Marchand-Brustel Y
and Tanti JF: Metformin and cancer therapy. Curr Opin Oncol.
24:103–108. 2012. View Article : Google Scholar
|
|
78
|
Iliopoulos D, Hirsch HA and Struhl K:
Metformin decreases the dose of chemotherapy for prolonging tumor
remission in mouse xenografts involving multiple cancer cell types.
Cancer Res. 71:3196–3201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hirsch HA, Iliopoulos D, Tsichlis PN and
Struhl K: Metformin selectively targets cancer stem cells, and acts
together with chemotherapy to block tumor growth and prolong
remission. Cancer Res. 69:7507–7511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Martin-Castillo B, Vazquez-Martin A,
Oliveras-Ferraros C and Menendez JA: Metformin and cancer: doses,
mechanisms and the dandelion and hormetic phenomena. Cell Cycle.
9:1057–1064. 2010. View Article : Google Scholar : PubMed/NCBI
|