Introduction
Liver cancer is the sixth most common malignant
disease worldwide but the third most frequent cause of
cancer-related death. Of these cases and deaths, approximately 50%
are estimated to occur in China. As the major histological subtype,
hepatocellular carcinoma (HCC) accounts for 70–85% of the total
liver cancer incidence worldwide. Both the morbidity and mortality
of HCC have increased during the past few decades and long-term
prognosis and survival rate of HCC patients after surgical
resection remain unsatisfactory because of the high rate of
recurrence and metastasis (1,2–6).
Hepatitis B or C viral infection, alcohol abuse and aflatoxin
intake are the main known risk factors for the development of HCC,
yet prevention and treatment of HCC require a better understanding
of the molecular mechanisms underlying the oncogenesis and cancer
progression of HCC (7–10).
Dual-specificity phosphatases (DUSPs) are a
heterogeneous group of protein phosphatases that can regulate the
activity of mitogen-activated protein kinases (MAPKs), which play a
critical role in the control of cell growth and survival in
physiological and pathological processes, including cancer
(11,12). DUSPs can be classified into six
major groups based on the presence of specific domains and sequence
similarity, including the MAPK phosphatases (MKPs), as well as a
group of small-size atypical DUSPs (13). As their name indicates, MKPs can
dephosphorylate MAPK proteins ERK, JNK and p38 with specificity.
The small-size atypical DUSPs are structurally and functionally
related to the MKPs, although they lack the N-terminal regulatory
domain. The MKPs and their alterations in cancer have been widely
investigated, while the involvement of the small atypical DUSPs in
cancer has been less studied (14–17).
In the past, atypical DUSPs have gene rally been grouped together
with the MKPs and characterized for their role in MAPK signaling
cascades, however, growing number of investigations indicate that
they can have vastly different substrate specificities and
physiological roles from those of the typical MKPs (18–20).
Some of these atypical DUSPs are emerging as potential regulators
of cell growth and apoptosis and several small-sized atypical DUSPs
have been directly related with human disease including cancer
(21–23). The active role for atypical DUSPs
in oncogenesis or resistance to cancer therapies makes them good
candidate targets for anti-cancer drugs. To date, some atypical
DUSPs are emerging as relevant targets for anti-cancer therapy and
drug-discovery efforts are undertaking to develop specific and
efficient DUSP enzyme inhibitors (24–27).
Despite previous studies that have been carried out
in this area, the expression of DUSP28, a member of the small-size
atypical DUSPs, is completely unknown in HCC. Our previous
genome-wide approach demonstrated that DUSP28 was markedly
upregulated in HCC clinical specimens compared to adjacent
non-cancerous livers (data not shown). In this current study, we
confirm that DUSP28 is frequently upregulated in HCC and can
significantly promote HCC cell proliferation and colony formation
in vitro. Further analysis reveals the potential role of
DUSP28 on HCC cell cycle transition. We indicate for the first time
that DUSP28 plays a critical role in HCC progression and may serve
as a candidate therapeutic target for HCC.
Materials and methods
Tissue samples
In this study, we collected 50 pairs of clinical
tissue samples from the First Affiliated Hospital of Nanjing
Medical University between January 2004 and May 2006. All the
tissue specimens, both cancerous and non-cancerous, were obtained
from resected specimens and were then rapidly frozen at −80°C for
storage until use. The diagnosis of HCC was validated by
pathological examination. Written informed consent was obtained
from all patients and the study was approved by the institutional
ethics committee of Nanjing Medical University.
Cell lines and culture conditions
The human HCC cell lines (Hep3B, HepG2, Huh7,
SK-Hep-1, Focus, YY-8103, PLC/PRF/5, LM3, LM6, MHCC-97H, MHCC-97L,
SMMC-7721) used in this study were obtained from the Chinese
National Human Genome Center at Shanghai. Each was cultured at
37°C, 95% air, 5% CO2 in Dulbecco’s modified Eagle’s
medium (DMEM) containing 10% fetal bovine serum (FBS) (both from
HyClone, Logan, UT, USA), 50 U/ml penicillin and 50 μg/ml
streptomycin.
Antibodies and reagents
Antibodies against Flag, DUSP2, cyclin D1, cyclin E,
p21, p27, and β-actin were from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Antibodies against ERK (total), ERK
(phosphorylated Thr202/Tyr204), JNK (total), JNK (phosphorylated
Thr183/Tyr185), p38 (total), p38 (phosphorylated Thr180/Tyr182)
were obtained from Cell Signaling Technology, Inc. (Danvers, MA,
USA). SB203580 was purchased from Sigma (St. Louis, MO, USA).
RNA extraction, semi-quantitative and
real-time PCR
Total RNA was extracted with TRIzol®
(Invitrogen Life Technologies, Carlsbad, CA, USA), following the
manufacturer’s protocol. Reverse transcription was performed in a
25-μl reaction volume using M-MLV Reverse Transcriptase (Promega
Corporation, Madison, WI, USA) with a total of 2 μg of RNA.
Semi-quantitative RT-PCR was performed and products were separated
on 1% agarose gel-containing ethidium bromide. Quantitative
real-time PCR was performed using a Thermal Cycler Dice Detection
System with the SYBR Premix Ex Taq™ (Perfect Real-Time) (Takara
Bio, Inc., Shiga, Japan). The sequences of primers specific to
DUSP28 and housekeeping genes were designed as follows: DUSP28
forward, 5′-CCTTCCAGATGGTGAAGAGC-3′ and reverse,
5′-GGTGAATGTGGGTGACACTG-3′; β-actin forward,
5′-AGAGCCTCGCCTTTGCCGATCC-3′ and reverse,
5′-CTGGGCCTCGTCGCCCACATA-3′.
Protein extraction and western blot
analysis
Total cellular proteins were extracted using cell
lysis buffer containing 50 mM Tris-HCl (pH 6.8), 2% SDS, 10%
2-mercaptoethanol, 10% glycerol, and protease inhibitor cocktail
(Sigma). Then protein concentration was determined using a BCA kit
(Thermo Fisher Scientific, Waltham, MA, USA). The protein (20 μg)
was subjected to electrophoresis by SDS-PAGE on a 10% gel and then
transferred to a polyvinylidene difluoride (PVDF) membrane. The
membrane was blocked with 5% non-fat dry milk and 0.1% Tween-20 in
PBS for 2 h at room temperature. After incubation with the
appropriate primary antibody overnight at 4°C, membranes were
washed and incubated with the IRDye 800CW or 680RD secondary
antibodies in TBST for 2 h at room temperature. The labeled protein
bands were detected using the Odyssey Infrared Imaging System
(Li-COR Biosciences, Lincoln, NE, USA). β-actin was used as a
loading control.
Plasmid construction
The full-length DUSP28 open reading frame (ORF) was
amplified from the cDNA of PLC/PRF/5 cell using PrimeStar PCR and
then the product was inserted into the expression vector
pcDNA3.1B-FLAG-GFP, obtained from the Chinese National Human Genome
Centre at Shanghai. The sequences of the cloning primers were as
follows: DUSP28-XhoI forward,
5′-TACTCGAGATGGGACCGGCAGAAGCTGGGCGCCG-3′; DUSP28-BamHI
reverse, 5′-GAGGATCCAGCCTCAGGGCCCAACCCTAAGGCTG-3′.
RNA interference
Two siRNAs against DUSP28 were chemically
synthesized (Shanghai GenePharma Co., Shanghai, China) and their
sequences are as follows: siRNA-294 forward,
5′-CUGCCUAGUCUACUGCAAGAACGdTdT-3′ and reverse,
5′-CGUUCUUGCAGUAGACUAGGCAGdTdT-3′; siRNA-438 forward,
5′-CUGGUCUCAGCUCCAGAAGUAUGdTdT-3′ and reverse,
5′-CAUACUUCUGGAGCUGAGACCAGdTdT-3′. The non-targeting nucleotides
were used as a negative control: siRNA-NC forward,
5′-UUCUCCGAACGUGUCACGUdTdT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAAdTdT-3′. The oligonucleotides encoding short
hairpin RNAs (shRNAs) for the continuous knockdown of endogenous
DUSP28 were synthesised and inserted into pSUPER (OligoEngine,
Seattle, WA, USA). Their sequences are as follows: shRNA-294
forward,
5′-GATCCCCCTGCCTAGTCTACTGCAAGAACGTTCAAGAGACGTTCTTGCAGTAGACTAGGCAGTTTTTGGAAA-3′
and reverse,
5′-AGCTTTTCCAAAAACTGCCTAGTCTACTGCAAGAACGTCTCTTGAACGTTCTTGCAGT
AGACTAGGCAGGGG-3′; shRNA-438 forward,
5′-GATCCCCCTGGTCTCAGCTCCAGAAGTATGTTCAAGAGACATA
CTTCTGGAGCTGAGACCAGTTTTTGGAAA-3′ and re verse,
5′-AGCTTTTCCAAAAACTGGTCTCAGCTCCAGAAGTATGTCTCTTGAACATACTTCTGGAGCTGAGACCAGGGG-3′;
shRNA-NC contained irrelevant nucleotides and served as a negative
control: shRNA-NC forward,
5′-GATCCCCTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTGGAAA-3′
and reverse,
5′-AGCTTTTCCAAAAATTCTCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAAGGG-3′.
Cell transfection
Cell transfection was performed with Lipofectamine
2000 (Invitrogen Life Technologies) according to the manufacturer’s
instructions. Cells were transfected with RNAi and plasmid at cell
density of 30–50% and 80–90%, respectively.
Cell proliferation
Transiently transfected HCC cells were plated at a
density of 2,000–5,000 cells/well into 96-well plates and cultured
for 6 days or 7 days. Cell growth was examined using the Cell
Counting kit-8 (Dojindo Laboratories, Kumamoto, Japan) according to
the manufacturer’s instructions. The absorbance value at a
wavelength of 450 nm was used as an indicator of cell
viability.
Colony formation
HCC cells were transfected and then plated at a
density of 10,000–50,000 cells/10-cm plate. Then G418 (Life
Technologies, Grand Island, NY, USA) was added to the medium at a
final concentration of 0.8–1 mg/ml for colony formation at 37°C in
5% CO2. After 2–3 weeks of selection, the remaining
colonies were washed twice with PBS, fixed with paraformaldehyde
and stained with Coomassie brilliant blue.
For the soft agar colony formation assay in 24-well
plates, 2,000–5,000 transfected cells were plated and grown on a
plate containing 1% base agar and 0.5% top agar. After cultured for
3–4 weeks, all colonies were photographed and counted under a
dissecting microscope.
Cell cycle analysis
Flow cytometry was performed to analyse the cell
cycle. Serum starvation was used to induce cell cycle
synchronization before cells were transfected with siRNA or plasmid
and then harvested as single cell suspensions. For DNA content
detection, cells were fixed in 70% ethanol, resuspended in PBS,
incubated with RNase A (10 mg/ml) and propidium iodide (10 μg/ml)
for 30 min in the dark, followed by flow cytometric analysis using
the FACSCalibur flow cytometer, CellQuest (BD Biosciences, Franklin
Lakes, NJ, USA).
Statistical analysis
Statistical analysis was performed by Student’s
t-test using GraphPad Prism 5 software. All results are presented
as means ± standard deviation (SD). P<0.05 was considered to be
statistically significant.
Results
Expression level of DUSP28 is
significantly upregulated in HCC
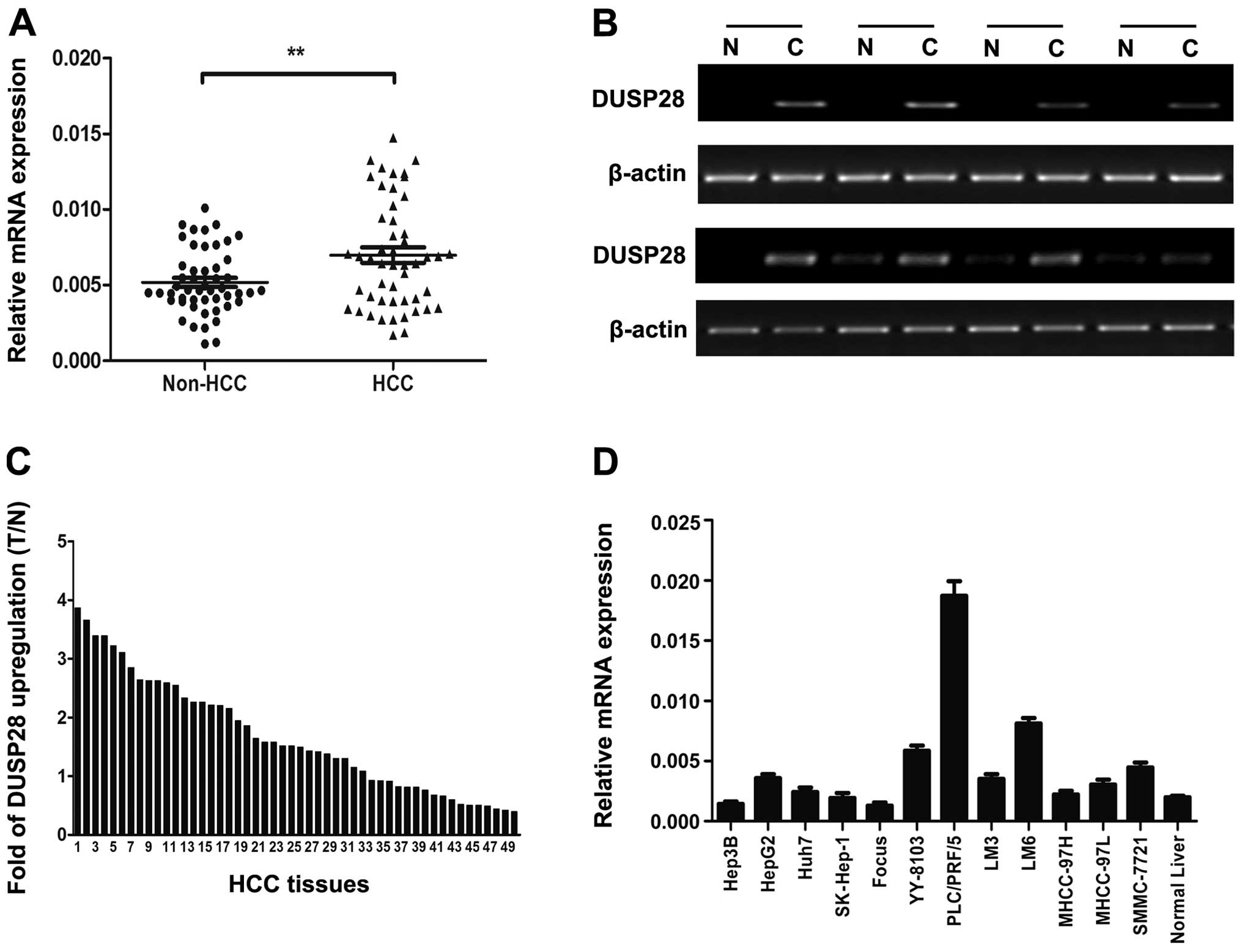
We first examined expression levels of DUSP28 in 50
paired HCC tissues by quantitative real-time PCR and
semi-quantitative RT-PCR. The results showed that DUSP28 mRNA
expression was significantly upregulated in the HCC tissues in
comparison with the corresponding adjacent non-cancerous livers
(Fig. 1A and B). Among these
samples, DUSP28 was shown to be elevated in 15/50 (30%) of the HCC
specimens at >2-fold higher levels (Fig. 1C). Moreover, DUSP28 expression
levels were assessed in set of 12 widely-used HCC cell lines and we
found that the expression of DUSP28 was higher in a majority of HCC
cell lines when compared to normal adult liver tissue (Fig. 1D).
Overexpression of DUSP28 promotes HCC
cell proliferation and colony formation in vitro
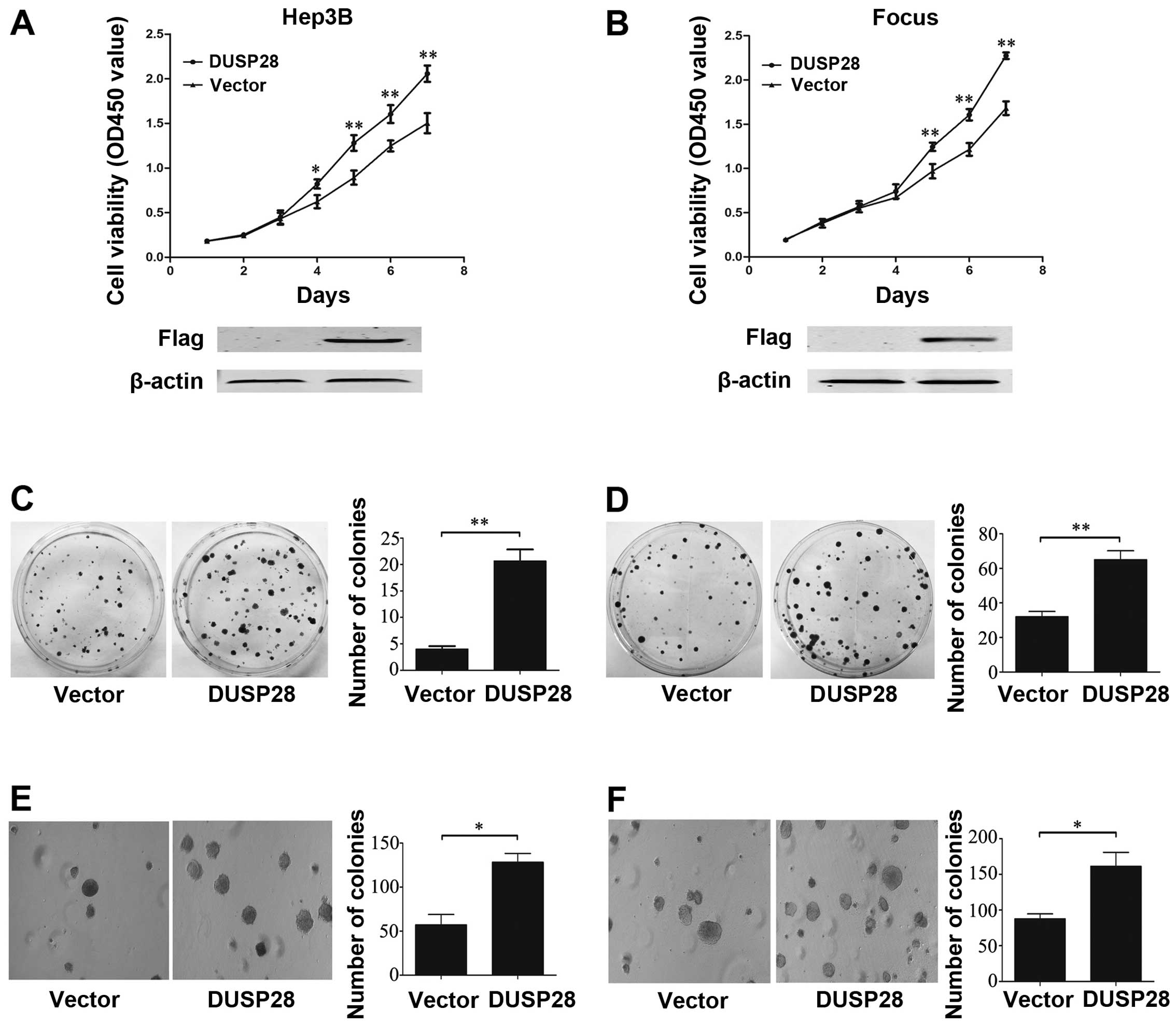
To evaluate the role of DUSP28 in HCC, we first
observed the effect of DUSP28 overexpression on HCC cells. So we
transiently transfected the recombinant plasmid pcDNA3.1-DUSP28
into Hep3B and Focus cell lines, which lack endogenous DUSP28
expression. The empty pcDNA3.1 vector was used as control. Western
blot analysis was performed to confirm that the transfected DUSP28
was expressed successfully in Hep3B and Focus cells. Our
observation revealed that DUSP28 overexpression significantly
promoted cell growth (Fig. 2A and
B) and colony formation (Fig. 2C
and D) when compared to that of the cells transfected with the
empty vector. Moreover, ectopic DUSP28 expression significantly
enhanced the anchorage-independent growth of Hep3B and Focus cell
lines in soft agar, as well as dramatically increased the number of
larger colonies relative to the cells transfected with the empty
vector (Fig. 2E and F). These data
suggest that DUSP28 overexpression plays an important role in
promoting cell proliferation, anchorage-dependent and -independent
colony formation in vitro.
Knockdown of DUSP28 inhibits HCC cell
proliferation and colony formation in vitro
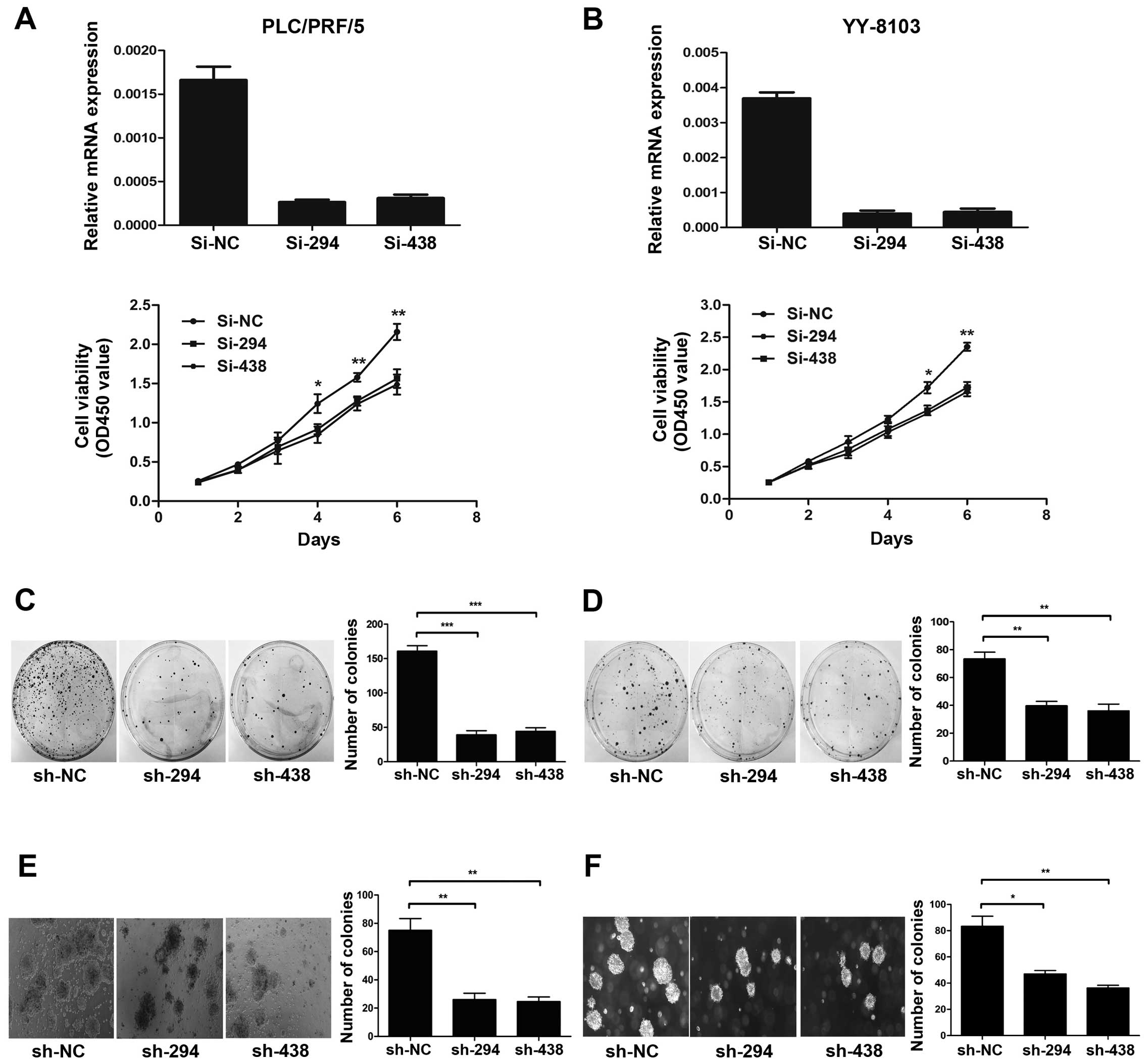
To further investigate the role of DUSP28 on cell
proliferation and colony formation, we used chemically synthesised
siRNAs (si-294 and si-438) to knock down endogenous DUSP28 in
PLC/PRF/5 and YY-8103 cells with relatively high expression of
DUSP28. The si-NC was used as a negative control. As expected, both
siRNAs were considered to be effective for DUSP28 knockdown and
were used subsequently. Cell proliferation assay showed that the
siRNAs-transfected cells exhibited significant growth suppression
in comparison with the si-NC-transfected cells (Fig. 3A and B). In order to examine the
effect of DUSP28 on colony formation, we constructed the
corresponding shRNA plasmids (pSUPER-sh-NC, pSUPER-sh-294 and
pSUPER-sh-438) and transfected them to the same HCC cell lines.
Results certified that both shRNAs significantly inhibited the
colony formation of PLC/PRF/5 and YY-8103 cells compared to the
control shRNA-NC-transfected cells (Fig. 3C and D). Furthermore, the
downregulation of DUSP28 restrained the anchorage-independent
colony formation of PLC/PRF/5 and YY-8103 cell lines in soft agar
and significantly reduced the number of larger colonies compared to
the cells transfected with the negative control shRNA (Fig. 3E and F). These collective data
indicate that endogenous DUSP28 knockdown can significantly
suppress cell growth, colony formation and soft agar colony
formation in HCC cells.
DUSP28 influences the G1/S cell cycle
transition
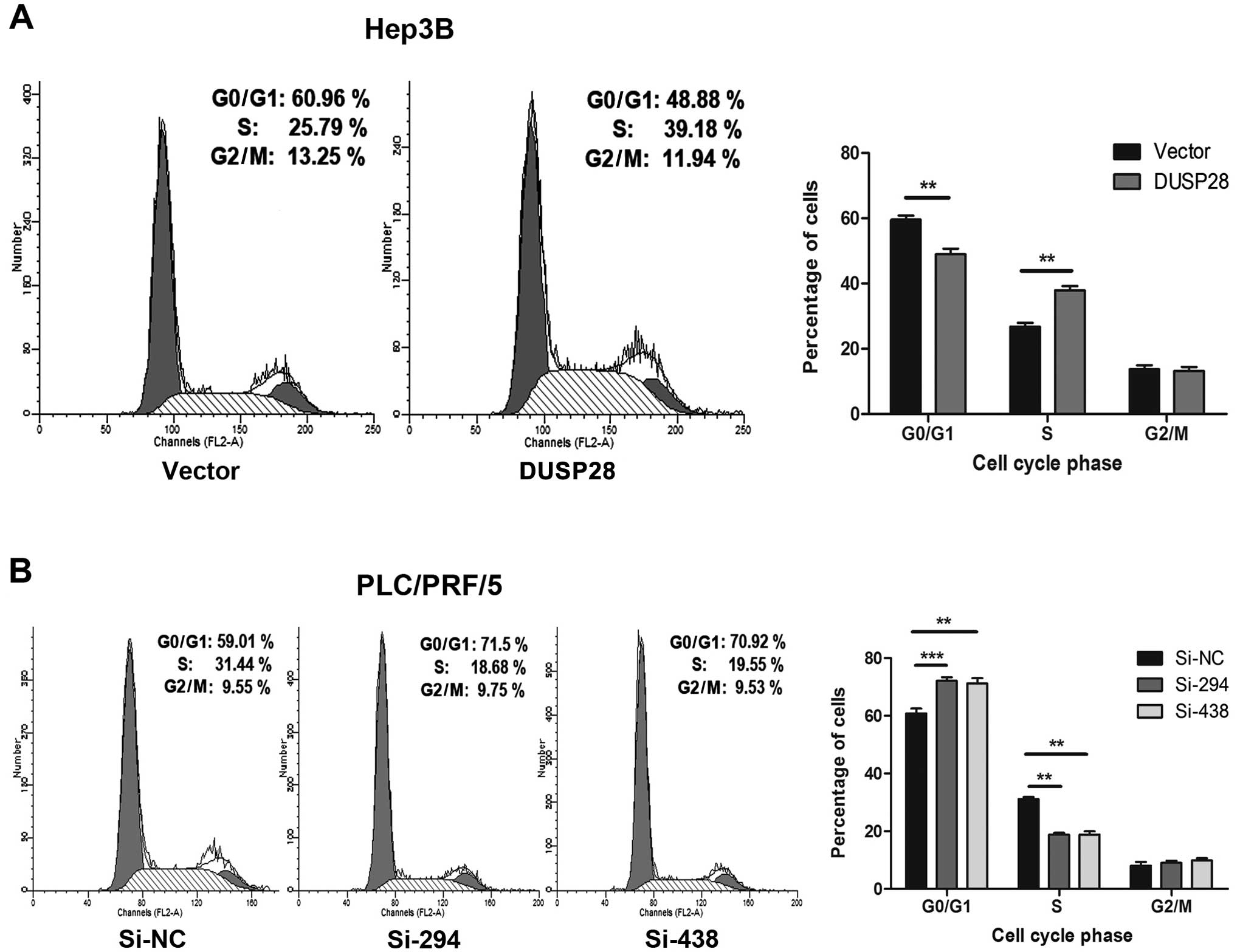
The fact that DUSP28 affects cell proliferation
suggested that it may influence the cell cycle. Therefore, we
performed a flow cytometric analysis and determined whether DUSP28
could affect cell cycle distribution. The results showed that the
proportion of cancer cells at the G0/G1 phase was significantly
decreased in Hep3B cells transfected with recombinant
pcDNA3.1-DUSP28 plasmid than in those treated with empty pcDNA3.1
vector, while the proportion in S phase was remarkably increased
(Fig. 4A). In contrast, the
fraction of cancer cells at the G0/G1 phase was significantly
increased upon DUSP28 downregulation with si-294 or si-438 in
PLC/PRF/5 cells than in those treated with the control si-NC, while
the proportion in S phase was notably decreased (Fig. 4B). In conclusion, these results
suggest that DUSP28 plays an essential role in the growth
regulation of HCC cells by modulating the G1/S transition.
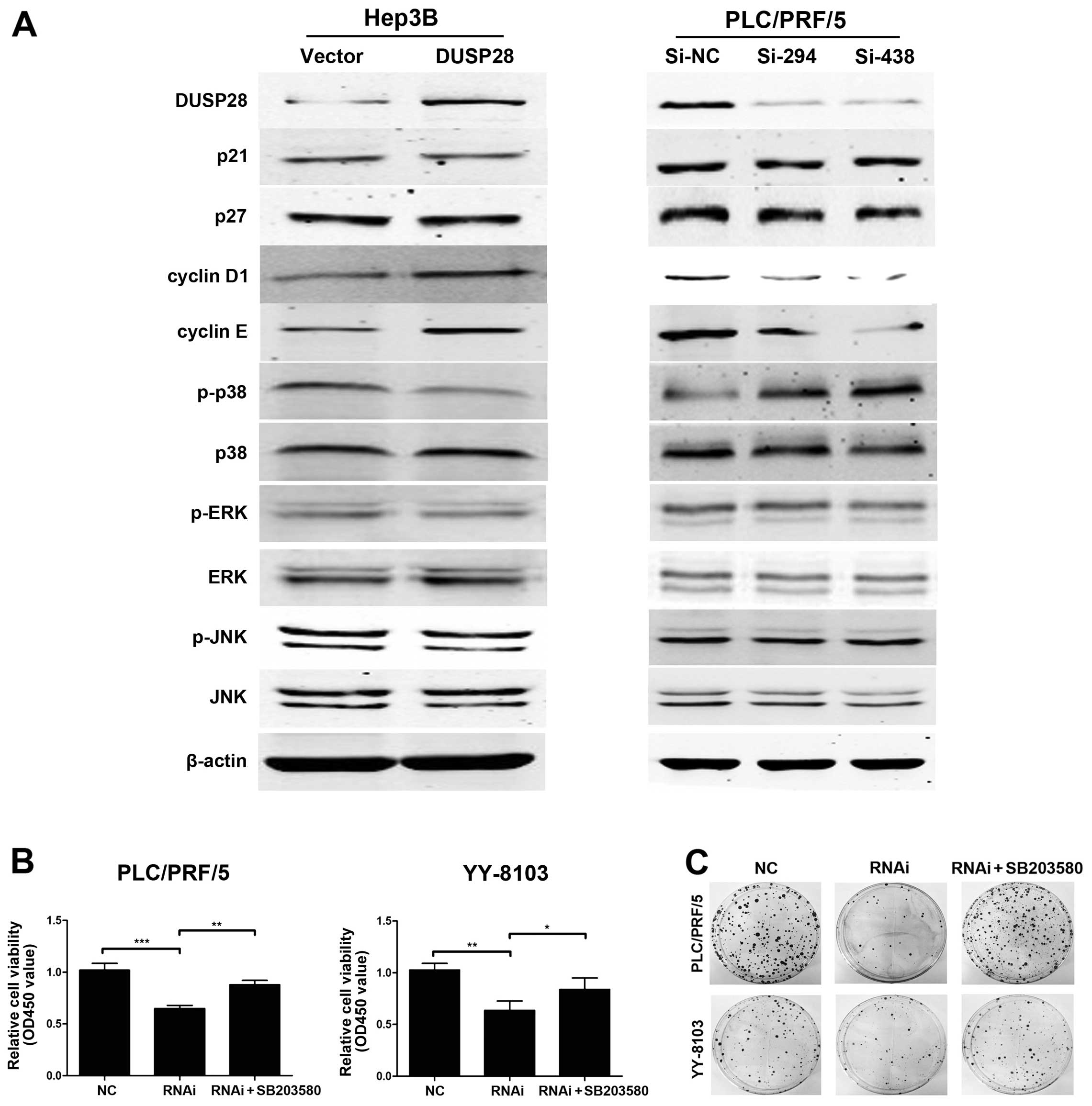
DUSP28 regulates the p38 MAPK signaling
pathway
To further clarify the mechanism by which DUSP28
affects the G1/S transition, western blot analysis was used to
assay several key molecules including p27, p21, cyclin D1 and
cyclin E, which are known to be involved in G1/S transition.
Interestingly, our data showed that DUSP28 overexpression led to an
increase of cyclin D1 and cyclin E in pcDNA3.1-DUSP28-infected
Hep3B cells when compared with that of the cells transfected the
empty vector, although no obvious changes were seen on p21 and p27
expression. In contrast, the opposite results were obtained when
PLC/PRF/5 cells were infected with si-294 or si-438, supporting the
observation of DUSP28 knockdown-mediated G1 arrest. Previous
studies demonstrated that p38 MAPK activation could reduce the
levels of cyclin D1, thus contributing to the induction of the G1/S
checkpoint. Moreover, increasing evidence suggest that p38 MAPK can
function as a suppressor of cell proliferation and tumorigenesis
(28,29). Therefore, we proposed that DUSP28
might also be involved in the p38 MAPK signaling pathway. As
confirmation, we assessed the levels of phosphorylated p38, as well
as ERK1/2 and JNK, which are the classical MAPKs. Expectedly, the
phosphorylation level of p38 was decreased in Hep3B cells
transfected with pcDNA3.1-DUSP28 and increased in PLC/PRF/5 cells
treated with si-294 or si-438. However, the phosphorylation level
of ERK1/2 and JNK, was not observably changed (Fig. 5A). To further confirm the
association between DUSP28 and p38, we used SB203580, a known p38
MAPK inhibitor, to determine whether p38 MAPK contributes to the
phenotypic changes of DUSP28 knockdown in HCC cells. The result
showed that p38 inhibitor SB203580 almost completely abolished the
inhibitory the role of DUSP28 knockdown in PLC/PRF/5 and YY-8103
cells (Fig. 5B and C). Therefore,
we infer that DUSP28 is involved in the activation of p38 MAPK
signaling.
Discussion
HCC is one of the most prevalent malignancies,
ranking as the sixth most common cancer worldwide and the third
most common cause of cancer mortality. The prognosis of HCC
patients remains very poor, so novel therapeutic approaches against
drug targets are urgently needed (1,30).
The DUSPs constitute a heterogeneous group of
cysteine-based protein tyrosine phosphatases. As a subgroup of
DUSPs, the small-size atypical DUSPs are generally smaller and lack
the CH2 domain in the N-terminus that is common to the typical MKPs
(13). A number of studies on
small atypical DUSPs in the past few decades has enhanced our
understanding of their function in cancer and indicated their
potential as anti-cancer therapy targets. For example, upregulation
of DUSP3 is detected in some human carcinomas, including prostate
and cervix carcinomas and it may act as a positive regulator of
cell growth in cancer cells. Thus, inhibitors of the activity of
DUSP3 could be putative anti-cancer drugs (31,32);
DUSP23 is upregulated in several human cancers, and it promotes
cell proliferation of human breast cancer cells by regulating the
G1/S transition, suggesting a carcinogenic role for DUSP23, which
makes it a promising drug target for anti-cancer therapy (33). Moreover, a recent study
demonstrates that DUSP21 is upregulated in HCC specimens and the
DUSP21 expression in HCC cells is essential for maintaining cell
proliferation and survival, making DUSP21 a potential therapeutic
target for HCC treatment (34).
Besides, several other small atypical DUSPs, including DUSP14 and
DUSP26, are also involved in cancer, raising the possibility of
them as targets for drug discovery (35,36).
Nevertheless, functional studies on some other small atypical
DUSPs, such as DUSP13b, DUSP18 and DUSP28, are limited, and their
relation with cancer especially HCC has not been confirmed. Due to
their similarity in structure with the small atypical DUSPs
mentioned above, it is possible that they may also play a role in
cancer progression.
In this study, we pay particular attention to
DUSP28, a member of the atypical DUSP family. Our previous
gene-expression profiles using the genome-wide method indicated
that DUSP28 was upregulated in HCC tissues compared with the
matched non-tumorous tissues (unpublished data). Therefore, we
suppose that DUSP28 may act as a possible oncogene in HCC. To
confirm this assumption, we evaluate the expression levels of
DUSP28 in additional 50 paired HCC samples and 12 HCC cell lines.
Our data indicated that DUSP28 was frequently upregulated in human
HCC tissues relative to matched non-tumorous samples. Besides, high
expression of DUSP28 was also found in HCC cell lines. These data
implied that DUSP28 might play a specific functional role in HCC
carcinogenesis. Then we further investigated the function of DUSP28
in HCC cell proliferation in vitro. As expected, in
vitro experiments revealed that DUSP28 overexpression
accelerated the proliferation and colony formation of HCC cells,
while DUSP28 knockdown led to a suppression of cell growth. Flow
cytometric analysis indicated that over expression of DUSP28
facilitated the G1/S transition in Hep3B cells. Likewise, knockdown
of DUSP28 in PLC/PRF/5 cells led to an arrest in G0/G1 phase.
In an attempt to explore the molecular mechanisms
involved in the functions of DUSP28, western blot analysis was
performed to detect several important molecules which are known to
be involved in G1/S transition. We observed a significant increase
of cyclin D1 and cyclin E in cells transfected with pcDNA3.1-DUSP28
plasmids, as well as an evident decrease of cyclin D1 and cyclin E
in cells with DUSP28 knockdown. Accumulated evidence suggests that
cyclin D1 and cyclin E are responsible for cell cycle progression
from G1 to S phase (37), and that
p38 activity can downregulate cyclin D1 expression, thus acting on
cell cycle checkpoints (28). The
role of the p38 MAPK pathway in the cell differentiation, growth
inhibition and apoptosis is well documented and p38 MAPK has been
defined as a tumor suppressor (38,39).
Therefore, we subsequently detected the phosphorylation level of
p38 in HCC cells. As expected, forced DUSP28 expression resulted in
a decreased phosphorylation level of p38 in HCC cells, while DUSP28
knockdown led to an increased phosphorylation level of p38.
Intriguingly, we did not observe obvious change on the
phosphorylation level of ERK and JNK, although they are also
possible substrates of DUSPs, indicating that DUSP28 might have a
high substrate specificity for p38. In addition, to further confirm
our observation, we used a known p38 MAPK inhibitor SB203580 in the
cell proliferation assay. As expected, the anti-proliferative
effect of DUSP28 knockdown in PLC/PRF/5 cells was significantly
rescued by the p38 inhibitor SB203580.
To sum up, our study shows significant upregulation
of DUSP28 in HCC tissues and cell lines. Ectopic expression of
DUSP28 promotes cell proliferation, colony formation, soft agar
colony formation of Hep3B and Focus cells, while knockdown of
DUSP28 has the anti-proliferative effect in PLC/PRF/5 and YY-8103
cells. Further molecular-mechanism investigations clarified that
DUSP28 was involved in the G1/S transition through the activation
of p38 MAPK signaling. These data suggest that DUSP28 plays a
significant role in HCC progression and targeting this molecule may
be a potential novel therapeutic strategy for HCC treatment.
Acknowledgements
This study was supported by the National Natural
Science Foundation, China (grant no. 81170415).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Shiraha H, Yamamoto K and Namba M: Human
hepatocyte carcinogenesis (review). Int J Oncol. 42:1133–1138.
2013.PubMed/NCBI
|
|
3
|
Perz JF, Armstrong GL, Farrington LA,
Hutin YJ and Bell BP: The contributions of hepatitis B virus and
hepatitis C virus infections to cirrhosis and primary liver cancer
worldwide. J Hepatol. 45:529–538. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang Y, Nagano H, Ota H, et al: Patterns
and clinicopathologic features of extrahepatic recurrence of
hepatocellular carcinoma after curative resection. Surgery.
141:196–202. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hwang S, Moon DB and Lee SG: Liver
transplantation and conventional surgery for advanced
hepatocellular carcinoma. Transplant Int. 23:723–727. 2010.
View Article : Google Scholar
|
|
6
|
Abdel-Wahab M, Sultan AM, Fathy OM, et al:
Factors affecting recurrence and survival after living donor liver
transplantation for hepatocellular carcinoma.
Hepatogastroenterology. 60:1847–1853. 2013.PubMed/NCBI
|
|
7
|
Sandhu DS, Baichoo E and Roberts LR:
Fibroblast growth factor signaling in liver carcinogenesis.
Hepatology. 59:1166–1173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guégan JP, Ezan F, Théret N, Langouët S
and Baffet G: MAPK signaling in cisplatin-induced death:
predominant role of ERK1 over ERK2 in human hepatocellular
carcinoma cells. Carcinogenesis. 34:38–47. 2013.PubMed/NCBI
|
|
9
|
Fuchs BC, Hoshida Y, Fujii T, et al:
Epidermal growth factor receptor inhibition attenuates liver
fibrosis and development of hepatocellular carcinoma. Hepatology.
59:1577–1590. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao W, Kim H, Feng M, et al: Inactivation
of Wnt signaling by a human antibody that recognizes the heparan
sulfate chains of glypican-3 for liver cancer therapy. Hepatology.
60:576–587. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeong DG, Wei CH, Ku B, et al: The
family-wide structure and function of human dual-specificity
protein phosphatases. Acta Crystallogr D Biol Crystallogr.
70:421–435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu GS: Role of mitogen-activated protein
kinase phosphatases (MKPs) in cancer. Cancer Metastasis Rev.
26:579–585. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Keyse SM: Dual-specificity MAP kinase
phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 27:253–261.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Haagenson KK and Wu GS: Mitogen activated
protein kinase phosphatases and cancer. Cancer Biol Ther.
9:337–340. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haagenson KK and Wu GS: The role of MAP
kinases and MAP kinase phosphatase-1 in resistance to breast cancer
treatment. Cancer Metastasis Rev. 29:143–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sekine Y, Tsuji S, Ikeda O, et al:
Regulation of STAT3-mediated signaling by LMW-DSP2. Oncogene.
25:5801–5806. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sekine Y, Ikeda O, Hayakawa Y, et al:
DUSP22/LMW-DSP2 regulates estrogen receptor-alpha-mediated
signaling through dephosphorylation of Ser-118. Oncogene.
26:6038–6049. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li JP, Fu YN, Chen YR and Tan TH: JNK
pathway-associated phosphatase dephosphorylates focal adhesion
kinase and suppresses cell migration. J Biol Chem. 285:5472–5478.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rahmouni S, Cerignoli F, Alonso A, et al:
Loss of the VHR dual-specific phosphatase causes cell-cycle arrest
and senescence. Nat Cell Biol. 8:524–531. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shang X, Vasudevan SA, Yu Y, et al:
Dual-specificity phosphatase 26 is a novel p53 phosphatase and
inhibits p53 tumor suppressor functions in human neuroblastoma.
Oncogene. 29:4938–4946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klinger S, Poussin C, Debril MB, Dolci W,
Halban PA and Thorens B: Increasing GLP-1-induced beta-cell
proliferation by silencing the negative regulators of signaling
cAMP response element modulator-alpha and DUSP14. Diabetes.
57:584–593. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Patterson KI, Brummer T, O’Brien PM and
Daly RJ: Dual-specificity phosphatases: critical regulators with
diverse cellular targets. Biochem J. 418:475–489. 2009.PubMed/NCBI
|
|
25
|
Prabhakar S, Asuthkar S, Lee W, et al:
Targeting DUSPs in glioblastomas - wielding a double-edged sword?
Cell Biol Int. 38:145–153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nunes-Xavier C, Romá-Mateo C, Ríos P, et
al: Dual-specificity MAP kinase phosphatases as targets of cancer
treatment. Anticancer Agents Med Chem. 11:109–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ríos P, Nunes-Xavier CE, Tabernero L, Köhn
M and Pulido R: Dual-specificity phosphatases as molecular targets
for inhibition in human disease. Antioxid Redox Signal.
20:2251–2273. 2014.PubMed/NCBI
|
|
28
|
Thornton TM and Rincon M: Non-classical
p38 map kinase functions: cell cycle checkpoints and survival. Int
J Biol Sci. 5:44–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ayub A, Ashfaq UA and Haque A: HBV induced
HCC: major risk factors from genetic to molecular level. Biomed Res
Int. 2013:8104612013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arnoldussen YJ, Lorenzo PI, Pretorius ME,
et al: The mitogen-activated protein kinase phosphatase vaccinia
H1-related protein inhibits apoptosis in prostate cancer cells and
is overexpressed in prostate cancer. Cancer Res. 68:9255–9264.
2008. View Article : Google Scholar
|
|
32
|
Henkens R, Delvenne P, Arafa M, et al:
Cervix carcinoma is associated with an up-regulation and nuclear
localization of the dual-specificity protein phosphatase VHR. BMC
cancer. 8:1472008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang JP, Tan CP, Li J, et al: VHZ is a
novel centrosomal phosphatase associated with cell growth and human
primary cancers. Mol Cancer. 9:1282010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Deng Q, Li KY, Chen H, et al: RNA
interference against cancer/testis genes identifies dual
specificity phosphatase 21 as a potential therapeutic target in
human hepatocellular carcinoma. Hepatology. 59:518–530. 2014.
View Article : Google Scholar
|
|
35
|
Bai L, Yoon SO, King PD and Merchant JL:
ZBP-89-induced apoptosis is p53-independent and requires JNK. Cell
Death Differ. 11:663–673. 2004.PubMed/NCBI
|
|
36
|
Yu W, Imoto I, Inoue J, Onda M, Emi M and
Inazawa J: A novel amplification target, DUSP26, promotes
anaplastic thyroid cancer cell growth by inhibiting p38 MAPK
activity. Oncogene. 26:1178–1187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bertoli C, Skotheim JM and de Bruin RA:
Control of cell cycle transcription during G1 and S phases. Nat Rev
Mol Cell Biol. 14:518–528. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hui L, Bakiri L, Mairhorfer A, et al:
p38alpha suppresses normal and cancer cell proliferation by
antagonizing the JNK-c-Jun pathway. Nat Genet. 39:741–749. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hui L, Bakiri L, Stepniak E and Wagner EF:
p38alpha: a suppressor of cell proliferation and tumorigenesis.
Cell Cycle. 6:2429–2433. 2007. View Article : Google Scholar : PubMed/NCBI
|