Introduction
We and other researchers have shown that the Jun
kinase (JNK) mediates transformation (1–4) and is
activated by genotoxic stress. Inhibition of this pathway is
postulated to i) inhibit the JNK contribution to the transformed
phenotype; and ii) to inhibit DNA repair and synthesis thereby
sensitizing tumor cells to cisplatin (P1atinol) and other
DNA-damaging chemotherapeutic agents. Cisplatin is a well-studied
chemotherapeutic agent that acts through DNA damage (5). It is widely used (particularly for
ovarian, bladder and testicular carcinomas), yet, is ineffective in
breast carcinoma (6). However, drug
resistance remains one of the biggest obstacles to successful
treatment of cancer and is believed to be the primary reason for
the treatment failure encountered in approximately half of all
cancers. Brain tumors with wild-type (wt) or mutant p53 status may
respond differently to radiation therapy. Actually, the p53 gene is
found to be mutated in more than 40% of gliomas (7) and it is believed that restoration of
p53 function would enhance the response to chemotherapeutic
treatments (6,8). The mechanism of resistance to
therapeutic drugs, most of which lead directly or indirectly to DNA
damage, is closely linked to the mechanisms of cancer, which we now
know to involve in part genetic changes that alter the cellular
response to DNA damage. Thus, many of the genetic changes,
including loss of p53, that affect the response to therapy may
affect tumor progression as well. Another important cellular
pathway, also triggered in response to DNA damage is the Jun
kinase/stress-activated protein kinase pathway (JNK/SAPK), one of
several distinct mitogen-activated protein kinase (MAPK) pathways
involved in signal transduction. JNK phosphorylates c-Jun, thereby
enhancing the transcriptional activity of c-Jun. The JNK/SAPK
pathway is induced by oncogene expression (9–12) and
is essential for transformation of rat embryo fibroblasts (13,14).
JNK activity is strongly induced in response to a variety of DNA
damaging treatments such as UV irradiation (15), cisplatin (16–19)
and camptothecin (4,20), and as we have shown, appears to
promote resistance of tumor cells to cisplatin through a mechanism
involving increased DNA repair. We examined the role of the JNK
pathway following treatment with cisplatin, in T98G glioblastoma
cells (which express mutant p53) transfected with JNK1- and
JNK2-small interfering RNAs (siRNAs). Therefore, the JNK/SAPK
pathway plays a p53-independent role in resistance to DNA damaging
agents in these cells, yet not to agents that do not damage DNA
(21). In each case the promoter
region contains one or more AP-1 or ATF2/CREB regulatory sequences.
Thus, several of the genes known to be involved in cisplatin-DNA
adduct repair may be upregulated upon activation of the JNK pathway
following damage to DNA by cisplatin, and the downregulation of one
or more of them in JNK-siRNA-treated cells could account for the
therapy sensitization effect we observed.
The activation of JNK and loss of p53 may represent
independent mechanisms by which tumor cells undergo progression to
accommodate genome instability and ensure survival while sustaining
potentially lethal genome destabilizing events. By promoting DNA
repair, the JNK pathway may help to limit genome instability and
the associated DNA damage to levels compatible with survival
limiting genome instability. In the present study, we investigated
the possibility of sensitization of glioma cells to cisplatin by
abrogation of Jun-N-terminal kinase activity. We compared the
response of basal JNK protein expression in untreated T98G cells
and in cells treated with JNK1-siRNA, JNK2-siRNA alone or in
combination with cisplatin. We found that disruption of either JNK1
or JNK2 by siRNAs sensitized T98G cells to cisplatin thereby
markedly decreasing the viability of the cells.
Materials and methods
Cell culture
The T98G brain tumor cell line, established from
human glioblastoma, was obtained from the American Type Culture
Collection (ATCC; Manassas, VA, USA). The cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (FBS), 1 mM
sodium pyruvate, 100 Um/l penicillin G, 100 μg/ml, 1 streptomycin,
2 mM glutamine, 1 mM MEM non-essential amino acids and 50 μM
2-mercaptoethanol in a 5% CO2 incubator at 37°C. The
cells were dissociated using 0.25% trypsin and 0.53 mM EDTA
solution and subcultured once in 3–5 days.
Reagents
Tris-borate-EDTA and acrylamide:bisacrylamide (29:1)
were obtained from Bio-Rad (Richmond, CA, USA). Lipofectamine was
obtained from Life Technologies, Inc., USA. Complete Mini EDTA-free
protease inhibitor cocktail tablets and Annexin V-Fluos were
purchased from Roche Diagnostics GmbH (Mannheim, Germany). Phorbol
12-myristate-13-acetate (TPA) and TNF-α were purchased from
Stratagene Inc. (La Jolla, CA, USA). Antibodies JNK (FL) (sc-571).
JNK-1 (C-17) (sc-474), JNK2 (N18) (sc-827) and β-actin (sc-1616)
were used at a concentration of 0.07 and 0.1 mg/ml, respectively
(in a total volume of 12 ml), and were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA).
siRNA transfection
T98G glioblastoma cells were cultured to 60–80%
confluency on 6-well plates. The cells were incubated with 60
nmol/l siRNAs (Santa Cruz) targeting JNK1 (sc-29380), JNK2
(sc-39101) or control ‘scrambled’ siRNA (sc-37007) and 6 μl siRNA
transfection reagent, according to the manufacturer’s
recommendation (Santa Cruz) in 1 ml serum-free medium (MEM) at 37°C
for 6 h. After transfection, cells were cultured in complete medium
for 42 h or until being used. The siRNAs were purchased from Santa
Cruz Biotechnology, Inc.
Western immunoblot analysis
After reaching 70–80% confluency, T98G cells were
starved overnight with serum-free medium, and then treated at the
indicated concentrations and time periods with the different drugs
in the absence of serum. Forty-eight hours after transfection,
cells were collected and washed twice by cold PBS, and each well
was treated with 50 ml lysis buffer (2 mmol/l Tris-HCl pH 7.4, 50
mmol/l NaCl, 25 mmol/l EDTA, 50 mmol/l NaF, 1.5 mmol/l
Na3VO4, 1% Triton X-100, 0.1% SDS,
supplemented with protease inhibitors 1 mmol/l phenylmethylsulfonyl
fluoride, 10 mg/l pepstatin, 10 mg/l aprotinin and 5 mg/l
leupeptin) (all from Sigma). Protein concentrations were determined
using the Bradford protein assay. Equal amounts of protein (40 mg)
were separated on a 15% SDS polyacrylamide gel and transferred to a
nitrocellulose membrane (Hybond C; Amersham, Freiburg, Germany).
Membranes were blocked in 5% non-fat dry milk in Tris-buffered
saline (TBS) for 1 h at room temperature and probed with rabbit
anti-JNKs antibodies (dilution 1:500; Santa Cruz Biotechnology,
Inc.) overnight at 4°C. After 3 washings with TBS containing 0.1%
Tween-20, membranes were incubated with anti-rabbit IgG horseradish
peroxidase (1:5,000; Santa Cruz Biotechnology, Inc.) and developed
by luminol-mediated chemiluminescence (Appylgen Technologies Inc.,
China). To confirm equal protein loading, membranes were reprobed
with a 1:1,000 dilution of an anti-actin antibody (Santa Cruz
Biotechnology, Inc.). Densitometric analyses were performed using
Scion Image software.
Soft agar assays
To evaluate the ability of individual cell lines to
grow in an anchorage-independent manner, cells were plated in soft
agar (Agarose-1000; Gibco-BRL) as previously described (22). In brief, a 2.5% agarose stock was
made in 1X PBS. The bottom 0.5% agar support was prepared in DMEM
containing 10% FBS. Cells were harvested, washed and mixed with the
top-agarose suspension at a final concentration of 0.3–0.33%, which
was then layered onto the bottom agar. The agar plates were
incubated at 37°C in a humidified incubator for 10 days. Each assay
was performed in triplicate. The number of colonies was then
counted.
DNA fragmentation assay
Cells were plated in 96-well plates 24 h before
treatment. After treatment, DNA fragmentation was evaluated by
examination of cytoplasmic histone-associated DNA fragments
(mononucleosomes and oligonucleosomes) using a Cell-Death Detection
ELISA kit (Roche Molecular Biochemicals, Indianapolis, IN, USA)
according to the manufacturer’s recommendations.
Flow cytometry
T98G glioblastoma cells (5×105) were
seeded in triplicate onto 6-well plates and cultured in RPMI-1640
supplemented with 100 ml/l FBS. Following transfection for 48 h,
the cells were collected and washed with ice-cold PBS, and fixed in
70% ethanol overnight at 4°C. The fixed cells were pelleted, washed
in PBS, resuspended in PBS containing 0.1 mg/ml of propidium iodide
and analyzed by flow cytometry.
Results
The JNKs form one subfamily of the MAPK group of
serine/threonine protein kinases. The JNKs were first identified by
their activation in response to a variety of extracellular stresses
and their ability to phosphorylate the N-terminal transactivation
domain of the transcription factor c-Jun. Our previous studies
(17,23–25)
revealed that specific inhibition of the JNK pathway in a variety
of human tumor cells sensitizes the cells to the cytotoxic effects
of cisplatin. In the present study, we characterized the ability of
siRNAs against JNK1 and/or JNK2 to induce drug sensitivity in T98G
glioblastoma cells. The viability of these cells in the presence of
cisplatin was compared to that of the parental and empty-vector
control cells.
Expression of JNK1 and JNK2 protein in
human T98G glioblastoma cell line
JNK1 and JNK2 are among the nuclear factors that
plays an important role in the regulation of several genes. To
determine the effect of blocking the expression of both JNK1 and
JNK2 by siRNAs, we first studied the protein expression of genes
encoding the JNK1 and JNK1 products in T98G cells. The cells were
treated with several inductors of JNKs, such as FCS 0.5%, FCS 10%,
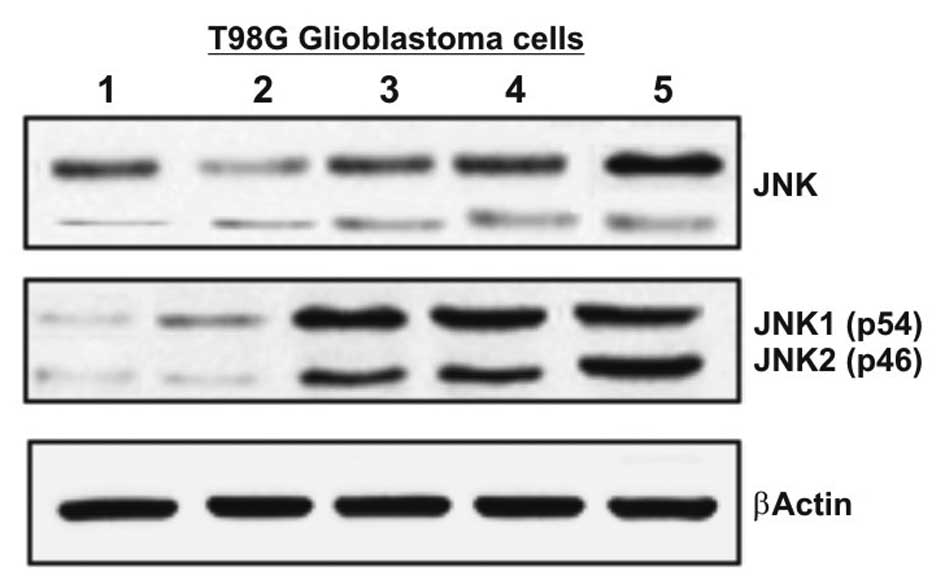
TPA (30 nM) and TNF-α (20 ng/ml). As showed in Fig. 1, the protein expression of JNK1 and
JNK2 was assessed by western blot analysis (Fig. 1). The basal protein levels of both
JNK1 and JNK2 were higher in the cells cultured in FBS 10%,
compared to the cells cultured in FBS 0.5%. Treatment with TPA and
TNF-α strongly activated JNK in all cases.
Knockdown of JNK1 and/or JNK2 expression
by specific siRNAs strongly decreases the protein levels of JNK
gene products and sensitizes cells to the genotoxic effect of
cisplatin
In order to study the role of JNKs in the growth
regulation of T98G cells, we treated the cells with specific siRNAs
to either JNK1 or JNK2. The cells were then treated with cisplatin
at concentrations of 100 μM and analyzed for protein expression.
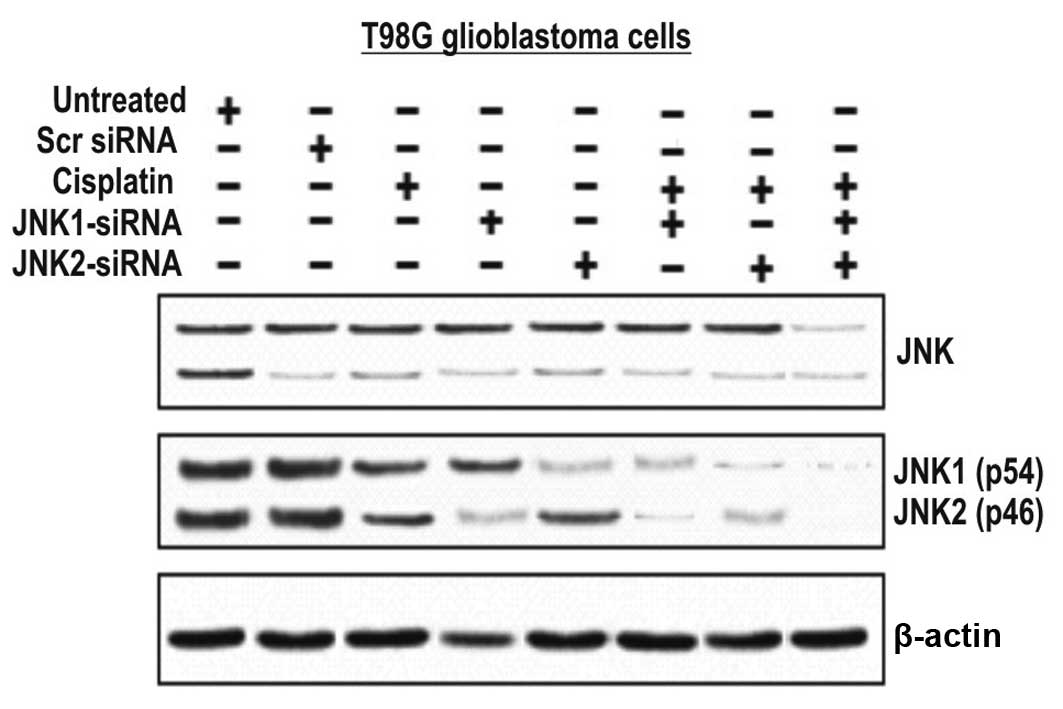
The effect of these assays was assessed by western blot analysis
(Fig. 2). JNK1/2-siRNAs and to a
lower extent cisplatin-treated cells, but not cells treated with
scrambled or the untreated cells, showed a marked reduction in
protein levels (Fig. 2). The
combination of either JNK1-siRNA or JNK2-siRNA with cisplatin
further increased the genotoxic effect of cisplatin, suggesting
that these two proteins appear to be important for T98G cell
survival.
Growth inhibition of T98G cells by siRNAs
against JNK1 and JNK2 alone or in combination with cisplatin
Another feature of malignant cells is that they can
grow under anchorage-independent conditions. The most common assay
to assess this is the ability of cells to grow and form colonies in
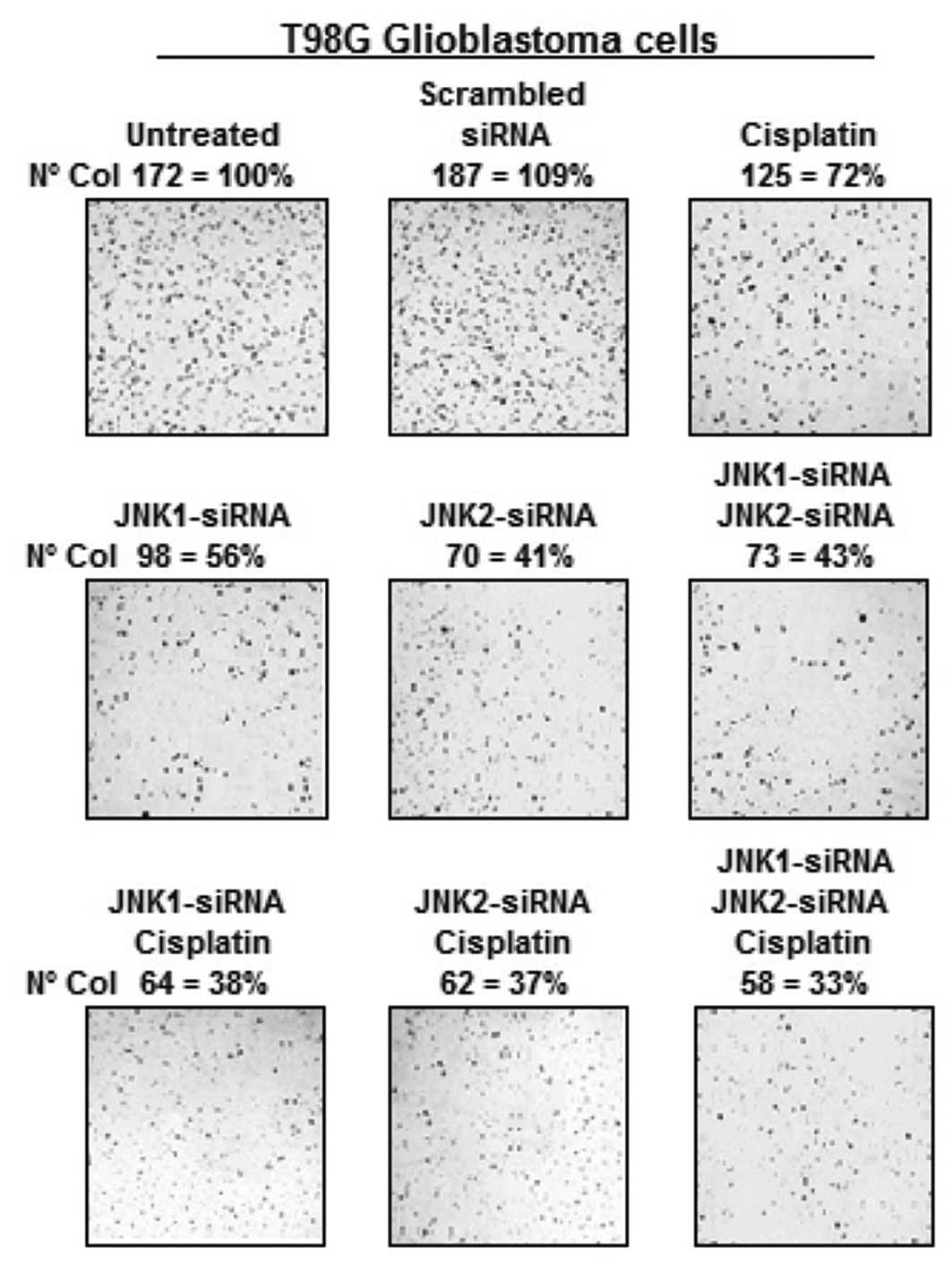
soft agar. As shown in Fig. 3, the
untreated T98G cells or cells treated with the scrambled siRNA
readily formed colonies in soft agar. In contrast, the T98G cells
treated with cisplatin exhibited a greatly reduced capacity to form
colonies (Fig. 3). Moreover, the
T98G cells treated with siRNA-JNK1 and/or siRNA-JNK2 exhibited a
further reduced capacity to form colonies. The combination of
cisplatin with siRNAs slightly enhanced the effects obtained with
JNK1- or JNK2-siRNA alone. Collectively, these assays clearly
demonstrate the ability of the siRNAs against JNK genes to suppress
phenotypes related to malignant potential.
Inhibition of JNK1 and JNK2 by siRNAs
sensitizes the T98G cells to cisplatin-induced apoptosis
JNKs are activated in response to cytokines or
environmental stress and can induce both pro-apoptotic and
pro-survival response (26), we
analyzed the role of JNK1 and JNK2 in cisplatin-induced cell
apoptosis in glioblastoma cells. Treatment of T98G glioblastoma
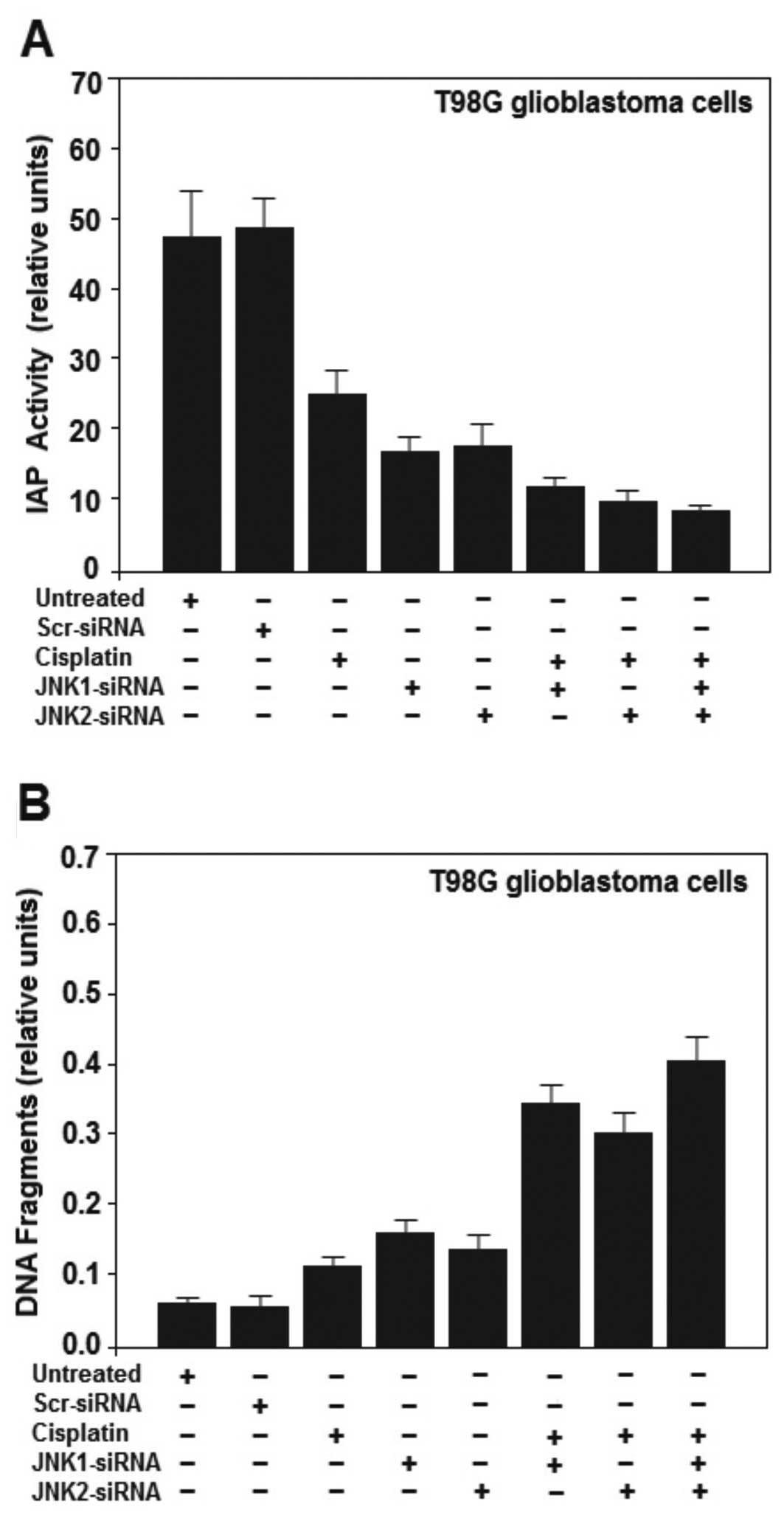
cells with siRNAs against JNK1 and/or JNK2 decreased IAP activity
(Fig. 4A). IAP is a family of
proteins involved in preventing cell death by apoptosis.
Furthermore, supporting the notion that inhibition of JNK sensitize
cells to cisplatin, we treated T98G cells with cisplatin (100 μM)
or with cisplatin together with cells previously treated with
JNK-siRNA or JNK2-siRNA. The activity of IAP proteins was further
decreased (Fig. 4A). The two IAP
proteins involved in death receptor signaling, cellular inhibitor
of apoptosis-1 (cIAP-1) and cIAP-2, undergo rapid cellular
elimination after binding to proteins via autoubiquitination and
subsequent proteasome-mediated degradation (27,28).
In addition, IAPs have been implicated as potential pro-metastatic
genes, particularly by promoting cell motility (29). Subsequently, we assessed the effect
of JNK knockdown on cisplatin-induced T98G cell death. JNK1 and
JNK2 promoted obvious cell survival as shown by decreased DNA
fragmentation in the untreated T98G cells (Fig. 4B) and this decrease was strongly
attenuated by inhibition of JNK1 and JNK2 using JNK1/2-siRNA
transfection. A further increase in DNA fragmentation was observed
in T98G cells treated with the siRNAs along with cisplatin (10 μM),
inducing significant apoptosis (Fig.
4B) in the T98G cells. The results suggest a survival role of
JNK1 and JNK2 in glioma brain cancer progression.
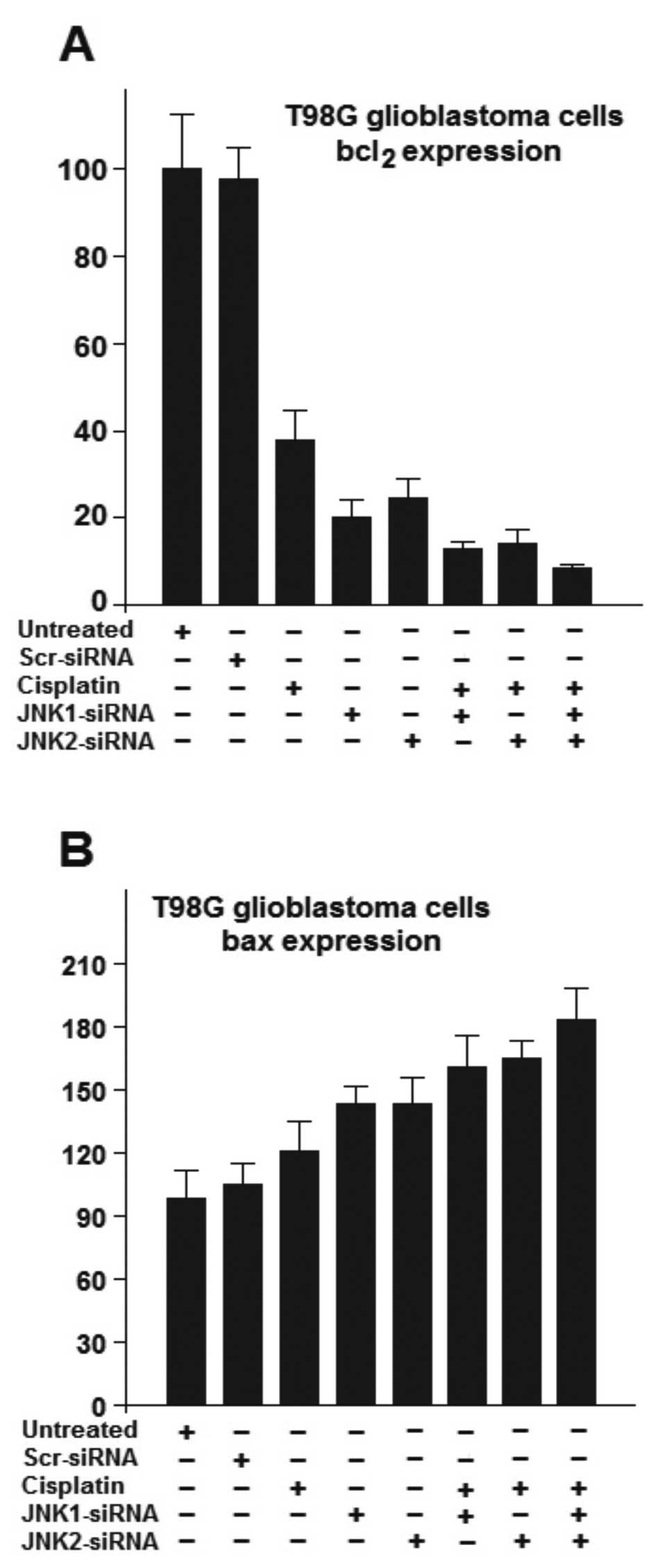
JNK silencing by siRNAs decreases bcl2
expression, yet strongly induces bax expression, sensitizing the
cells to cisplatin-mediated apoptosis
Since other pathways may also influence the survival
or death of T98G glioblastoma cells, we further analyzed the
expression of two genes involved in apoptosis or survival of cells.
We investigate the expression of bcl2 and bax gene products in the
T98G cells treated with siRNAs against JNK1 or JNK2 in the presence
or absence of cisplatin to investigate the expression of these
genes.
The Bcl-2 family proteins regulate a distal step in
an evolutionarily conserved pathway for programmed cell death
(30,31). Several members of the Bcl-2 protein
family can form physical interactions with each other in a
complicated network of homodimers and heterodimers (32–34).
Although many details remain unclear at present, in general, the
ratio between anti-apoptotic proteins such as bcl-2 relative to
pro-cell death proteins such as Bax determines the ultimate
sensitivity of cells to various apoptotic stimuli (35).
As shown in Fig. 5A,
the basal levels of the anti-apoptotic gene bcl2, was strongly
suppressed in the T98G cells treated with cisplatin. Upon treatment
with either JNK1-siRNA or JNK2-siRNA, bcl2 levels decreased to a
greater extend. The results also showed a relatively small
difference in the regulation of bcl2 gene product by JNK1 and JNK2.
Nevertheless, stronger inhibition was reached following treatment
with the combination of JNK1-siRNA, JNK2-siRNA and cisplatin
(Fig. 5A). As shown in Fig. 5B, the basal level of bax gene
expression was altered after treatment of T98G cells with either
cisplatin, JNK1-siRNA or JNK2-siRNA. Nevertheless, stronger
induction of bax expression was reached in cells treated with a
combination of either JNK1-siRNA or JNK2-siRNA and cisplatin
(Fig. 5B). The effects of cisplatin
in both cases (Fig. 5A and B) were
substantially increased in the cells previously treated with siRNAs
against JNK, clearly indicating that siRNA against either JNK1 or
JNK2 sensitized the T98G cells to cisplatin-mediated apoptosis.
Discussion
In the present study, we examined how small
interfering RNAs (siRNAs) against c-Jun-N-terminal kinase (JNK)
affect cell proliferation, DNA repair and susceptibility to
apoptosis in T98G glioblastoma cells treated or not with cisplatin.
JNK activity is strongly induced in a variety of DNA damaging
treatments such as UV irradiation (36), cisplatin (16–19),
camptothecin (4) and promotes
resistance of tumor cells to cisplatin through a mechanism
involving increased DNA repair. Previous studies have shown that a
mutant version of c-Jun (mJun), a direct target of the JNK pathway,
can inhibit cell proliferation in several types of tumor cells
(37,38). In this case, mJun can enter into
AP-1 complexes but fails to be activated by JNK due to the alanine
replacement at the two critical sites of serine phosphorylation
(39). In addition, the effect of
blocking JNK pathway expression by siRNA in prostate and breast
cancer cells has been demonstrated in several studies (17,22,24–26).
In the present study, we examined the role of the JNK pathway
following treatment with siRNAs in the presence or absence of
cisplatin in T98G glioblastoma cancer cells. We showed that siRNAs
against JNKs (siRNA-JNK1/2) induced decreased DNA repair and
sensitizes the T98G glioblastoma cells to the DNA damaging drug
cisplatin. These results were associated with reduced cell survival
and loss of anchorage-independent colony formation ability. The
results indicate that effective inhibition of the JNK pathway
significantly sensitizes glioblastoma cells to cisplatin, a
compound of proven clinical value whose spectrum of application is
limited by resistance phenomena, further supporting the notion that
the JNK pathway may promote survival by limiting genome
instability. The loss of p53 in T98G glioblastoma cells would
further enhance survival owing to a downregulated apoptotic
response. Nevertheless, restoration of p53 in T98G cells through
gene transfer results in partial G1 arrest or apoptosis (40). Our results demonstrated that cells
treated with JNK-siRNAs exhibit an increased apoptosis and elevated
DNA fragmentation compared with untreated T98G cells. Thus, the
success or failure of DNA repair in T98G cells may probably play a
role in determining the consequences of blocking JNK expression in
these tumor cells, suggesting that downregulation of JNK expression
could be used to enhance the therapeutic effect of cisplatin in
glioblastoma cells. As we have proposed, activation of JNK and loss
of p53 may represent independent mechanisms by which tumor cells
undergo progression, induce genome instability and ensure survival.
By promoting DNA repair, the JNK pathway may help to limit genome
instability and the associated DNA damage to levels compatible with
survival. This result, combined with inhibition of JNK through
siRNAs against JNK1/2, would therefore constitute a tumor-specific
therapeutic intervention, since such a strategy exploits the DNA
damage resulting from the intrinsic genome instability unique to
cancer cells. Such strategies would tend to have synergistic
antitumor effects and enhance the potential of DNA damaging
chemotherapies as well. Our observations indicate that effective
inhibition of the JNK pathway utilizing siRNAs significantly
sensitized T98G glioblastoma cells to cisplatin.
Acknowledgements
This study was supported by the Intramural Regular
Research Grant from the Universidad de Tarapacá, UTA-6710-14.
References
|
1
|
Liu Y, Gorospe M, Holbrook NJ and Anderson
CW: Post-translational mechanisms leading to mammalian gene
activation in response to genotoxic stress. DNA Damage and Repair.
Nickoloff JA and Hoekstra MF: 2. Humana Press Inc; Totowa, NJ: pp.
263–298. 1998, View Article : Google Scholar
|
|
2
|
Whitmarsh AJ and Davis RJ: Transcription
factor AP-1 regulation by mitogen-activated protein kinase signal
transduction pathways. J Mol Med. 74:589–607. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tournier C, Hess P, Yang DD, Xu J, Turner
TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA and Davis RJ:
Requirement of JNK for stress-induced activation of the cytochrome
c-mediated death pathway. Science. 288:870–874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Parra E and Ferreira J: The effect of
siRNA-Egr-1 and camptothecin on growth and chemosensitivity of
breast cancer cell lines. Oncol Rep. 22:1159–1165. 2010.
|
|
5
|
Dorigo O, Turla ST, Lebedeva S and Gjerset
RA: Sensitization of rat glioblastoma multiforme to cisplatin in
vivo following restoration of wild-type p53 function. J Neurosurg.
88:535–540. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ru P, Steele R, Hsueh EC and Ray RB:
Anti-miR-203 upregulates SOCS3 expression in breast cancer cells
and enhances cisplatin chemosensitivity. Genes Cancer. 2:720–727.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Keshava R, Jothi M, Gope M and Gope R:
Functional modulation of the p53 gene and its protein in human
brain tumors. Ann Neurosci. 15:32008.
|
|
8
|
Bullock AN and Fersht AR: Rescuing the
function of mutant p53. Nat Rev Cancer. 1:68–76. 2001. View Article : Google Scholar
|
|
9
|
Raitano AB, Halpern JR, Hambuch TM and
Sawyers CL: The Bcr-Abl leukemia oncogene activates Jun kinase and
requires Jun for transformation. Proc Natl Acad Sci USA.
92:11746–11750. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dickens M, Rogers JS, Cavanagh J, Raitano
A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL and Davis RJ: A
cytoplasmic inhibitor of the JNK signal transduction pathway.
Science. 277:693–696. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi CS, Tuscano JM, Witte ON and Kehrl JH:
GCKR links the Bcr-Abl oncogene and Ras to the stress-activated
protein kinase pathway. Blood. 93:1338–1345. 1999.PubMed/NCBI
|
|
12
|
Atfi A, Prunier C, Mazars A, Défachelles
AS, Cayre Y, Gespach C and Bourgeade MF: The oncogenic TEL/PDGFR β
fusion protein induces cell death through JNK/SAPK pathway.
Oncogene. 18:3878–3885. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bost F, McKay R, Dean N and Mercola D: The
JUN kinase/stress-activated protein kinase pathway is required for
epidermal growth factor stimulation of growth of human A549 lung
carcinoma cells. J Biol Chem. 272:33422–33429. 1997. View Article : Google Scholar
|
|
14
|
Nielsen C, Thastrup J, Bøttzauw T,
Jäättelä M and Kallunki T: c-Jun NH2-terminal kinase 2
is required for Ras transformation independently of activator
protein 1. Cancer Res. 67:178–185. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu S, Loke HN and Rehemtulla A:
Ultraviolet radiation-induced apoptosis is mediated by Daxx.
Neoplasia. 4:486–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mansouri A, Ridgway LD, Korapati AL, Zhang
Q, Tian L, Wang Y, Siddik ZH, Mills GB and Claret FX: Sustained
activation of JNK/p38 MAPK pathways in response to cisplatin leads
to Fas ligand induction and cell death in ovarian carcinoma cells.
J Biol Chem. 278:19245–19256. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parra E and Ferreira J: Modulation of the
response of prostate cancer cell lines to cisplatin treatment using
small interfering RNA. Oncol Rep. 30:1936–1942. 2013.PubMed/NCBI
|
|
18
|
Persons DL, Yazlovitskaya EY, Cui W and
Pelling JC: Cisplatin-induced activation of mitogen-activated
protein kinases in ovarian carcinoma cells: inhibition of
extracellular signal-regulated kinase activity increases
sensitivity to cisplatin. Clin Cancer Res. 5:1007–1014.
1999.PubMed/NCBI
|
|
19
|
Sánchez-Perez I, Murguía JR and Perona R:
Cisplatin induces a persistent activation of JNK that is related to
cell death. Oncogene. 16:533–540. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Costa-Pereira AP, McKenna SL and Cotter
TG: Activation of SAPK/JNK by camptothecin sensitizes
androgen-independent prostate cancer cells to Fas-induced
apoptosis. Br J Cancer. 82:1827–1834. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Woo RA, McLure KG, Lees-Miller SP,
Rancourt DE and Lee PW: DNA-dependent protein kinase acts upstream
of p53 in response to DNA damage. Nature. 394:700–704. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Still IH, Hamilton M, Vince P, Wolfman A
and Cowell JK: Cloning of TACC1, an embryonically expressed,
potentially transforming coiled coil containing gene, from the 8p11
breast cancer amplicon. Oncogene. 18:4032–4038. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Parra E, Gutiérrez L and Ferreira J:
Increased expression of p21Waf1/Cip1 and JNK with costimulation of
prostate cancer cell activation by an siRNA Egr-1 inhibitor. Oncol
Rep. 30:911–916. 2013.PubMed/NCBI
|
|
24
|
Parra E: Inhibition of JNK-1 by small
interfering RNA induces apoptotic signaling in PC-3 prostate cancer
cells. Int J Mol Med. 30:923–930. 2012.PubMed/NCBI
|
|
25
|
Parra E and Ferreira J: Knockdown of the
c-Jun-N-terminal kinase expression by siRNA inhibits MCF-7 breast
carcinoma cell line growth. Oncol Rep. 24:1339–1345. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Cell Biol. 19:142–149. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vucic D, Dixit VM and Wertz IE:
Ubiquitylation in apoptosis: a post-translational modification at
the edge of life and death. Nat Rev Mol Cell Biol. 12:439–452.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bertrand MJ, Milutinovic S, Dickson KM, Ho
WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ and
Barker PA: cIAP1 and cIAP2 facilitate cancer cell survival by
functioning as E3 ligases that promote RIP1 ubiquitination. Mol
Cell. 30:689–700. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fulda S: Regulation of cell migration,
invasion and metastasis by IAP proteins and their antagonists.
Oncogene. 33:671–676. 2014. View Article : Google Scholar
|
|
30
|
Gascoyne RD, Krajewska M, Krajewski S,
Connors JM and Reed JC: Prognostic significance of Bax protein
expression in diffuse aggressive non-Hodgkin’s lymphoma. Blood.
90:3173–3178. 1997.PubMed/NCBI
|
|
31
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar
|
|
32
|
Zha H, Aimé-Sempé C, Sato T and Reed JC:
Proapoptotic protein Bax heterodimerizes with Bcl-2 and
homodimerizes with Bax via a novel domain (BH3) distinct from BH1
and BH2. J Biol Chem. 271:7440–7444. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reed JC: Mechanisms of Bcl-2 family
protein function and dysfunction in health and disease. Behring
Inst Mitt. 97:72–100. 1996.PubMed/NCBI
|
|
34
|
Metcalfe AD, Gilmore A, Klinowska T,
Oliver J, Valentijn AJ, Brown R, Ross A, MacGregor G, Hickman JA
and Streuli CH: Developmental regulation of Bcl-2 family protein
expression in the involuting mammary gland. J Cell Sci.
112:1771–1783. 1999.PubMed/NCBI
|
|
35
|
Basu A and Haldar S: The relationship
between Bcl2, Bax and p53: consequences for cell cycle progression
and cell death. Mol Hum Reprod. 4:1099–1109. 1998. View Article : Google Scholar
|
|
36
|
Fritz G and Kaina B: Activation of c-Jun
N-terminal kinase 1 by UV irradiation is inhibited by wortmannin
without affecting c-jun expression. Mol Cell Biol. 19:1768–1774.
1999.PubMed/NCBI
|
|
37
|
Smeal T, Binetruy B, Mercola DA, Birrer M
and Karin M: Oncogenic and transcriptional cooperation with Ha-Ras
requires phosphorylation and c-Jun on serines 63 and 73. Nature.
354:494–496. 1991. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brown PH, Chen TK and Birrer MJ: Mechanism
of action of a dominant-negative mutant of c-Jun. Oncogene.
9:791–799. 1994.PubMed/NCBI
|
|
39
|
Shaulian E and Karin M: AP-1 in cell
proliferation and survival. Oncogene. 20:2390–2400. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang LC, Clarkin KC and Wahl GM:
Sensitivity and selectivity of the DNA damage sensor responsible
for activating p53-dependent G1 arrest. Proc Natl Acad
Sci USA. 93:4827–4832. 1996. View Article : Google Scholar
|