Introduction
Angiogenesis is a physiological process that new
blood vessels grow from existing microvasculature in ischemic
tissues. Angiogenesis plays a critical role in tumor progression by
supplying nutrients, oxygen and removing metabolic waste for tumor
growth and metastasis (1).
Microvessel density (MVD), reflecting angiogenesis, has been proven
to be an important prognostic factor in a wide range of tumors
(2–4).
Endothelial hyperproliferation is a critical step in
the process of angiogenesis and is triggered by a diverse of
biochemical and microenvironment factors. Hypoxia occurs as a
result of an imbalance between oxygen supply and consumption due to
exponential cellular proliferation in tumor. In order to adapt to a
hostile environment, hypoxia changes the phenotype of endothelial
cells by activating a series of genes, including vascular
endothelial growth factor (VEGF), platelet-derived growth factor-B
(PDGF-B) and transforming growth factor (TGF) (5). Considering that the activation of
vascular endothelial cell proliferation and resistance to apoptosis
are vital characteristics of angiogenesis, induction of cell
apoptosis and cell cycle arrest may be a promising treatment
strategy for cancer.
Statins, also known as
3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase inhibitors, are
a class of compounds used to lower cholesterol and its isoprenoid
intermediates, accompanied by elevation of HDL-cholesterol. Both
HDL particles and lower cholesterol level are protective factors
for blood vessels. A finding shows that cholesterol efflux from
endothelial cells to high-density lipoprotein (HDL) regulates
angiogenesis, which implies the key role of cholesterol metabolism
in proper angiogenesis (6).
Nevertheless, the non-cholesterol dependent effect of statins on
endothelial function, vascular inflammation, cell proliferation,
immuno-modulation (7) and
oxidation (8) is of even greater
concern.
A substantial body of data suggests that statins are
able to prevent angiogenesis by modulating endothelial cells.
Fluvastatin has apoptosis-inducing effect in endothelial cells by
the activation of caspase-3 and the presence of extensive DNA
fragmentation (9). Pravastatin
inhibits endothelial proliferation through inducing G1
arrest and decreasing cyclin D1, cyclin E and cyclin-dependent
kinase 2 expression (10). Statins
exert a biphasic dose-dependent effect on angiogenesis by
stimulating angiogenesis at low concentrations and inducing
endothelial apoptosis with high-dose treatment (11).
Vascular endothelial cell is a key component in
angiogenesis and is also an attractive target of statins. Notably,
so far the endothelial effects of statins in hypoxia have not been
clearly elucidated. Therefore, we conducted the present study to
test the effects of fluvastatin on cell growth in hypoxic human
umbilical vein endothelial cells (HUVEC) and identify the
underlying mechanisms, providing evidence for future development in
cancer treatment.
Materials and methods
Reagents
Fluvastatin was purchased from Sigma-Aldrich Inc.
(St. Louis, MO, USA). Stock solution of 10 mg/ml was prepared by
dissolving fluvastatin powder in dimethyl sulfoxide (DMSO)
(Invitrogen, Carlsbad, CA, USA). The previous stock solution (2 μl)
was diluted with 98 μl of phosphate-buffer solution (PBS) to give a
fresh secondary solution (0.2 mg/ml) each time it was required.
Working solutions of different concentrations were obtained
directly in the culture media and DMSO controls were used in the
study at a range of concentrations between 1.355×10−6
and 6.938×10−4 (vol/vol) to overcome the possible
interference of DMSO.
Cells and cell culture
HUVEC were obtained from the American Type Culture
Collection (ATCC, Manassas, VA, USA) and cultured in F-12K medium
(Gibco, Grand Island, NY, USA) supplemented with 10% (vol/vol)
heat-inactivated fetal bovine serum (Gibco) and 0.05 mg/ml
endothelial cell growth supplement (ECGS). Cell cultures were
maintained at 37°C in a humidified atmosphere incubator (Hera cell
150; Heraeus, Langenese, Germany) of 5% CO2 in air.
Induction of hypoxic condition was performed in a modular incubator
(Galaxy R, RS Bitotech, Alloa, UK) flushed with 1% O2,
5% CO2 and 94% N2.
Cell viability assay
HUVEC were seeded in 96-well plates at a density of
2×103/well. Cells were exposed to normoxia or hypoxia
and treated with fluvastatin or DMSO at different concentrations of
0.125, 0.25, 0.5, 1, 2, 4, 8 and 16 μM for 48 h. Cell Counting
Kit-8 (CCK-8) (Dojindo Laboratories, Tokyo, Japan) was used to
measure cell viability. Cells were incubated with 10 μl CCK-8
solution at 37°C for 4 h following the manufacturer’s instructions.
Optical density (OD) values at 450 nm were obtained using an ELx800
Universal Microplate Reader (BioTek Instruments, Inc., Winooski,
VT, USA).
Cell apoptosis assay
Cell apoptosis was quantified using the Annexin
V/propidium iodide (PI) detection kit (Beyotime, Shanghai, China)
and analyzed by flow cytometry. Cells (2×105/well) were
plated in 6-well dishes and incubated with 0.03125, 0.0625, 0.125,
0.25, 0.5, 1, 2, 4, 8 and 16 μM fluvastatin or DMSO exposed to
hypoxia. After 48-h treatment, collected cells were incubated in
400 μl binding buffer with 5 μl Annexin V-FITC and 5 μl PI in dark
for 15 min at room temperature (RT). The cells were analyzed by
flow cytometry (BD Biosciences, San Jose, CA, USA) to identify the
early apoptotic (Annexin V positive and PI negative) and late
apoptotic (Annexin V positive and PI positive) cells.
Cell cycle assay
HUVEC were harvested and fixed in 70% cold ethanol
at 4°C overnight. Cells were washed with cold PBS and resuspended
in 400 μl staining buffer with 25 μl PI solution and 10 μl RNase A
(Beyotime) at 37°C in a water bath. Flow cytometry data were
analyzed with BD FACSDiva 6.1.
Human apoptosis antibody array
The expression of apoptosis-related proteins was
detected using a human apoptosis antibody array kit (RayBiotech
Inc., Norcross, GA, USA) according to the protocols. This glass
chip contains duplicate spots of 43 apoptosis-related proteins.
Briefly, the slides were incubated with blocking buffer at RT for
30 min. Lysates of HUVEC treated with fluvastatin or DMSO were then
added and kept at 4°C overnight. After thorough rinsing, the chip
was incubated with biotin-conjugated antibodies at RT for 2 h
followed by addition of streptavidin. The chip was scanned using
the Axon GenePix laser scanner (Molecular Devices Corp., Union
City, CA, USA) and the images were quantified by GenePix Pro 6.0
software.
Real-time PCR
Total RNA was extracted using TRIzol (Takara,
Dalian, China). The semi-quantitative assessment of cellular mRNA
expression were performed using reverse transcription polymerase
chain reaction (RT-PCR) kit (Takara) and SYBR Green real-time
quantitative PCR (qPCR) kit (Takara). The signals were collected
and analyzed by ABI Prism Fast 7500 system (Applied Biosystems,
Foster City, CA, USA). The amplification conditions were: preheat
at 95°C for 30 sec, 40 cycles of 95°C for 5 sec, 60°C for 34 sec
followed by 95°C for 15 sec and 60°C for 1 min. Primers were:
β-actin F-5′-AGCGAGCATCCCCCAAAGTT-3′ and
R-5′-GGGCACGAAGGCTCATCATT-3′; IGFBP-6 F-5′-CATGCCGTAGACATCTGGAC-3′
and R-5′-GGTAGAAGCCTCGATGGTCA-3′; BAX F-5′-AAGCTGAGCGAGTGTCTCAAG-3′
and R-5′-CAAAGTAGAAAAGGGCGACAAC-3′; BCL-2
F-5′-ATGTGTGTGGAGAGCGTCAAC-3′ and R-5′-AGAGACAGCCAGGAGAAATCAAAC-3′;
p27 F-5′-CGGGACTTGGAGAAGCACT-3′ and R-5′-AGTAGAACTCGGGCAAGCTG-3′;
p53 F-5′-CCCCTCCTGGCCCCTGTCATCTTC-3′ and
R-5′-GCAGCGCCTCACAACCTCCGTCAT-3′; CCNB
F-5′-CTGGATAATGGTGAATGGACAC-3′ and R-5′-CGATGTGGCATACTTGTTCTTG-3′;
CCND F-5′-CGGAGGAGAACAAACAGATCAT-3′ and
R-5′-AGGCGGTAGTAGGACAGGAAGT-3′; BIRC F-5′-CACCGCATCTCTACATTCAAGA-3′
and R-5′-CAAGTCTGGCTCGTTCTCAGT-3′; VEGF
F-5′-TTCTGAGTTGCCCAGGAGAC-3′ and R-5′-TGGTTTCAATGGTGTGAGGA-3.
Western blot analysis
Cell lysates of HUVEC treated with fluvastatin or
DMSO under hypoxia were analyzed by western blotting using
antibodies to glyceraldehyde-3-phosphate dehydrogenase (GAPDH),
bax, bcl-2, caspase-9, caspase-3, cytochrome c, p53, cyclin
B1, cyclin D1, survivin and VEGF. The antibodies listed above were
purchased from Proteintech Group (1:1,000 dilution for western
blots). In addition, the antibodies to voltage-dependent
anion-selective channel (VDAC), poly(ADP-ribose) polymerase (PARP),
insulin-like growth factor binding protein-6 (IGFBP-6), p27 were
obtained from Abcam Inc. (1:1,000 dilution for western blots).
Mitochondrial proteins were extracted using cell mitochondria
isolation kit (Beyotime). Blots were reprobed with GAPDH and VDAC
to confirm equal loading of cell lysate and mitochondrial proteins.
The intensity of protein bands was quantified by ImageJ and
normalized to GAPDH and VDAC in the analyses.
Statistical analysis
Statistical analysis was performed in GraphPad Prism
5.0 software (GraphPad Software Inc., San Diego, CA, USA).
Comparison between two groups was analyzed by the Student’s t-test
and multi-group analysis of variances was carried out by two-way
ANOVA with Bonferroni post test. Apoptotic cells following
fluvastatin treatment increased with different concentrations.
IC50 was analyzed using a non-linear regression of log
(inhibitor) vs. response (variable slope). All experiments were
repeated at least four times and the data are expressed as the mean
± SEM. P<0.05 is considered to be statistically significant
(*P<0.05, **P<0.01 or
***P<0.001, respectively, in the figures).
Results
Fluvastatin inhibits hypoxia-induced
HUVEC proliferation and accelerates apoptosis
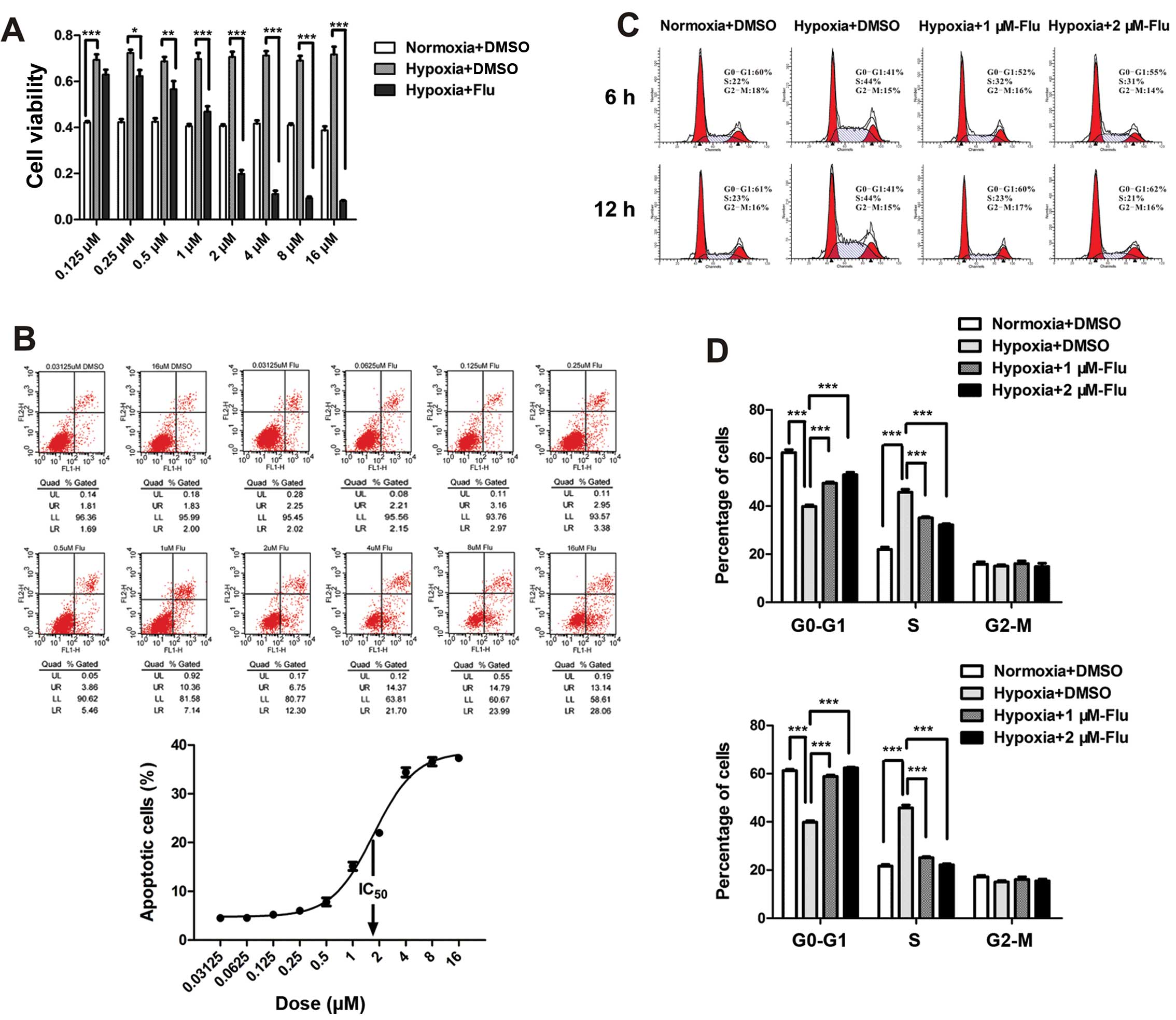
The effects of fluvastatin on cell viability,
apoptosis and cell cycle were evaluated by the CCK-8 test, Annexin
V/PI detection and PI staining. Hypoxia significantly increased
cell viability of HUVEC and fluvastatin completely reverse the
enhancement compared with DMSO controls. The inhibition rate rose
with the increasing concentrations ranging from 0.125 to 16 μM
(Fig. 1A).
To investigate the apoptosis-inducing effect and the
half maximal inhibitory concentration (IC50) of statin,
HUVEC were incubated with fluvastatin at 10 concentrations ranging
from 0.03125 to 16 μM under hypoxia for 48 h. Apoptosis rate was
assessed by the sum percentage of early and late apoptotic cells.
The data showed that the maximum apoptotic effect was ~40% and
IC50 value was between 1 and 2 μM (Fig. 1B). Therefore, 1 and 2 μM were
chosen as concentrations of fluvastatin in the following
experiments. It is worth noting that necrotic cells (Annexin V
negative and PI positive) were not observed in the experiment
(Fig. 1B).
The effect of fluvastatin on cell cycle of HUVEC was
expressed by the percentage of cells in
G0-G1, S, G2-M phase. A
significant increase in S-phase cells was observed following
hypoxia. Fluvastatin reversed the transition of G1/S
compared with DMSO controls in a time-dependent manner (Fig. 1C and D). The number of
G2-M phase cells was not affected after 12 h of
fluvastatin treatment.
The results suggest that fluvastatin mitigates
hypoxia-induced abnormal proliferation of HUVEC through inducing
cell apoptosis and preventing cell cycle progression.
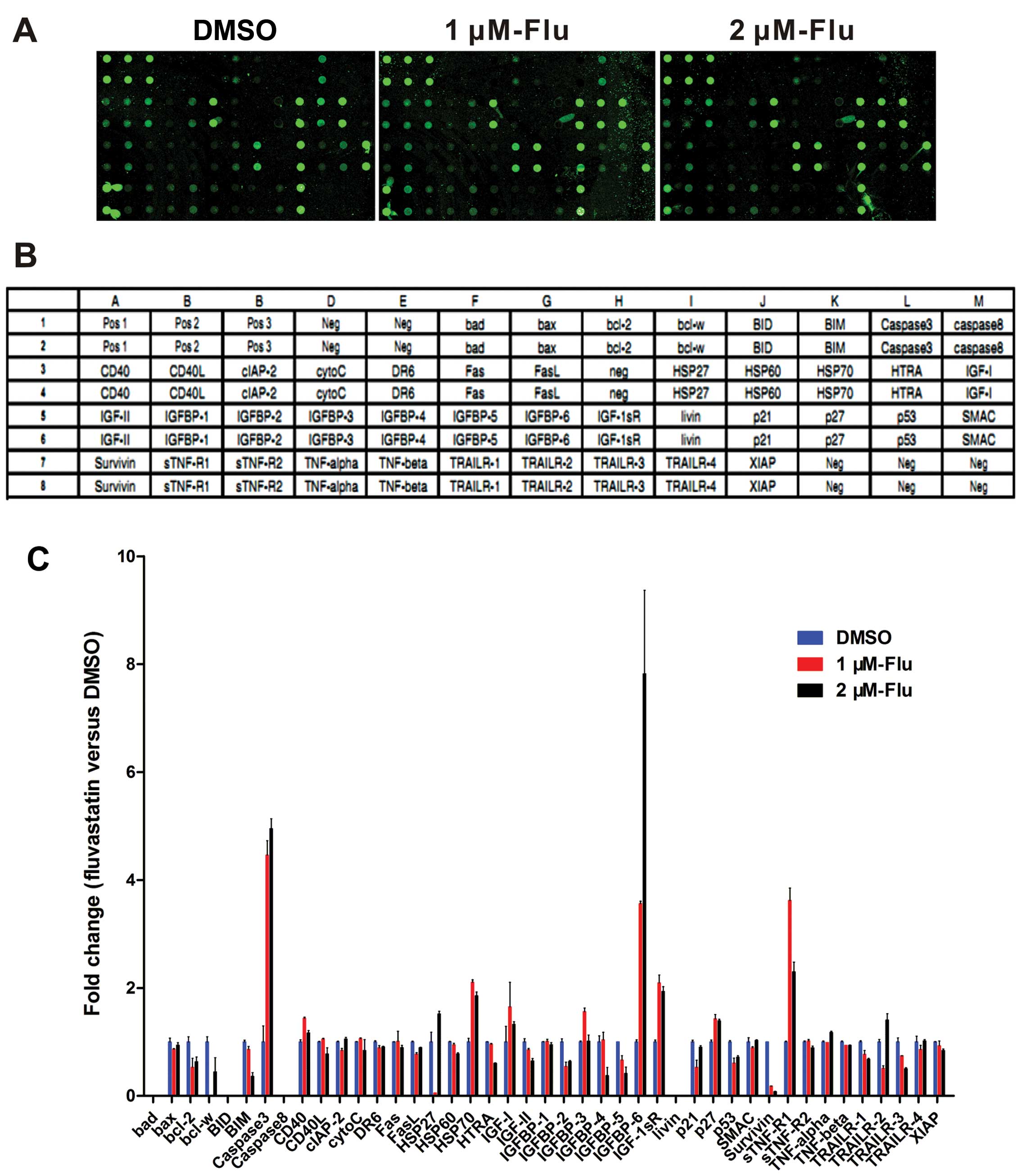
Analysis of human apoptosis antibody
array
To make clear the molecular profile of decreased
cell growth, human apoptosis antibody array was exploited to
analyze the apoptosis-related proteins expression in HUVEC with
fluvastatin treatment at 1 and 2 μM exposed to hypoxia for 48 h
(Fig. 2A and B). The data showed
that there is an obvious increase of caspase-3, heat shock protein
(HSP) 70, IGFBP-6, IGF-1sR, p27 and soluble tumor necrosis factor
receptor-1 (sTNF-R1) levels while bcl-2, bim, p53 and survivin were
downregulated (Fig. 2C).
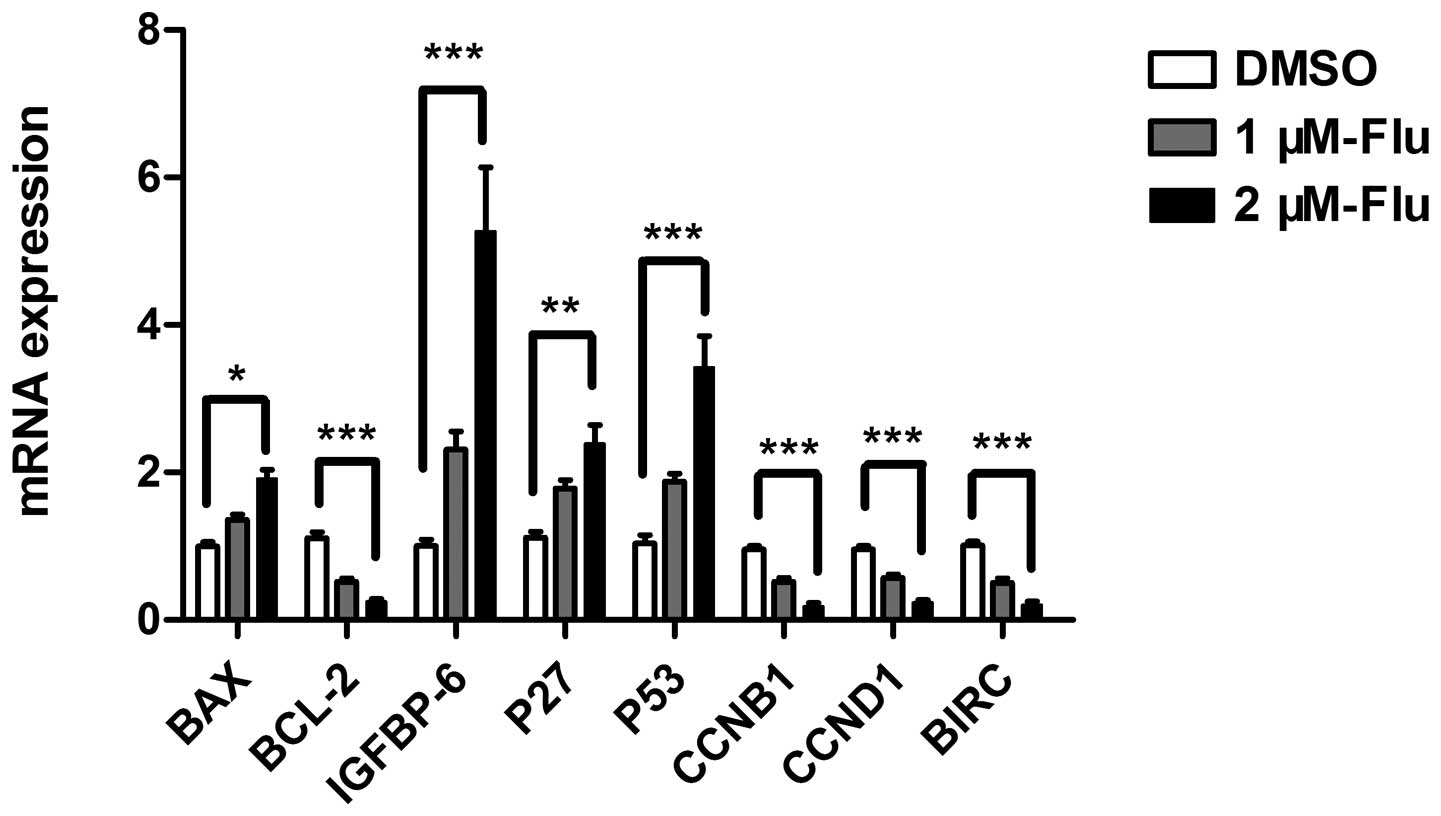
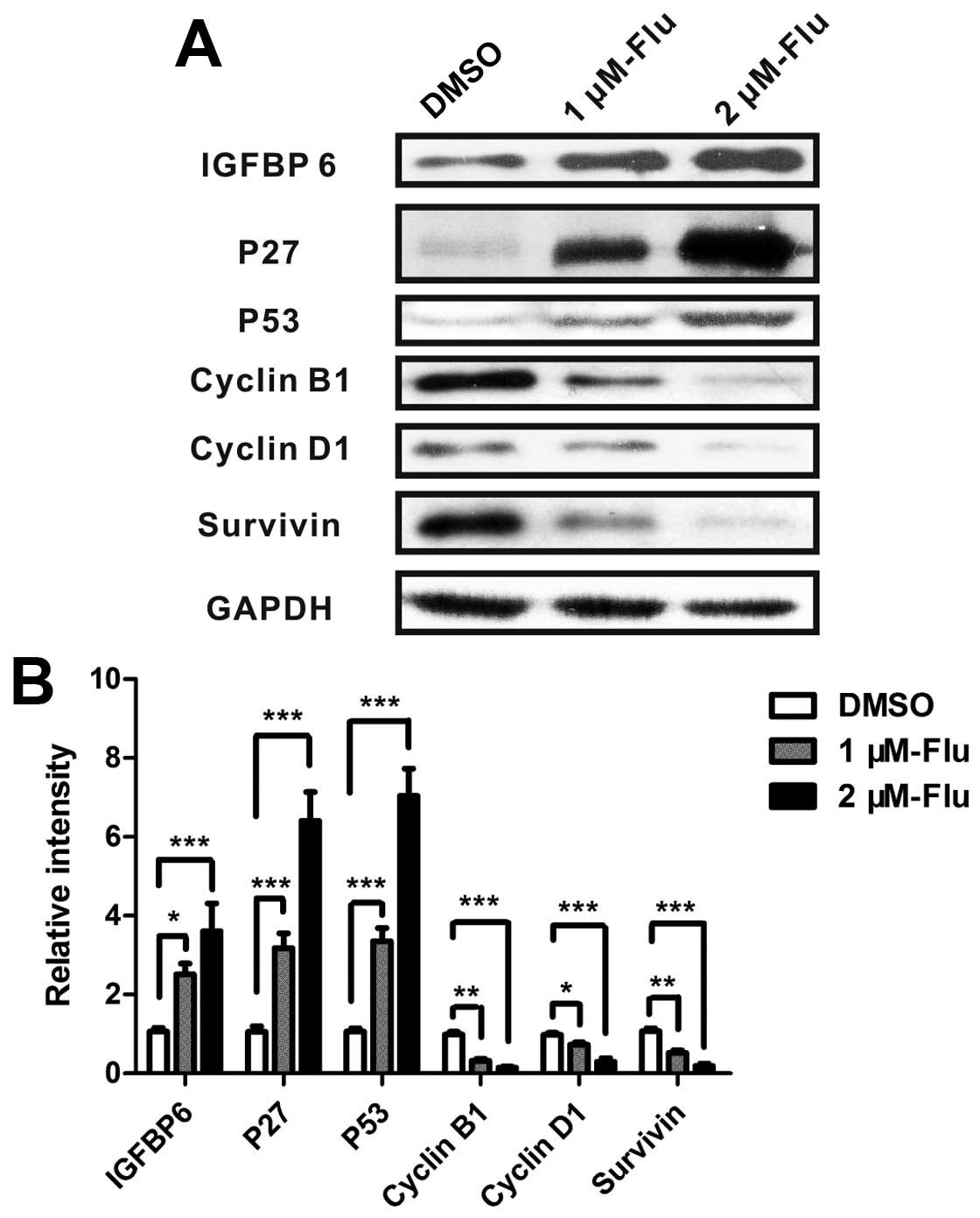
Gene expression of apoptotic mediators
and proliferative proteins
The regulation of cell cycle is a key element
controlling cell proliferation. Cell cycle checkpoints are
responsible for ensuring cell division in a steady and correct way.
Cyclin B1 and cyclin D1 are, respectively, required for
G2/M and G1/S transition, both of which were
explored in the present study. According to the results of antibody
array and cell cycle analysis, we performed real-time PCR to detect
mRNA expression of bax, bcl-2, bim, HSP 70, IGFBP-6, IGF-1sR, p27,
p53, sTNF-R1, cyclin B1, cyclin D1 and survivin. The results
confirmed gene expression changes of bax, bcl-2, IGFBP-6, p27,
cyclin B1, cyclin D1, survivin and showed a significant
upregulation of p53 (Fig. 3).
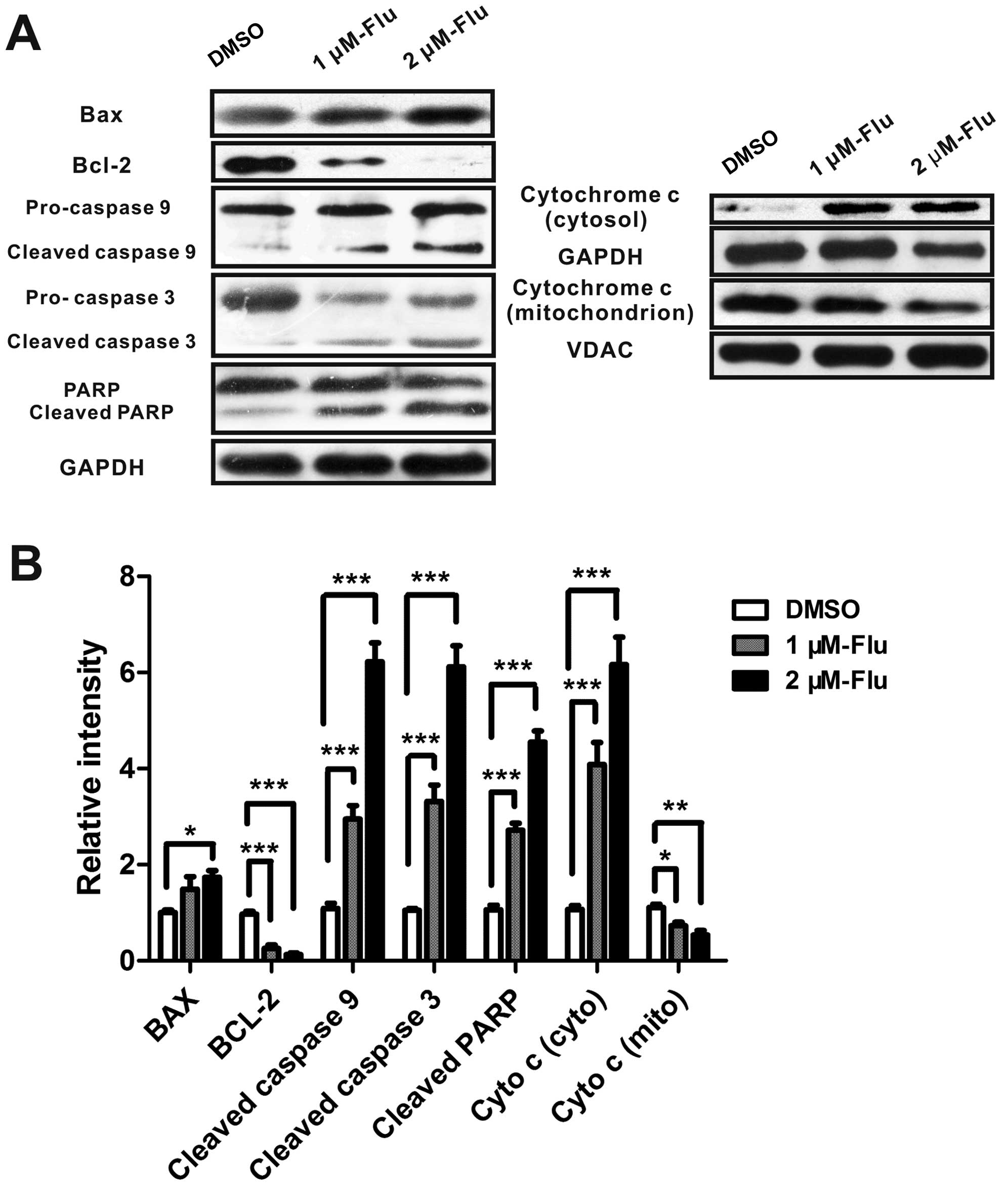
Fluvastatin activates mitochondrial
pathway of apoptosis
Mitochondrial pathway is initiated by the altered
expression of pro-apoptotic molecules and anti-apoptotic proteins.
The increase of bax/bcl-2 ratio promotes the release of
mitochondrial cytochrome c which binds to apoptotic protease
activating factor-1 (Apaf-1), ATP and pro-caspase-9 to form a
complex, called an apoptosome. The apoptosome cleaves caspase-9,
and caspase-3 then activating PARP, leading to cell death (12). Because bax, bcl-2 and caspase-3
were mediated by fluvastatin in protein array and real-time PCR
analyses, we next examined whether fluvastatin treatment modulated
bax, bcl-2, cytochrome c, caspase-9, caspase-3, and PARP
expression in HUVEC using western blotting. As expected, the
protein level of bax was augmented and bcl-2 was significantly
inhibited, implying the activation of mitochondrial apoptosis.
Cytochrome c was released from mitochondria and cleaved
caspase-3, caspase-9 and PARP were induced, indicating that
fluvastatin induced cell apoptosis mainly by the mitochondrial
pathway (Fig. 4).
Regulation of proteins involved in the
inhibition of cell proliferation
Analyses of real-time PCR suggested that
proliferation-related proteins IGFBP-6, cyclin B1, cyclin D1, p27,
p53 and survivin were involved in the regulation of cell growth
following fluvastatin treatment. Therefore, the levels of proteins
were measured by western blotting and the results showed that
fluvastatin significantly enhanced IGFBP-6, p27 and p53 and reduced
cyclin B1, cyclin D1 and survivin (Fig. 5).
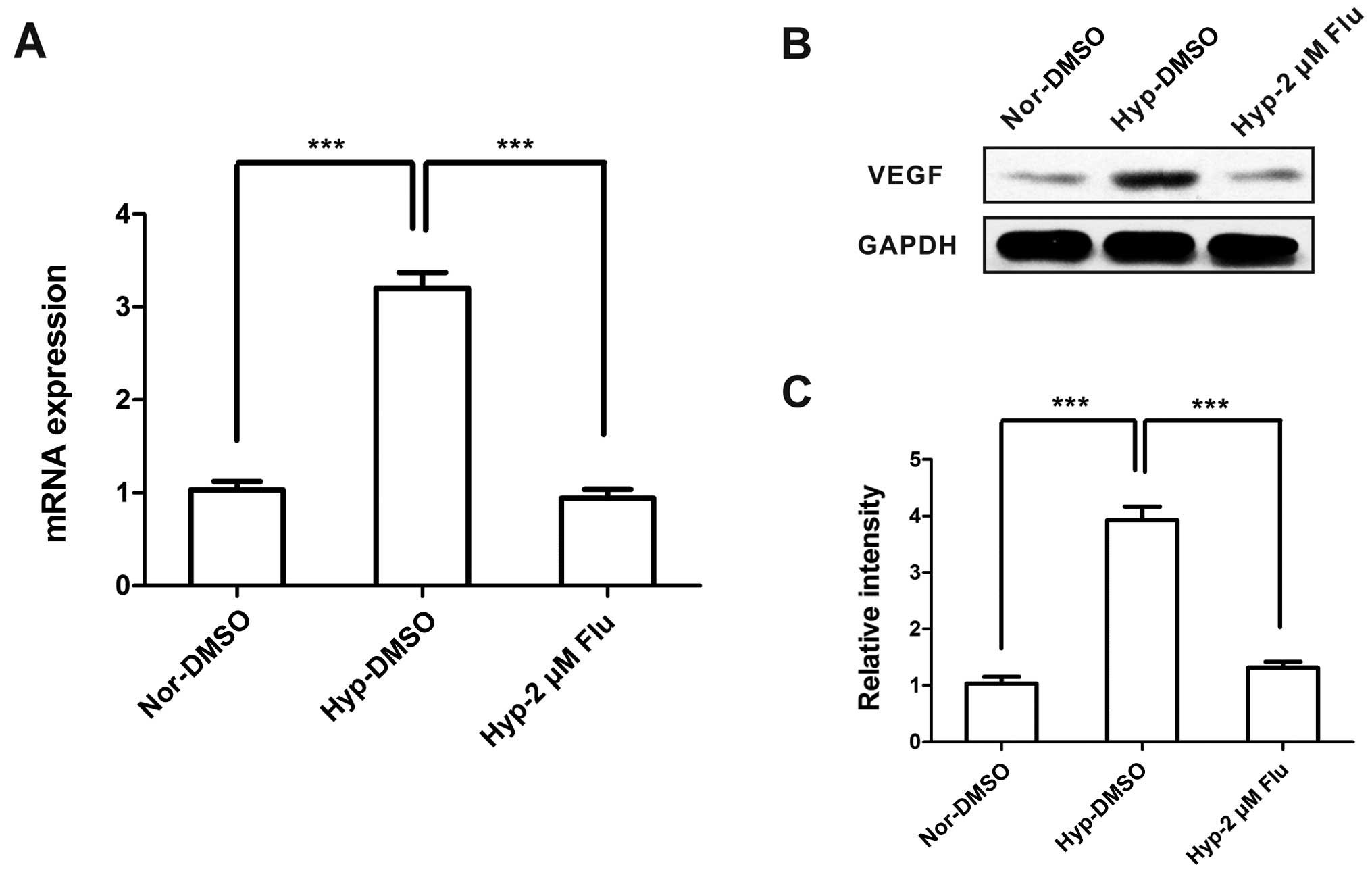
Inhibition of VEGF caused by fluvastatin
under hypoxia
VEGF is considered as a highly specific mitogen for
endothelial cells and a common biological biomarker of angiogenesis
(13). To investigate the role of
statins on VEGF expression under hypoxia, we examined VEGF under
conditions of normoxia and hypoxia with or without fluvastatin.
Hypoxia increased the mRNA and protein expression of VEGF, however,
VEGF level was dramatically decreased to normoxic level after
fluvastatin treatment (Fig.
6).
Discussion
Endothelial cells exhibit an immense proliferative
activity response to hypoxia. Hypoxic cells can maintain energy
generation and biomolecule production to survive and even
proliferate through adjusting the way of glucose (14) and glutamine metabolisms (15) in the adaptation process. Moreover,
a number of mitogenic factors and pathways are activated to sustain
the proliferation phenotype in endothelial cells, including VEGF,
endothelin-1, PDGFB, IGFBP3, placental growth factor (PGF)
(16) and mammalian target of
rapamycin (mTOR) signaling (17).
In the present study, hypoxia exerted a prominent proliferative
effect on HUVEC by regulating G1/S transition.
Statins, as the first-line agents in
hypercholesteremia therapy, have benefits in hypertensive patients
and are able to lower the risk of developing cardiovascular events
(18). Since hypertension is a
common complication of traditional anti-angiogenic therapy in
patients with cancer (19), the
supply of statins is an attractive strategy in cancer treatment.
Plenty of preclinical studies have revealed that the combination of
statins and other chemotherapeutics enhance the effectiveness of
cancer treatment (20). Long-term
use of statins for more than one year is proven to be associated
with a decreased risk of endometrial cancer (21) and a 36% reduction of colorectal
cancer-specific mortality (22).
Our research demonstrated that statins inhibited
cell growth by inducing cell apoptosis and regulating cell cycle
under hypoxia in a dose- and time-dependent manner. Previously it
was reported that statins block hypoxia-induced endothelial
proliferation through decreasing Ca2+ and preventing ROS
generation (23). However, the
underlying molecular mechanisms are poorly understood.
Statins block the synthesis of mevalonate by
competitively inhibiting HMG-CoA reductase. Mevalonate is a
precursor of coenzyme Q10, a component which plays a key role in
mitochondrial respiration and facilitates mitochondrial bioenergy
transfer (24). Inhibition of the
mevalonate pathway engaged by statin is shown to disrupt
mitochondrial surveillance (25).
The results of antibody array revealed the involvement of bcl-2 and
caspase-3, implying that mitochondrial pathway may be implicated in
the regulation of cell growth by fluvastatin. Therefore, we
performed qPCR and western blot experiments to test the hypothesis.
As expected, data showed that fluvastatin significantly increased
bax/bcl-2 ratio and triggered release of cytochrome c from
mitochondria and then induced the cleavage of caspase-3, caspase-9
and eventually PARP.
Cytochrome c is able to transfer electrons
between cytochrome complexes III and IV, in order to maintain
proper energy conversion. Mitochondrial dysfunction caused by
mitochondrial complex III inhibitor significantly suppresses the
hypoxia-induced VEGF expression in human proximal tubular
epithelial cells (hPTECs) (26).
Based on the central role of VEGF in hypoxic cell proliferation, we
measured the expression of VEGF in HUVEC with fluvastatin or DMSO
treatment. Consistent with the study of Loboda et al
(27) in human microvascular
endothelial cells (HMEC-1), our results suggested that VEGF was
augmented under hypoxia and attenuated to normoxic level following
fluvastatin treatment. However, the underlying association of VEGF
and mitochondrial apoptosis warrants further investigation.
Fluvastatin was revealed to prevent endothelial
cells from leaving G1 (9) and cause G2/M arrest in
leukemic natural killer cells (28). In the present study, fluvastatin
induced the accumulation of G0-G1 phase cells
and correspondingly reduced the proportion of cells in S phase
time-dependently, slowing down the progression of cell mitosis.
The altered expression was confirmed in hypoxic
HUVEC treated with fluvastatin or DMSO, including cell cycle
regulators IGFBP-6, cyclin B1, cyclin D1, p27, p53, as well as
proliferation-related protein survivin. IGFBP-6 was identified as
oncosuppressor in nasopharyngeal carcinoma (29) and inhibits cell proliferation in
connective tissue disease (30).
Cells overexpressing IGFBP-6 are blocked at the G1/S
transition compared with scramble cells (31). Cyclin B1 and cyclin D1 are critical
cell cycle regulatory proteins. Cyclin B1 mRNA and protein levels
were strongly inhibited following fluvastatin treatment while
G2-M phase cells were not accumulated. Cell cycle
checkpoints arrest in response to DNA damage contributes to allow
time for DNA repair and defects in the G2-M arrest are
associated with damaged cell mitosis and final cell death (32), which may help explain the above
described results. P27 and p53 are referred as tumor suppressors
because of their major function on cell cycle inhibition (33,34).
P27 negatively regulates G1 progression and strongly
interacts with cyclin D1. P53 controls both the G1 and
G2 checkpoints and also mediates cell apoptosis in
response to DNA damage through transcriptionally activating bax and
repressing bcl-2 (35). Survivin
is highly expressed in the majority of carcinomas and has an
important bearing on cell apoptosis and proliferation (36). DNA damage is defined to be related
with p53-survivin signaling pathway to induce cell apoptosis and
cell cycle arrest (37),
suggesting that a potential cross talk between different signaling
pathways existed in the effect of statins on HUVEC.
The above study is a protein array-based screen for
specific proteins involved in the process. However, the expression
for some proteins in the apoptosis array analysis is not consistent
with the results from qPCR and western blot experiments, such as
bax and p53. The discrepancy may be attributed to a tendency to
generate false negative signals in a high-throughput method
(38).
This is the first report comprehensively describing
the mechanism underlying the effect of statins on vascular
endothelial cell survival under hypoxia. Our data revealed that
fluvastatin induced apoptosis through mitochondrial pathway and
inhibited cell proliferation by increasing IGFBP-6, p27, p53 and
decreasing cyclin B1, cyclin D1, survivin, VEGF levels. Statin
treatment may have potential for effective cancer therapy.
Acknowledgements
The present study was supported by grants from the
Natural Science Foundation of China (81270106), the Ministry of
Science and Technology of China (2012BAI05B00) and the Ministry of
Public Health (201002008).
References
|
1
|
Nishida N, Yano H, Nishida T, Kamura T and
Kojiro M: Angiogenesis in cancer. Vasc Health Risk Manag.
2:213–219. 2006. View Article : Google Scholar
|
|
2
|
Uzzan B, Nicolas P, Cucherat M and Perret
GY: Microvessel density as a prognostic factor in women with breast
cancer: a systematic review of the literature and meta-analysis.
Cancer Res. 64:2941–2955. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bremnes RM, Camps C and Sirera R:
Angiogenesis in non-small cell lung cancer: the prognostic impact
of neoangiogenesis and the cytokines VEGF and bFGF in tumours and
blood. Lung Cancer. 51:143–158. 2006. View Article : Google Scholar
|
|
4
|
Zhao HC, Qin R, Chen XX, et al:
Microvessel density is a prognostic marker of human gastric cancer.
World J Gastroenterol. 12:7598–7603. 2006.PubMed/NCBI
|
|
5
|
Vaupel P: The role of hypoxia-induced
factors in tumor progression. Oncologist. 9(Suppl 5): S10–S17.
2004. View Article : Google Scholar
|
|
6
|
Fang L, Choi SH, Baek JS, et al: Control
of angiogenesis by AIBP-mediated cholesterol efflux. Nature.
498:118–122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang CY, Liu PY and Liao JK: Pleiotropic
effects of statin therapy: molecular mechanisms and clinical
results. Trends Mol Med. 14:37–44. 2008. View Article : Google Scholar
|
|
8
|
Grosser N, Hemmerle A, Berndt G, et al:
The antioxidant defense protein heme oxygenase 1 is a novel target
for statins in endothelial cells. Free Radic Biol Med.
37:2064–2071. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Newton CJ, Ran G, Xie YX, et al:
Statin-induced apoptosis of vascular endothelial cells is blocked
by dexamethasone. J Endocrinol. 174:7–16. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Asakage M, Tsuno NH, Kitayama J, et al:
3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor
(pravastatin) inhibits endothelial cell proliferation dependent on
G1 cell cycle arrest. Anticancer Drugs. 15:625–632. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weis M, Heeschen C, Glassford AJ and Cooke
JP: Statins have biphasic effects on angiogenesis. Circulation.
105:739–745. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hoeben A, Landuyt B, Highley MS, Wildiers
H, Van Oosterom AT and De Bruijn EA: Vascular endothelial growth
factor and angiogenesis. Pharmacol Rev. 56:549–580. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Polet F and Feron O: Endothelial cell
metabolism and tumour angiogenesis: glucose and glutamine as
essential fuels and lactate as the driving force. J Intern Med.
273:156–165. 2013. View Article : Google Scholar
|
|
15
|
Wise DR, Ward PS, Shay JE, et al: Hypoxia
promotes isocitrate dehydrogenase-dependent carboxylation of
alpha-ketoglutarate to citrate to support cell growth and
viability. Proc Natl Acad Sci USA. 108:19611–19616. 2011.
View Article : Google Scholar
|
|
16
|
Manalo DJ, Rowan A, Lavoie T, et al:
Transcriptional regulation of vascular endothelial cell responses
to hypoxia by HIF-1. Blood. 105:659–669. 2005. View Article : Google Scholar
|
|
17
|
Humar R, Kiefer FN, Berns H, Resink TJ and
Battegay EJ: Hypoxia enhances vascular cell proliferation and
angiogenesis in vitro via rapamycin (mTOR)-dependent signaling.
FASEB J. 16:771–780. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Osende JI, Ruiz-Ortega M, Blanco-Colio LM
and Egido J: Statins to prevent cardiovascular events in
hypertensive patients. The ASCOT-LLA study. Nephrol Dial
Transplant. 19:528–531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pande A, Lombardo J, Spangenthal E and
Javle M: Hypertension secondary to anti-angiogenic therapy:
experience with bevacizumab. Anticancer Res. 27:3465–3470.
2007.PubMed/NCBI
|
|
20
|
Osmak M: Statins and cancer: current and
future prospects. Cancer Lett. 324:1–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lavie O, Pinchev M, Rennert HS, Segev Y
and Rennert G: The effect of statins on risk and survival of
gynecological malignancies. Gynecol Oncol. 130:615–619. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cardwell CR, Hicks BM, Hughes C and Murray
LJ: Statin use after colorectal cancer diagnosis and survival: a
population-based cohort study. J Clin Oncol. 32:3177–3183. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schaefer CA, Kuhlmann CR, Weiterer S, et
al: Statins inhibit hypoxia-induced endothelial proliferation by
preventing calcium-induced ROS formation. Atherosclerosis.
185:290–296. 2006. View Article : Google Scholar
|
|
24
|
Deichmann R, Lavie C and Andrews S:
Coenzyme q10 and statin-induced mitochondrial dysfunction. Ochsner
J. 10:16–21. 2010.
|
|
25
|
Liu Y, Samuel BS, Breen PC and Ruvkun G:
Caenorhabditis elegans pathways that surveil and defend
mitochondria. Nature. 508:406–410. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Satoh M, Fujimoto S, Horike H, et al:
Mitochondrial damage-induced impairment of angiogenesis in the
aging rat kidney. Lab Invest. 91:190–202. 2011. View Article : Google Scholar
|
|
27
|
Loboda A, Jazwa A, Jozkowicz A, Molema G
and Dulak J: Angiogenic transcriptome of human microvascular
endothelial cells: effect of hypoxia, modulation by atorvastatin.
Vascul Pharmacol. 44:206–214. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Crosbie J, Magnussen M, Dornbier R,
Iannone A and Steele TA: Statins inhibit proliferation and
cytotoxicity of a human leukemic natural killer cell line. Biomark
Res. 1:332013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kuo YS, Tang YB, Lu TY, Wu HC and Lin CT:
IGFBP-6 plays a role as an oncosuppressor gene in NPC pathogenesis
through regulating EGR-1 expression. J Pathol. 222:299–309. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Raykha C, Crawford J, Gan BS, Fu P, Bach
LA and O’Gorman DB: IGF-II and IGFBP-6 regulate cellular
contractility and proliferation in Dupuytren’s disease. Biochim
Biophys Acta. 1832:1511–1519. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grellier P, De Galle B and Babajko S:
Expression of insulin-like growth factor-binding protein 6
complementary DNA alters neuroblastoma cell growth. Cancer Res.
58:1670–1676. 1998.PubMed/NCBI
|
|
32
|
DiPaola RS: To arrest or not to
G2-M Cell-cycle arrest: commentary re: A.K. Tyagi, et
al: Silibinin strongly synergizes human prostate carcinoma DU145
cells to doxorubicin-induced growth inhibition, G2-M
arrest, and apoptosis. Clin Cancer Res 8: 3512–3519, 2002. Clin
Cancer Res. 8:3311–3314. 2002.PubMed/NCBI
|
|
33
|
Toyoshima H and Hunter T: p27, a novel
inhibitor of G1 cyclin-Cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yin Y, Tainsky MA, Bischoff FZ, Strong LC
and Wahl GM: Wild-type p53 restores cell cycle control and inhibits
gene amplification in cells with mutant p53 alleles. Cell.
70:937–948. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Basu A and Haldar S: The relationship
between BcI2, Bax and p53: consequences for cell cycle progression
and cell death. Mol Hum Reprod. 4:1099–1109. 1998. View Article : Google Scholar
|
|
36
|
Sarela AI, Verbeke CS, Ramsdale J, Davies
CL, Markham AF and Guillou PJ: Expression of survivin, a novel
inhibitor of apoptosis and cell cycle regulatory protein, in
pancreatic adenocarcinoma. Br J Cancer. 86:886–892. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou M, Gu L, Li F, Zhu Y, Woods WG and
Findley HW: DNA damage induces a novel p53-survivin signaling
pathway regulating cell cycle and apoptosis in acute lymphoblastic
leukemia cells. J Pharmacol Exp Ther. 303:124–131. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maslov S and Sneppen K: Protein
interaction networks beyond artifacts. FEBS Lett. 530:255–256.
2002. View Article : Google Scholar : PubMed/NCBI
|