Introduction
In the past decades peritoneal dialysis (PD) has
become an established alternative therapy to hemodialysis for the
treatment of end-stage renal disease. The number of patients who
receiving PD therapy has increased progressively worldwide,
particularly in Asian countries. However, peritoneal fibrosis is an
inevitable consequence of PD and is one of the most important
causes of ultrafiltration failure (1–3).
Previous studies have found that the epithelial-mesenchymal
transition (EMT) of human peritoneal mesothelial cells (HPMCs) is
an early event during PD and is associated with high peritoneal
transport. HPMC EMT is a key process leading to peritoneal fibrosis
and function deterioration (4–7).
During dialysis, the peritoneum is exposed to continuous
inflammatory stimuli, including hyperosmotic, hyperglycemic and
acidic dialysis solutions, as well as peritonitis and
hemoperitoneum. These factors may cause acute and chronic
inflammation of the peritoneum, and may progressively lead to
fibrosis, angiogenesis and hyalinizing vasculopathy (8–10).
The mechanism by which high glucose levels elicit EMT has been a
major focus of current research on fibrosis.
Integrin-linked kinase (ILK) is a protein discovered
in 1996 using a yeast two-hybrid screen in which the cytoplasmic
tail of β1 integrin was used as the bait (11). ILK consists of an N-terminal domain
that contains four ankyrin repeats, a central pleckstrin
homology-like domain and a C-terminal kinase domain (12). ILK is a protein that has an
important role in extracellular matrix (ECM)-mediated signaling. It
is a key molecule in the mediation of several biological functions,
including cell-matrix interactions, angiogenesis and invasion. It
is also involved in the EMT of podocytes and renal interstitial
fibrosis (13,14). Aberrant regulation of ILK is
implicated in the pathogenesis of various proteinuric kidney
diseases, including diabetic nephropathy, congenital nephritic
syndrome and experimental models of glomerular disease (15–17).
In addition, ILK has been shown to be an important intracellular
mediator that controls EMT in tubular epithelial cells (18). Furthermore, it has been proposed
that ILK regulates the phosphorylation of Akt at Ser 473 and the
phosphorylation of glycogen synthase kinase 3β (GSK-3β) in various
cell types (19,20), which suggests that ILK may have a
role in EMT.
In the present study, a preliminary experiment
revealed that there was a large and complex network of molecular
signals in EMT. Among these key molecules, ILK, a downstream factor
of transforming growth factor β1 (TGF-β1), participates in multiple
signaling pathways, including the integrin, TGF-β1/Sma- and
Mad-related protein (Smad), mitogen-activated protein kinases
(MAPK), connective tissue growth factor (CTGF), phosphoinositide
3-kinase (PI3K)/AKT and extracellular signal-regulated kinase 1/2
(ERK1/2) pathways (21–24). To date, to the best of our
knowledge, there have been no published data on ILK expression
patterns in HPMCs. Therefore, in this study, the role of ILK in the
regulation of HPMC EMT induced by high glucose conditions was
investigated.
Materials and methods
Cell culture
HPMCs were provided by Dr Pierre Ronco (Tenon
Hospital, Paris, France). The isolation, primary culture and
immortalization of HPMC cell line were performed as previously
described (25,26). The cell line was established
following infection of a fully characterized primary culture of
human peritoneal mesothelial cells with an amphotropic recombinant
retrovirus that encodes SV40 large-T Ag under the control of the
Moloney virus long terminal repeat and resistance to the antibiotic
G418. Briefly, cells were cultured in low-glucose Dulbecco’s
modified Eagle medium supplemented with 10% fetal bovine serum
(FBS; Gibco®-BRL, Carlsbad, CA, USA) and 100 U/ml
penicillin-streptomycin (Solarbio, Beijing, China) at 37°C in an
incubator with 5% CO2. The present study was approved by
the ethics committee of Central South University (Changsha, Hunan,
China).
Experimental design
Cells were seeded at 80–90% confluence in
FBS-containing medium. HPMCs were cultured for 24 h in medium
containing 5.6 mmol/l D-glucose and 10% FBS prior to being exposed
to the various experimental conditions. To study the effect of
different concentrations of glucose, the HPMCs were cultured in a
medium containing 0, 30, 60 and 90 mmol/l D-glucose for 24 h. To
study the effect of time, HPMCs were exposed to high glucose levels
(60 mmol/l) for 0, 12, 24 and 48 h. Cells were then used for
further analysis.
Immunofluorescence
Conditioned cells were grown on chamber slides. The
cells were then washed twice with phosphate-buffered saline (PBS)
and fixed with 2–4% paraformaldehyde for 15 min. Cells were
permeabilized with 0.3% Triton X-100 (Beijing Dingguochangsheng
Biotechnology Co., Ltd., Beijing, China) for 10 min and then
blocked with 5% bovine serum albumin (Proliant Biologicals Inc.,
Ankeny, IA, USA) for 1 h. The primary antibodies, including mouse
monoclonal anti-human vimentin antibody (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) and goat monoclonal anti-human
E-cadherin antibody (Santa Cruz Biotechnology, Inc.) were incubated
with the cells overnight at 4°C. The cells were then incubated with
secondary antibodies of fluorescein isothiocyanate-labeled rabbit
anti-goat immunoglobulin G (IgG; 1:200; ZSGB-BIO, Beijing, China)
and goat anti-mouse IgG (1:200; Zymed, San Francisco, CA, USA) for
1 h at room temperature. The cells were washed with PBS and
4′,6-diamidino-2-phenylindole (Santa Cruz Biotechnology, Inc.) was
used to stain the nuclei. Cells were then analyzed using a confocal
microscope (Zeiss LSM 510; Carl Zeiss, Oberkochen, Germany).
Quantitative polymerase chain reaction
(qPCR)
Total RNA was extracted using an RNAiso Plus reagent
(Takara Bio, Inc., Shiga, Japan), and cDNA was synthesized using a
RevertAid™ First Strand cDNA Synthesis kit (MBI Fermentas, Amherst,
NY, USA). qPCR was performed using an ABI PRISM® 7300
real-time PCR system (Applied Biosystems®, Carlsbad, CA,
USA). Primers were based on human sequences, and the amplification
efficiencies of all of the genes were similar. The primers used for
qPCR are shown in Table I.
 | Table IPrimer sequences used for qPCR. |
Table I
Primer sequences used for qPCR.
| Genes | Primer
sequences |
|---|
| E-cadherin |
5′-TCATGAGTGTCCCCCGGTAT-3′
5′-TCTTGAAGCGATTGCCCCAT-3′ |
| Vimentin |
5′-GCTACGTGACTACGTCCACC-3′
5′-TAGTTGGCGAAGCGGTCATT-3′ |
| FN |
5′-AACTGGTAACCCTTCCACACCC-3′
5′-AGCTTCTTGTCCTACATTCGGC-3′ |
| ILK |
5′-CAACACGGAGAACGACCTCA-3′
5′-GTGTCATCCCCACGGTTCAT-3′ |
| GSK-3β |
5′-GGAACTCCAACAAGGGAGCA-3′
5′-TTCGGGGTCGGAAGACCTTA-3′ |
| Cyclin D1 |
5′-GATGCCAACCTCCTCAACGA-3′
5′-GGAAGCGGTCCAGGTAGTTC-3′ |
| COL-1 |
5′-GCCAAGACGAAGACATCCCA-3′
5′-GGCAGTTCTTGGTCTCGTCA-3′ |
| GAPDH |
5′-CAATGACCCCTTCATTGACC-3′
5′-GACAAGCTTCCCGTTCTCAG-3′ |
Western blot analysis
Total protein was extracted from HPMCs and separated
using sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(Beijing Dingguochangsheng Biotechnology Co.). The polyvinylidene
difluoride membrane (Millipore, Billerica, MA, USA) was blocked
using 5% nonfat milk in Tris-buffered saline with 1% Tween 20
(Amresco Inc., Solon, Ohio, USA) for 2 h at room temperature. The
membrane was subsequently incubated with the primary antibodies
[mouse monoclonal anti-human vimentin, mouse monoclonal anti-human
fibronectin (FN), goat monoclonal anti-human E-cadherin, rabbit
polyclonal anti-human ILK, rabbit monoclonal anti-human p-GSK-3β,
mouse monoclonal anti-human cyclin D1 and rabbit polyclonal
anti-human collagen, type I (COL-1)] overnight at 4°C, followed by
incubation with horseradish peroxidase (HRP)-conjugated secondary
antibodies (rabbit anti-goat, goat anti-rabbit and goat anti-mouse
antibodies) for 1 h. The signals were detected using a Kodak
Digital Imaging System (4000MM; Kodak, Rochester, NY, USA).
Small interfering RNA (siRNA)
transfection
siRNA against ILK was purchased from Qiagen
(Valencia, CA, USA) whilst negative control siRNA was purchased
from Dharmacon (siGENOME® Non-Targeting siRNA #1; Thermo
Fisher Scientific, Waltham, MA, USA). At 60% confluence, cells were
washed with Opti-MEM® (Gibco®-BRL) and
transfected with targeted or control siRNA (at a final
concentration of 25 nM) in accordance with the manufacturer’s
instructions. Six hours post-transfection, Opti-MEM was replaced
with normal medium and cells were further incubated for 24 or 48 h.
The sequences of siRNA for ILK were as follows:
5′-CCUCUACAAUGUACUACAUTT dTdT and 3′-AUGUAGUACAUUGUAGAGGTT
dGdT.
Statistical analysis
All of the data are expressed as the mean ± standard
deviation and were analyzed by one-way analysis of variance using
SPSS 13.0 statistical software (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
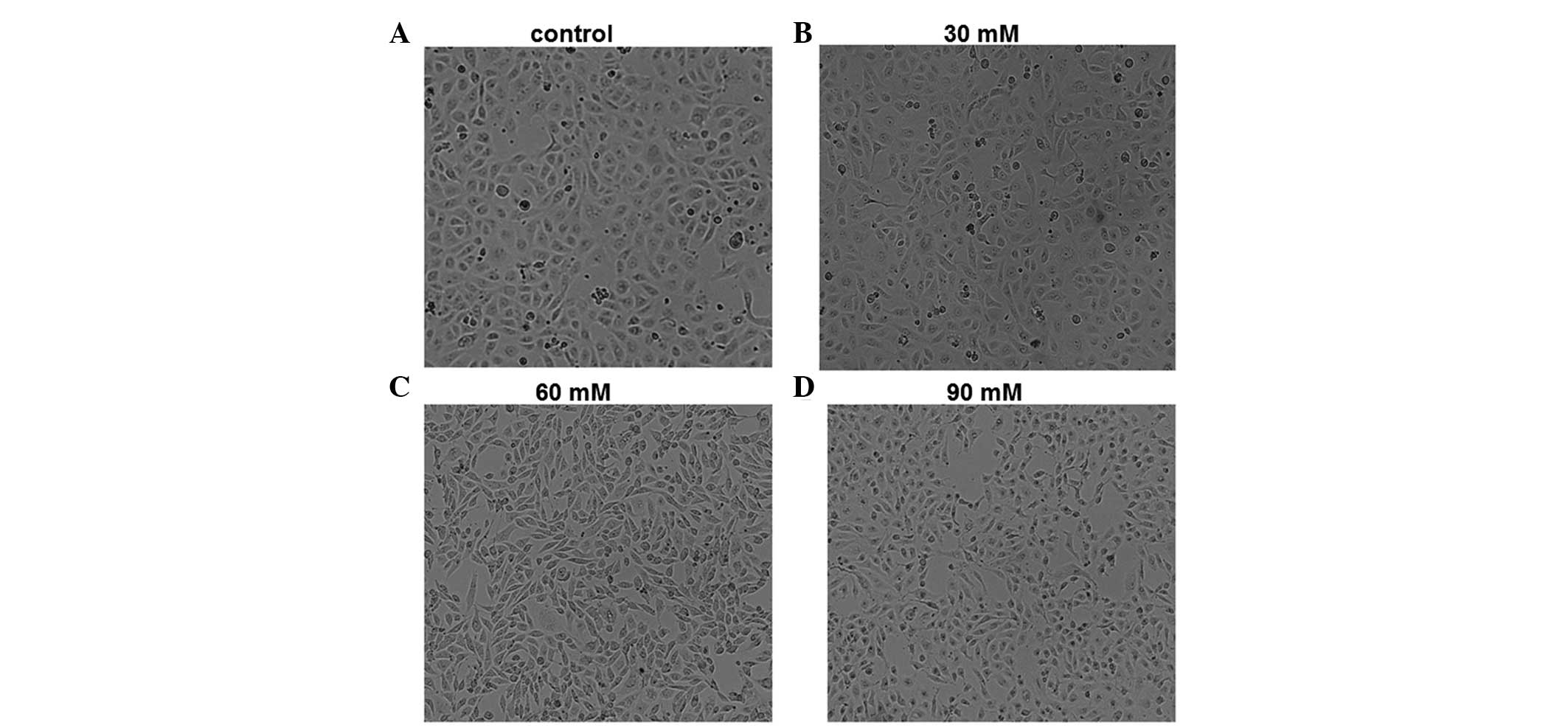
High glucose levels induce morphological
changes in HPMCs
To determine the effect of high-glucose incubation
on the morphology of HPMCs, cells were incubated with 30, 60 and 90
mM glucose for 24 h and observed using phase-contrast microscopy.
Representative images are shown in Fig. 1. In the control group not treated
with glucose, HPMCs were tightly packed with polygonal or oval
morphology and were arranged in a paving stone-like manner.
Following incubation with 30 mM glucose for 24 h, certain cells
demonstrated an elongated morphology. However, following incubation
with 60 mM glucose for 24 h, HPMCs exhibited a series of phenotypic
changes compared with the control cells, including elongation,
branching and loss of the paving stone appearance. The phenotypic
changes were similar but more evident in cells incubated with 90 mM
glucose compared with those incubated with 30 mM for 24 h.
Therefore, cell morphology of HPMCs was markedly affected by
glucose incubation, and these morphological changes were induced in
a dose-dependent manner.
High glucose levels induce EMT in
HPMCs
To investigate the effect of high-glucose incubation
on peritoneal EMT, an in vitro EMT study was performed in
HPMCs. In order to induce EMT in the HPMCs, cells were incubated
with 60 mM glucose for 0, 12, 24 and 48 h, respectively. The
markers for EMT were then analyzed using western blot analysis and
qPCR. The detected markers included FN, COL-1, E-cadherin and
vimentin. Representative results from the western blot analysis are
shown in Fig. 2A. Following
incubation for 24 h, the expression level of E-cadherin, an
epithelial phenotype marker, was downregulated and reached the
lowest level at 48 h. The expression level of vimentin, a
mesenchymal marker, was almost undetectable at 0 h of incubation;
however, expression was upregulated from as early as 12 h and
peaked at 48 h. FN expression also increased from 12 h of
incubation. COL-1 protein levels changed in a time-dependent
manner, increasing from 12 h of incubation and peaking at 48 h of
incubation. The representative qPCR results are shown in Fig. 2B. The qPCR results indicate that
E-cadherin mRNA expression levels decreased in a time-dependent
manner, and significant differences were observed at 24 h
(P<0.05). FN mRNA expression levels increased in time-dependent
manner. Significant differences were observed at 12 h (P<0.05).
COL-1 and vimentin mRNA expression levels were both significantly
increased at 12 h (P<0.05) and 24 h (P<0.01). These results
suggest that incubation at high glucose levels increases the
expression of the ECM components and induces EMT in HPMCs in a
time-dependent manner.
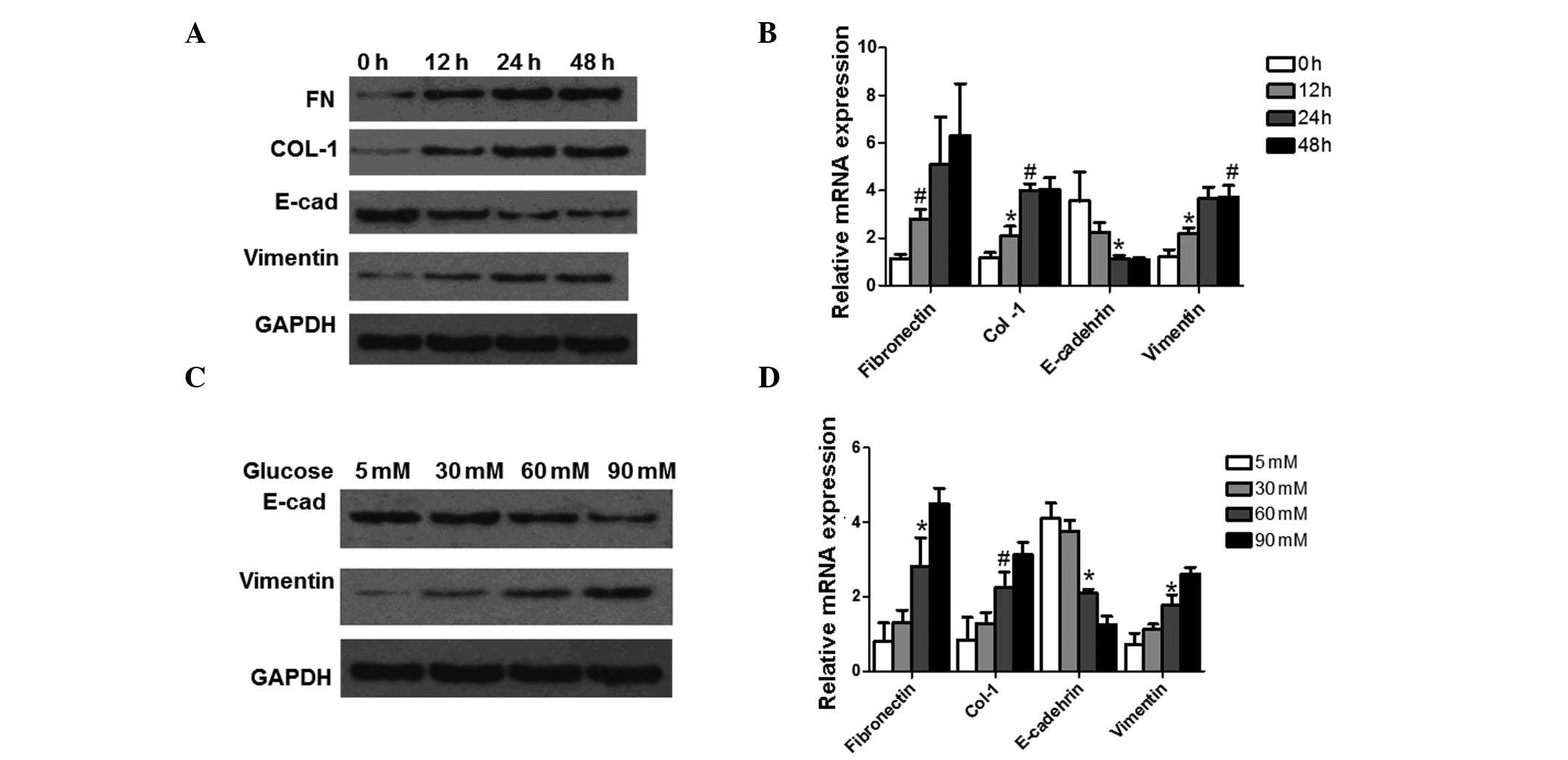 | Figure 2Expression analysis of EMT marker
proteins at the protein and mRNA level following treatment with
high levels of glucose. The analyzed proteins were FN, COL-1,
E-cadherin and vimentin. GAPDH was used as an internal control. (A
and B) Cells were incubated with 60 mM glucose for 0, 12, 24 and 48
h, respectively, prior to analysis. Representative results from the
western blot analysis and qPCR are shown. (C and D) Cells were
incubated with 5, 30, 60 and 90 mM glucose for 24 h prior to
analysis. Representative results from the western blot analysis and
qPCR are shown. Experiments were performed three times and data are
expressed as the mean ± standard deviation. *P<0.05
and #P<0.01, compared with cells incubated for 0 h.
EMT, epithelial to mesenchymal transition; FN, fibronectin; COL-1,
collagen, type I; qPCR, quantitative polymerase chain reaction;
E-cad, E-cadherin. |
To further determine the effects of high glucose
levels on peritoneal EMT, HPMCs were incubated in media containing
5, 30, 60 and 90 mM glucose for 24 h. The protein expression levels
of E-cadherin and vimentin were analyzed using western blot
analysis and the mRNA expression levels of FN, COL-1, E-cadherin
and vimentin were determined using qPCR. Representative results
from the western blot analysis are shown in Fig. 2C. Glucose treatment downregulated
E-cadherin expression, with E-cadherin expression being lowest
following treatment with 90 mM glucose. The expression of vimentin
was almost undetectable following treatment with 5 mM glucose;
however, expression increased following treatment with 30 and 60 mM
glucose and peaked at 90 mM glucose. qPCR yielded similar results.
As shown in Fig. 2D, mRNA
expression levels of E-cadherin decreased in a dose-dependent
manner and there was a significant decrease in expression levels
following treatment with 60mM glucose compared with expression
following treatment with 5 mM glucose (P<0.05). The mRNA
expression levels of vimentin, COL-1 and FN also increased in a
dose-dependent manner. Significant differences were observed at
60mM of treatment (P<0.05; P<0.01). These results suggest
that expression of ECM components and the EMT of HPMCs were induced
by glucose in dose-dependent manner.
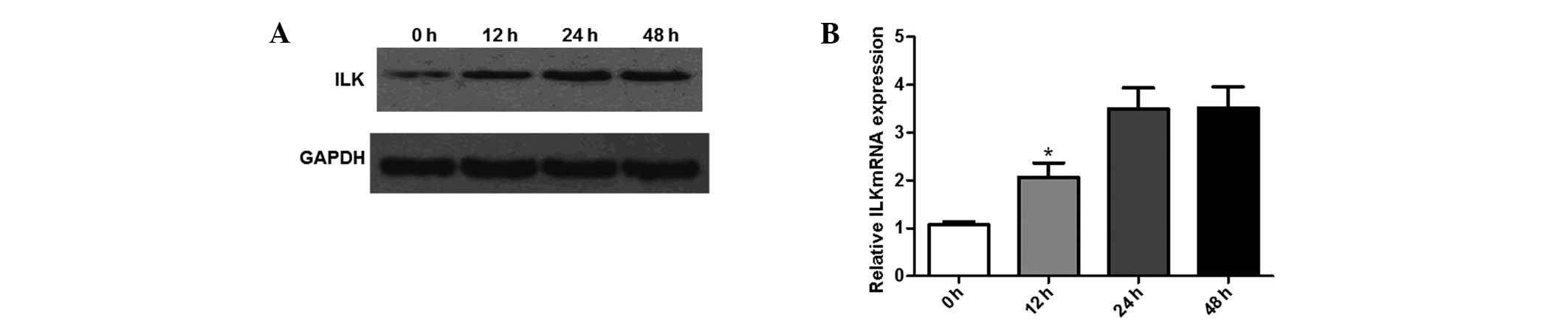
ILK expression is increased during EMT in
HPMCs
In order to examine the changes in ILK expression
following incubation with glucose in HPMCs, western blot analysis
and qPCR were performed. Prior to analysis, cells were incubated
with 60 mM glucose for 0, 12, 24 and 48 h, respectively. As shown
in Fig. 3A, ILK expression was
upregulated in response to incubation at high glucose levels. The
upregulation was observed from 12 h and peaked following 24 h of
treatment. This was further confirmed by the results from the qPCR.
As shown in Fig. 3B, ILK mRNA
expression levels increased between 12 and 48 h of treatment. These
results indicate that ILK expression was induced during EMT in
HPMCs.
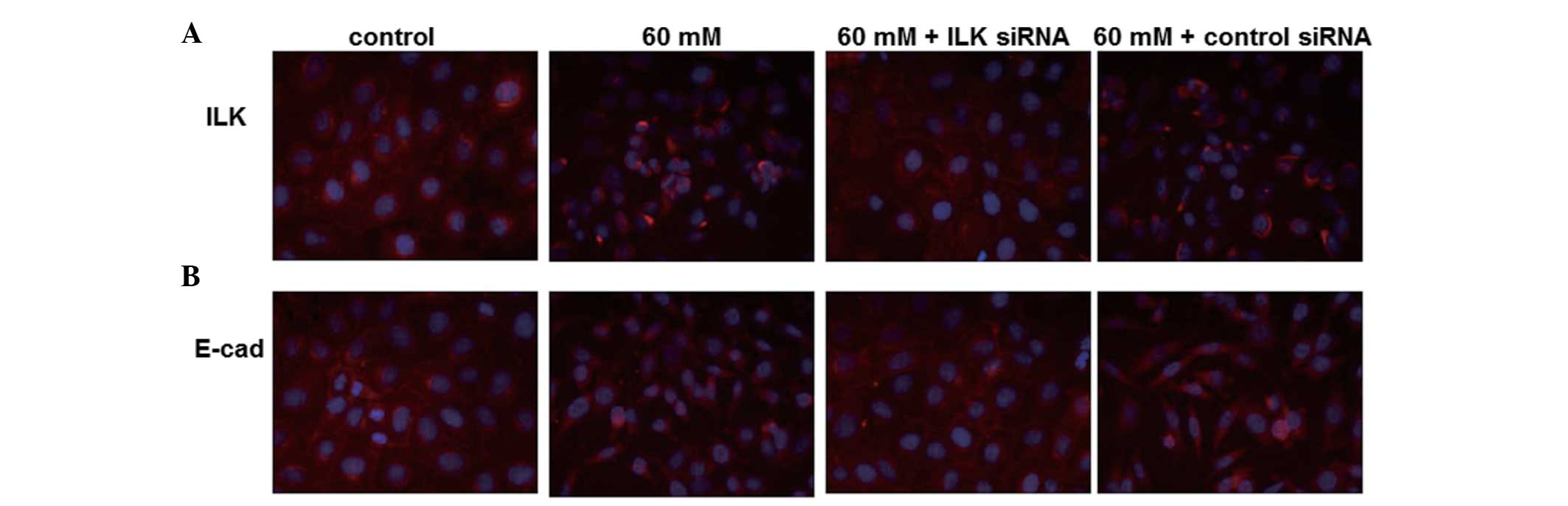
Distribution of ILK and E-cadherin in
HPMCs induced by high glucose levels
In order to further elucidate the observed changes
in ILK expression, the distribution of ILK in the normal HPMCs and
during the development of fibrosis in the HPMCs was investigated
using an immunofluorescence assay. Under normal glucose conditions,
differentiated HPMCs exhibited a typical spreading cobblestone-like
morphology with processes and expressed little ILK in the cytoplasm
(Fig. 4A). In the HPMCs induced by
high glucose levels (60 mM), however, a significant increase in ILK
staining was observed, and the staining was equally distributed in
the cytoplasm. HPMCs were then transfected with ILK-specific siRNAs
to knock down ILK expression. In cells with ILK-knockdown, it was
observed that ILK staining was abolished and the cells showed an
epithelial morphology.
The distribution of E-cadherin in HPMCs was also
observed using an immunofluorescence assay. As shown in Fig. 4B, strong staining for E-cadherin
was visible in the plasma membrane of normal HPMCs. However,
E-cadherin staining largely disappeared in the HPMCs induced by
high glucose levels. By contrast, the E-cadherin staining markedly
increased in response to ILK siRNA. In addition, the staining
further confirmed the phenotypic transition of HPMCs from a spindle
morphology with processes to a cobblestone appearance under high
glucose conditions. This result further indicates that ILK was
involved in HPMC EMT induced by high glucose levels.
Knockdown of ILK expression blocks EMT of
HPMCs and suppresses high glucose-induced downstream signaling
To further characterize the potential function of
ILK in HPMCs, the effect of the knockdown of endogenous ILK on the
expression levels of EMT-associated proteins and downstream signal
transduction proteins were investigated. siRNA was used to knock
down endogenous ILK expression. Protein and mRNA expression of
E-cadherin, vimentin, FN, ILK, phosphorylated GSK-3β (p-GSK-3β) and
cyclin D1 were detected using western blot analysis and qPCR,
respectively. GAPDH was used as an internal control. The expression
levels of EMT-associated proteins were first analyzed. As shown in
Fig. 5A and B, transfection of
HPMCs with specific siRNA resulted in a significant reduction in
endogenous ILK expression at the protein and mRNA level,
respectively (P<0.01). The results from the western blot
analysis and the qPCR analysis demonstrated that knockdown of ILK
significantly restored E-cadherin expression (P<0.05). However,
downregulation of ILK significantly reduced vimentin and FN
expression (P<0.05), preventing FN overproduction in response to
stimulation by high glucose levels. These findings further show
that EMT of HPMCs may be inhibited by ILK-knockdown.
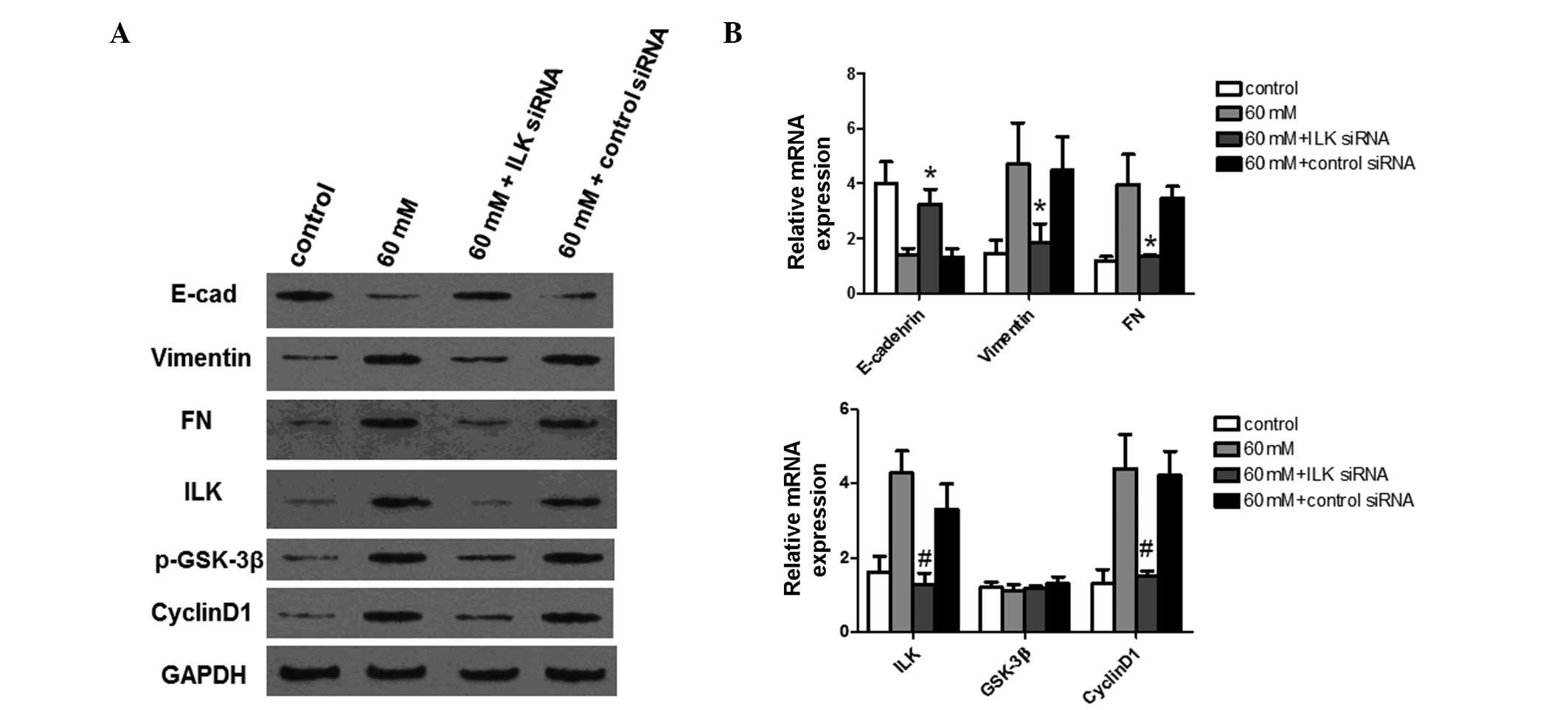 | Figure 5Expression analysis of EMT-associated
proteins and downstream signal transduction proteins following
ILK-knockdown. ILK expression was knocked down using siRNA. Control
and ILK-knockdown cells were treated with 60 mM glucose for 24 h
prior to analysis. E-cadherin, vimentin, FN, ILK, GSK-3β and cyclin
D1 protein and mRNA expression levels were detected using (A)
western blot analysis and (B) quantitative polymerase chain
reaction, respectively. GAPDH was used as an internal control.
Experiments were performed in triplicate and data are presented as
the mean ± standard deviation. *P<0.05 and
#P<0.01, compared with the cells treated with 60 mM
glucose. EMT, epithelial to mesenchymal transition; ILK,
integrin-linked kinase; siRNA, small interfering RNA; FN,
fibronectin; GSK-3β, glycogen synthase kinase 3β; p-GSK-3β,
phos-phorylated GSK-3β; E-cad, E-cadherin. |
GSK-3β is a known ILK downstream target. It was then
investigated whether inhibition of ILK activity by siRNA leads to a
reduction in GSK-3β phosphorylation induced by high glucose
conditions. As shown in Fig. 5A,
phosphorylation of GSK-3β induced by high glucose levels was
inhibited by ILK-knockdown. However, the expression of GSK-3β at
the mRNA level was not significantly affected (Fig. 5B), suggesting that ILK-knockdown by
siRNA specifically blocks GSK-3β phosphorylation in HPMCs. Cyclin
D1 is a key downstream target of activated canonical Wnt signaling.
As demonstrated by the results from the western blot analysis and
qPCR, cyclin D1 expression was also increased by high glucose
treatment, but was significantly decreased following ILK-knockdown
(P<0.01). In combination, these results suggest that ILK may
inhibit EMT in HPMCs through phosphorylation of GSK-3β.
Discussion
In this study, high glucose levels were found to
increase ILK expression in HPMCs. Under normal glucose conditions,
HPMCs exhibited a spreading arborized morphology, whereas they had
a cobblestone morphology under high glucose conditions.
Furthermore, in the high-glucose environment, E-cadherin expression
was found to be decreased, accompanied by an increased expression
of the mesenchymal marker vimentin. These findings demonstrate that
HPMCs underwent EMT in high glucose conditions.
It is known that ILK has a key role at the interface
between the ECM, integrins, actin-based cytoskeleton and cellular
phenotype in kidney diseases (27). In addition, ILK acts as an adaptor
protein that interacts with nephrin and α-actinin-4 to form a
ternary complex, which is essential for the maintenance of podocyte
function and glomerular filter integrity (28). Previous studies have suggested that
TGF-β1 and high glucose levels induce HPMCs to undergo EMT
(1,2). Additionally, ILK inhibition with a
small molecule inhibitor, QLT-0267, prevented EMT of podocytes and
ameliorated proteinuria, suggesting that ILK was a key
intracellular mediator in EMT. Of note, the inhibitor had no
adverse effect on normal kidney structure and function (29). The disruption of the interaction
between integrin and ILK may contribute to podocyte dysfunction,
leading to EMT. In accordance with these results, in the present
study it was demonstrated that high glucose levels induced ILK
overexpression and EMT in HPMCs. Previous studies have demonstrated
that particularly interesting new cysteine-histidine rich protein 1
(PINCH-1), an adaptor protein that interacts with ILK, is
dysregulated in the fibrotic kidney following obstructive injury.
The concomitant induction of PINCH-1 (30) and ILK (18) suggests that the PINCH-1-ILK complex
may have a fundamental role in mediating tubular EMT. Tissue-type
plasminogen activator (tPA) exerts its fibrogenic function by a
novel signal transduction pathway. It binds to the membrane
receptor low-density lipoprotein receptor-related protein 1 (LRP-1)
and triggers the receptor tyrosine phosphorylation that, in turn,
recruits β1 integrin and subsequently activates ILK signaling
(31). ILK induction by TGF-β1 is
clearly dependent upon intact Smad signaling in tubular epithelial
cells, since overexpression of inhibitory Smad-7 abolishes Smad-2
phosphorylation and ILK induction. ILK signaling is also implicated
in Snail expression. The kinase-dead form of ILK largely abolishes
TGF-β1-induced tubular EMT and inhibition of ILK expression by
hepatocyte growth factor (HGF), blocks tubular EMT and reduces
renal fibrosis. ILK strategically bridges the integrins and actin
cytoskeleton-associated proteins, including calponin homology
domain-containing ILK binding protein and paxillin, and transmits
signal exchanges between the intracellular and extracellular
compartments (32–34). Consistently, a previous study also
demonstrated that ILK induces an invasive phenotype in brain tumor
cell lines via activator protein 1 (AP-1)-dependent upregulation of
matrix metalloproteinase-9 (MMP-9) expression (35). Similarly, ILK directly
phosphorylates GSK-3 on Ser 9, causing its inhibition, which leads
to the stabilization of β-catenin and stimulation of the activity
of AP-1 and cyclic adenosine monophosphate response element binding
protein (20,36,37).
Since GSK-3β is a downstream substrate for Akt, ILK also induces
GSK-3β phosphorylation indirectly via the Akt pathway (14). The results of the present study
found that the inhibition of ILK by siRNA inhibited ILK expression
and attenuated EMT, suggesting that ILK is involved in the EMT of
HPMCs, which is consistent with the results found in previous
studies.
It has been reported that overexpression of active
ILK in podocytes induces translocation of β-catenin to the cell
nucleus, as well as nuclear colocalization of β-catenin with
lymphoid enhancer-binding factor 1 (LEF-1) (38). In patients with primary focal
segmental glomerulosclerosis (FSGS), the activation of ILK
activated the Wnt signaling pathway and GSK-3β in damaged podocytes
(37). The results of the present
study showed that incubation of HPMCs at high glucose levels led to
the acquisition of fibrotic characteristics and initiation of EMT.
Additionally, high glucose conditions were demonstrated to
upregulate ILK expression in a GSK-3β-dependent manner. In cultured
podocytes, ILK has been shown to orchestrate a wide array of
functions, including membrane proximal initiation of signal
transduction via Akt, GSK-3β and β-catenin (15,16)
and regulating cell phenotype and survival (15,16,39).
A previous study demonstrated that Dp71f modulates GSK3-β
recruitment to the β1-integrin adhesion complex in PC12 cells
(40).
Previous studies have shown that ILK inhibition
blocks phosphorylated Akt (p-Akt), p-GSK-3β, β-catenin and Snail
expression, as well as MMP9 and Twist expression (41). Changes in ECM composition may alter
the balance between apoptosis and survival (42). ILK links the ECM with the
intracellular compartment through interaction with the cytoplasmic
domains of β1 and β3 integrins (43). As a central part of the
ILK/PINCH/parvin complex, ILK connects the ECM with the actin
cytoskeleton and transmits signals to the inner region of the cell
through its serine/threonine kinase activity. The kinase activity
of ILK is stimulated by integrins and soluble mediators, including
growth factors and chemokines, and is regulated in a PI3K-dependent
manner (44,45). EMT is mediated through several
transcription repressors, including Snail, Slug, Twist, MMP-2,
MMP-9 and zinc finger E-box-binding homeobox 1 (ZEB1), which induce
EMT by suppressing the transcription of the E-cadherin gene, an
epithelial cell marker and a potent suppressor of cell invasion and
metastasis (46–48). Furthermore, it was demonstrated in
the present study that downregulation of ILK also inhibits
phosphorylation of GSK-3β in HPMCs. ILK regulates phosphorylation
of its downstream targets Akt and GSK-3β on Ser 473 and Ser 9,
respectively (49). GSK-3β, which
is known as the ‘guardian of the epithelial state’, regulates Snail
degradation through its phosphorylation on Ser 246. ILK and Akt are
known to phosphorylate and inactivate GSK-3β, which in turn
suppresses phosphorylation of Snail and leads to EMT (50,51).
Maseki et al (52)
demonstrated that EMT was mediated by activation of the
Akt/GSK-3β/Snail pathway. Akt phosphorylates GSK-3β at Ser 9,
leading to inactivation of its kinase activity. GSK-3β also
interacts with ILK, and ILK phosphorylates GSK-3β at Ser 9 in an
Akt-independent manner (53).
The phosphorylation of GSK-3β by ILK signaling
pathways leads to the activation of transcription factors,
including AP1 and the β-catenin/lymphocyte enhancer factor, which
in turn stimulate MMP-9 and cyclin-D1, respectively (42). The results of the present study
demonstrate that inhibition of ILK activity results in p-GSK-3β
downregulation. Therefore, the results from the present study
further confirm the previous findings, and suggest that ILK may
have a crucial role in EMT through phosphorylation of GSK-3β. ILK
overexpression in rat intestinal epithelial cells resulted in
stimulation of the G1/S cyclin-Cdk complex and subsequent cell
cycle progression (54).
Furthermore, the dominant-negative mutant of ILK was shown to
induce G1 cell cycle arrest in prostate cancer cells (55). This may indicate that the parallel
cell cycle regulatory events seen in diseased podocytes (i.e.,
collapsing focal segmental glomerulosclerosis) may at least in part
be downstream of ILK (56,57).
In conclusion, the results of the present study have
demonstrate for the first time, to the best of our knowledge, that
ILK has an important role in the high glucose level-induced EMT of
HPMCs. Knockdown of ILK inhibits EMT of HPMCs, with an increase in
E-cadherin and a decrease in vimentin expression levels.
Furthermore, ILK-knockdown suppresses phosphorylation of GSK-3β,
and thus inhibits its downstream pathway. Therefore, ILK, a newly
identified peritoneal EMT regulator, may be used as a diagnostic
marker for peritoneal fibrosis and may be a potential therapeutic
target for the treatment of peritoneal fibrosis.
Acknowledgements
This study was supported by the 2010–2012 Key
Projects for Clinical Disciplines of the Subordinate Hospital of
the Ministry of Health.
References
|
1
|
Aroeira LS, Aguilera A, Selgas R, et al:
Mesenchymal conversion of mesothelial cells as a mechanism
responsible for high solute transport rate in peritoneal dialysis:
role of vascular endothelial growth factor. Am J Kidney Dis.
46:938–948. 2005. View Article : Google Scholar
|
|
2
|
Yáñez-Mó M, Lara-Pezzi E, Selgas R, et al:
Peritoneal dialysis and epithelial-to-mesenchymal transition of
mesothelial cells. N Engl J Med. 348:403–413. 2003.
|
|
3
|
Kim YL: Update on mechanisms of
ultrafiltration failure. Perit Dial Int. 29(Suppl 2): S123–S127.
2009.PubMed/NCBI
|
|
4
|
Loureiro J, Aguilera A, Selgas R, et al:
Blocking TGF-β1 protects the peritoneal membrane from
dialysate-induced damage. J Am Soc Nephrol. 22:1682–1695. 2011.
|
|
5
|
Margetts PJ, Bonniaud P, Liu L, et al:
Transient overexpression of TGF-{beta}1 induces epithelial
mesenchymal transition in the rodent peritoneum. J Am Soc Nephrol.
16:425–436. 2005.
|
|
6
|
Vargha R, Endemann M, Kratochwill K, et
al: Ex vivo reversal of in vivo transdifferentiation in mesothelial
cells grown from peritoneal dialysate effluents. Nephrol Dial
Transplant. 21:2943–2947. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang X, Nie J, Jia Z, et al: Impaired
TGF-beta signalling enhances peritoneal inflammation induced by
E. coli in rats. Nephrol Dial Transplant. 25:399–412. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee HB and Ha H: Mechanisms of
epithelial-mesenchymal transition of peritoneal mesothelial cells
during peritoneal dialysis. J Korean Med Sci. 22:943–945. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zeisberg M and Duffield JS: Resolved: EMT
produces fibroblasts in the kidney. J Am Soc Nephrol. 21:1247–1253.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hannigan GE, Leung-Hagesteijn C,
Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC and Dedhar
S: Regulation of cell adhesion and anchorage-dependent growth by a
new beta 1-integrin-linked protein kinase. Nature. 379:91–96. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Legate KR, Montañez E, Kudlacek O and
Fässler R: ILK, PINCH and parvin: the tIPP of integrin signaling.
Nat Rev Mol Cell Biol. 7:20–31. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai HY, Zheng M, Lv LL, Tang RN, Ma KL,
Liu D, Wu M and Liu BC: The roles of connective tissue growth
factor and integrin-linked kinase in high glucose-induced
phenotypic alterations of podocytes. J Cell Biochem. 113:293–301.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Y, Tan X, Dai C, Stolz DB, Wang D and
Liu Y: Inhibition of integrin-linked kinase attenuates renal
interstitial fibrosis. J Am Soc Nephrol. 20:1907–1918. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
de Teixeira VP, Blattner SM, Li M, et al:
Functional consequences of integrin-linked kinase activation in
podocyte damage. Kidney Int. 67:514–523. 2005.PubMed/NCBI
|
|
16
|
Kretzler M, Teixeira VP, Unschuld PG, et
al: Integrin-linked kinase as a candidate downstream effector in
proteinuria. FASEB J. 15:1843–1845. 2001.PubMed/NCBI
|
|
17
|
Guo L, Sanders PW, Woods A and Wu C: The
distribution and regulation of integrin-linked kinase in normal and
diabetic kidneys. Am J Pathol. 159:1735–1742. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Yang J, Dai C, et al: Role for
integrin-linked kinase in mediating tubular epithelial to
mesenchymal transition and renal interstitial fibrogenesis. J Clin
Invest. 112:503–516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Persad S, Attwell S, Gray V, Mawji N, Deng
JT, Leung D, et al: Regulation of protein kinase B/Akt-serine 473
phosphorylation by integrin-linked kinase: critical roles for
kinaseactivity and amino acids arginine 211 and serine 343. J Biol
Chem. 276:27462–27469. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Delcommenne M, Tan C, Gray V, Rue L,
Woodgett J and Dedhar S: Phosphoinositide-3-OH kinase-dependent
regulation of glycogen synthase kinase 3 and protein kinase B/AKT
by the integrin-linked kinase. Proc Natl Acad Sci USA.
95:11211–11216. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vi L, de Lasa C, DiGuglielmo GM, et al:
Integrin-linked kinase is required for TGF-β1 induction of dermal
myofibroblast differentiation. J Invest Dermatol. 131:586–593.
2011.
|
|
22
|
Li Y, Yang J, Luo JH, et al: Tubular
epithelial cell dedifferentiation is driven by the helix-loop-helix
transcriptional inhibitor Id1. J Am Soc Nephrol. 18:449–460. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smeeton J, Zhang X, Bulus N, et al:
Integrin-linked kinase regulates p38 MAPK-dependent cell cycle
arrest in ureteric bud development. Development. 137:3233–3243.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu XC, Liu BC, Zhang XL, et al: Role of
ERK1/2 and PI3-K in the regulation of CTGF-induced ILK expression
in HK-2 cells. Clin Chim Acta. 382:89–94. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stylianou E, Jenner LA, Davies M, Coles GA
and Williams JD: Isolation, culture and characterization of human
peritoneal mesothelial cells. Kidney Int. 37:1563–1570. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rougier JP, Moullier P, Piedagnel R and
Ronco PM: Hyperosmolality suppresses but TGF beta 1 increases MMP9
in human peritoneal mesothelial cells. Kidney Int. 51:337–347.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blattner SM and Kretzler M:
Integrin-linked kinase in renal disease: connecting cell-matrix
interaction to the cytoskeleton. Curr Opin Nephrol Hypertens.
14:404–410. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dai C, Stolz DB, Bastacky SI, St-Arnaud R,
Wu C, Dedhar S and Liu Y: Essential role of integrin-linked kinase
in podocyte biology: Bridging the integrin and slit diaphragm
signaling. J Am Soc Nephrol. 17:2164–2175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kang YS, Li Y, Dai C, Kiss LP, Wu C and
Liu Y: Inhibition of integrin-linked kinase blocks podocyte
epithelial-mesenchymal transition and ameliorates proteinuria.
Kidney Int. 78:363–373. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li Y, Dai C, Wu C and Liu Y: PINCH-1
promotes tubular epithelial-to-mesenchymal transition by
interacting with integrin-linked kinase. J Am Soc Nephrol.
18:2534–2543. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hu K, Wu C, Mars WM and Liu Y: Tissue-type
plasminogen activator promotes murine myofibroblast activation
through LDL receptor-related protein 1-mediated integrin signaling.
J Clin Invest. 117:3821–3832. 2007.
|
|
32
|
Guo L and Wu C: Regulation of fibronectin
matrix deposition and cell proliferation by the PINCH-ILK-CH-ILKBP
complex. FASEB J. 16:1298–1300. 2002.PubMed/NCBI
|
|
33
|
Tu Y, Huang Y, Zhang Y, Hua Y and Wu C: A
new focal adhesion protein that interacts with integrin-linked
kinase and regulates cell adhesion and spreading. J Cell Biol.
153:585–598. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nikolopoulos SN and Turner CE: Molecular
dissection of actopaxin-integrin-linked kinase-Paxillin
interactions and their role in subcellular localization. J Biol
Chem. 277:1568–1575. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Troussard AA, Costello P, Yoganathan TN,
Kumagai S, Roskelley CD and Dedhar S: The integrin linked kinase
(ILK) induces an invasive phenotype via AP-1 transcription
factor-dependent upregulation of matrix metalloproteinase 9
(MMP-9). Oncogene. 19:5444–5452. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
D’Amico M, Hulit J, Amanatullah DF, et al:
The integrin-linked kinase regulates the cyclin D1 gene through
glycogen synthase kinase 3beta and cAMP-responsive element-binding
protein-dependent pathways. J Biol Chem. 275:32649–32657.
2000.PubMed/NCBI
|
|
37
|
Joshi MB, Ivanov D, Philippova M, Erne P
and Resink TJ: Integrin-linked kinase is an essential mediator for
T-cadherin-dependent signaling via Akt and GSK3beta in endothelial
cells. FASEB J. 21:3083–3095. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sellin JH, Umar S, Xiao J and Morris AP:
Increased beta-catenin expression and nuclear translocation
accompany cellular hyperproliferation in vivo. Cancer Res.
61:2899–2906. 2001.PubMed/NCBI
|
|
39
|
Yang Y, Guo L, Blattner SM, Mundel P,
Kretzler M and Wu C: Formation and phosphorylation of the
PINCH-1-integrin linked kinase-alpha-parvin complex are important
for regulation of renal glomerular podocyte adhesion, architecture,
and survival. J Am Soc Nephrol. 16:1966–1976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cortés JC, Montalvo EA, Muñiz J, Mornet D,
Garrido E, Centeno F and Cisneros B: Dp71f modulates GSK3-beta
recruitment to the beta1-integrin adhesion complex. Neurochem Res.
34:438–444. 2009.PubMed/NCBI
|
|
41
|
Kalra J, Sutherland BW, Stratford AL, et
al: Suppression of Her2/neu expression through ILK inhibition is
regulated by a pathway involving TWIST and YB-1. Oncogene.
29:6343–6356. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
del Nogal M, Luengo A, Olmos G, Lasa M,
Rodriguez-Puyol D, Rodriguez-Puyol M and Calleros L: Balance
between apoptosis or survival induced by changes in
extracellular-matrix composition in human mesangial cells: a key
role for ILK-NfκB pathway. Apoptosis. 17:1261–1274. 2012.
|
|
43
|
Papachristou DJ, Gkretsi V, Rao UN, et al:
Expression of integrin-linked kinase and its binding partners in
chondrosarcoma: association with prognostic significance. Eur J
Cancer. 44:2518–2525. 2008. View Article : Google Scholar
|
|
44
|
McDonald PC, Fielding AB and Dedhar S:
Integrin-linked kinase - essential roles in physiology and cancer
biology. J Cell Sci. 121:3121–3132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fielding AB, Dobreva I, McDonald PC,
Foster LJ and Dedhar S: Integrin-linked kinase localizes to the
centrosome and regulates mitotic spindle organization. J Cell Biol.
180:681–689. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shin SY, Rath O, Zebisch A, Choo SM, Kolch
W and Cho KH: Functional roles of multiple feedback loops in
extracellular signal-regulated kinase and Wnt signaling pathways
that reg-ulate epithelial-mesenchymal transition. Cancer Res.
70:6715–6724. 2010. View Article : Google Scholar
|
|
47
|
Acloque H, Thiery JP and Nieto MA: The
physiology and pathology of the EMT. Meeting on the
epithelial-mesenchymal transition. EMBO Rep. 9:322–326. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xing Y, Qi J, Deng S, Wang C, Zhang L and
Chen J: Small interfering RNA targeting ILK inhibits metastasis in
human tongue cancer cells through repression of
epithelial-to-mesenchymal transition. Exp Cell Res. 319:2058–2072.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
McDonald PC, Oloumi A, Mills J, et al:
Rictor and integrin-linked kinase interact and regulate Akt
phosphorylation and cancer cell survival. Cancer Res. 68:1618–1624.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Doble BW and Woodgett JR: Role of glycogen
synthase kinase-3 in cell fate and epithelial-mesenchymal
transitions. Cells Tissues Organs. 185:73–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
McPhee TR, McDonald PC, Oloumi A and
Dedhar S: Integrin-linked kinase regulates E-cadherin expression
through PARP-1. Dev Dyn. 237:2737–2747. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Maseki S, Ijichi K, Tanaka H, et al:
Acquisition of EMT phenotype in the gefitinib-resistant cells of a
head and neck squamous cell carcinoma cell line through
Akt/GSK-3β/snail signalling pathway. Br J Cancer. 106:1196–1204.
2012.PubMed/NCBI
|
|
53
|
Wu C and Dedhar S: Integrin-linked kinase
(ILK) and its interactors: a new paradigm for the coupling of
extracellular matrix to actin cytoskeleton and signaling complexes.
J Cell Biol. 155:505–510. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Radeva G, Petrocelli T, Behrend E,
Leung-Hagesteijn C, Filmus J, Slingerland J and Dedhar S:
Overexpression of the integrin-linked kinase promotes
anchorage-independent cell cycle progression. J Biol Chem.
272:13937–13944. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tan C, Costello P, Sanghera J, Dominguez
D, Baulida J, de Herreros AG and Dedhar S: Inhibition of integrin
linked kinase (ILK) suppresses beta-catenin-Lef/Tcf-dependent
transcription and expression of the E-cadherin repressor, snail, in
APC−/− human colon carcinoma cells. Oncogene. 20:133–140.
2001.PubMed/NCBI
|
|
56
|
Barisoni L, Mokrzycki M, Sablay L, Nagata
M, Yamase H and Mundel P: Podocyte cell cycle regulation and
proliferation in collapsing glomerulopathies. Kidney Int.
58:137–143. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Petermann AT, Pippin J, Hiromura K, et al:
Mitotic cell cycle proteins increase in podocytes despite lack of
proliferation. Kidney Int. 63:113–122. 2003. View Article : Google Scholar : PubMed/NCBI
|