Introduction
Breast cancer is one of the most common cancers
affecting women worldwide, accounting for almost 23% of all cancers
diagnosed in women (1–2). Although early detection, precise
resection using wide margins and systematic adjuvant therapy have
improved survival, breast cancer remains one of the leading causes
of death among women (3).
Conventional chemotherapy is still one of the major approaches to
the treatment of breast cancer, particularly for cases of breast
cancer in stages 2–4, and is particularly beneficial in estrogen
receptor-negative (ER−) disease (4). Most chemotherapy medications work by
destroying fast-growing and/or fast-replicating cancer cells,
either by causing DNA damage upon replication or by other
mechanisms.
Autophagy, also termed as type II programmed cell
death (PCD), is a genetically programmed, evolutionarily conserved
process that degrades long-living cellular proteins and organelles,
including the endoplasmic reticulum, Golgi apparatus and
mitochondria (5). Autophagy is
important in the normal development and cellular response to
environmental stimuli. In addition, autophagy participates in
numerous diseases, including bacterial and viral infections,
neurodegenerative disorders, cardiovascular diseases and cancers
(5). Autophagy exhibits either a
protumorigenic or antitumorigenic function, depending on the cell
type, developmental stage of cancer and stimulator.
Mechanistically, autophagy begins with sequestering cytoplasmic
proteins or organelles in a membrane vacuole to form an
autophagosome. Autophagosome formation is mediated by a set of
evolutionarily conserved autophagy-related proteins (ATG proteins)
(6). Previously, we demonstrated
that stimulation of autophagy could be a potential strategy for the
treatment of breast cancer (5).
In the present study, we investigated whether
autophagy could be involved in chemotherapy against breast cancer
and whether this participation could be protumorigenic or
antitumorigenic. Moreover, mechanisms involved in this autophagic
cell death by chemotherapeutics were also investigated.
Materials and methods
Cell line and cultures
The human breast cancer cell lines MDA-MB-231 and
MDA-MB-453 were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA). Cells were maintained in RPMI-1640
medium, supplemented with 10% fetal calf serum (both from Gibco,
Grand Island, NY, USA), 100 U/ml penicillin and 100 mg/ml
streptomycin. The cultures were incubated at 37°C in a humidified
atmosphere with 5% CO2. Cells were passaged every 2–3
days to obtain exponential growth.
Reagents
3-Methyladenine (3-MA) was purchased from Sigma (St.
Louis, MO, USA). Epirubicin was purchased from Pfzer (Shanghai,
China). Docetaxel and methotrexate (MTX) were purchased from
Jiangsu Hengrui Medicine Co., Ltd. (Liangyungang, Jiangsu, China).
Cyclophosphamide (CTX) was purchased from Baxter Oncology GmbH
(Kantstrasse, Halle, Germany). Fluorouracil (5-FU) was purchased
from Shanghai Xudong Haipu Pharmaceutical Co., Ltd. (Shanghai,
China). Cisplatin was purchased from Nanjing Pharmaceutical Factory
Co., Ltd. (Nanjing, Jiangsu, China).
MTT assay
Cellular growth was evaluated by methyl thiazolyl
tetrazolium (MTT) assay (7). Cells
were seeded into 24-well tissue culture plates at 5×104
cells/well. After treatment, MTT (Sigma, St. Louis, MO, USA) was
added to each well to a final concentration of 0.5 mg/ml, followed
by incubation at 37°C for 4 h. The medium was then removed, and 800
µl of dimethylsulfoxide (DMSO) was added/well. The
absorbance in each well was measured at 490 nm using a microplate
ELISA reader (Bio-Rad Laboratories, Hercules, CA, USA). The
inhibition rate was calculated as follows: Inhibition rate = [(mean
control absorbance − mean experimental absorbance)/mean control
absorbance] × 100 (%). The concentration that caused 50% growth
inhibition (IC50) was calculated by the modified
Kärber’s method (7) according to
the formula: IC50 = lg−1[Xk-i (Σp − 0.5)], in
which Xk represents the logarithm of the highest drug
concentration; i, is that of the ratio of the adjacent
concentration; and Σp, is the sum of the percentage of growth
inhibition at various concentrations.
Autophagy assay using the DsRed-LC3
reporter
Autophagy is dependent on the presence of
autophagosomes and autolysosomes (8). LC3, the mammalian ontology of ATG8, is
a credible marker for autophagosomes (9). During the formation of autophagosomes,
LC3 will form punctate structures within the cytoplasm that
correspond to autophagic vesicles (8,9). To
develop an autophagy reporter, we fused the LC3 cDNA to the C
terminus of DsRed as previously described (10). To produce stable cell lines
continuously reporting autophagy activity, recombinant lentiviruses
expressing the DsRed-LC3 reporter were generated and used to infect
the cells. The LC3 puncta were visualized with an Olympus BX51
microscope (Tokyo, Japan). DNA was counterstained using DAPI
(Sigma).
Cell cycle analysis
Cell cycle analysis using propidium iodide (PI) was
performed as previously described (7). Prior to treatment, cells were
synchronized in the cell cycle by serum starvation for 24 h. After
treatment, the cells were fixed in 80% ice cold ethanol, and
incubated with 0.5% Triton X-100 solution containing 1 mg/ml RNase
A at 37°C for 30 min. PI (Sigma) was added to a final concentration
of 50 µg/ml followed by a 30-min incubation in the dark.
Cellular DNA content was analyzed by a fluorescence-activated cell
sorter (FACS; Becton-Dickinson, Franklin Lakes, NJ, USA). Data were
processed using ModFit LT software (Verity Software House, Inc.,
Topsham, ME, USA).
Real-time PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
protocol. After spectrophotometric quantification, 1 µg
total RNA in a final volume of 20 µl was used for reverse
transcription with PrimeScript RT reagent kit (Takara, Otsu, Shiga,
Japan) according to the manufacturer’s protocol. Aliquots of cDNA
corresponding to equal amounts of RNA were used for quantification
of mRNA by real-time PCR using the LightCycler 96 real-time
quantitative PCR detection system (Roche Diagnostics, Indianapolis,
IN, USA). The reaction system (25 µl) contained the
corresponding cDNA, forward and reverse primers, and SYBR-Green PCR
Master Mix (Roche Diagnostics). All data were analyzed using
β-actin gene expression as an internal standard. The specific
primers are presented in Table
I.
 | Table IPrimers. |
Table I
Primers.
| Genes | Sense (5′–3′) | Antisense
(3′–5′) | Product size
(bp) |
|---|
| AMBRA1 |
TCGGCAACAACATCATCGTC |
CTGGGAAGGGAAACAGGAAT | 253 |
| ATG3 |
ACTGATGCTGGCGGTGAAG |
GTGGCAGATGAGGGTGATTTT | 208 |
| ATG4A |
GGGATGTATGCTACGCTGTGG |
CACCCATTTGTGCCATTTGAT | 182 |
| ATG4B |
TAGGCCGAGATTGGAGGTG |
GCGCTATCTGGTGAATGGAGT | 114 |
| ATG4C |
TTTCCCTCTTGAGACATTCCAC |
GGTGATTTCTTCAGAAGCTCGTTT | 130 |
| ATG4D |
AGCCGAGTGGAAGTCTGTGGT |
AGCAGGAAGTCATCTTGGTAGCC | 177 |
| ATG5 |
ATCAGGTTTGGTGGAGGCA |
GGTTTAATGATGGCAGTGGAGG | 140 |
| ATG7 |
CCAAGGTCAAAGGACGAAGAT |
GTACGGTCACGGAAGCAAAC | 156 |
| ATG9A |
CCTTACCTCACCCGCAGTTC |
GGCAGCAAAGTATTTCCATCC | 241 |
| ATG9B |
CAGCCGAACACCAAAGTCAT |
GCTTCCCTTCCCTCTGTAAATC | 239 |
| ATG10 |
ACACTATTACGCAACAGGAACATC |
CTCAGAGGTAGATTCAGCCCAAC | 184 |
| ATG12 |
ACCCATTGCTCCTACTTGTTACTAT |
TTTCTGCCTGGTGGACTGC | 256 |
| ATG13 |
TGTCATTGCTGCTGAAGTCCC |
CCCACTGTCCCAACACGAACT | 169 |
| ATG14 |
GTGAGCCGAGATTGTGCCAT |
GGTAATAATGCCTGTTAGGACTCTTTC | 267 |
| ATG16L1 |
GGGATTTCTGAAGATTTGACTGAG |
ACCGACTTTGGAAGGACGAG | 248 |
| ATG16L2 |
ACCGGACAGTGAAGGAGTGG |
GGATCTTCTGGTCATTGTGGC | 129 |
| Beclin1 |
GCTGGATGATGAGCTGAAGAGT |
GTGCCAGATGTGGAAGGTTG | 109 |
| DRAM1 |
TCGTCAGCCGCCTTCATTAT |
CGAAACATCCCACCAATCC | 262 |
| GABARAP |
TCAAACACCACCTCCCTTATTC |
TGCCAACTCCACCATTACCC | 94 |
| GABARAPL1 |
GCCTGATCTGGACAAGAGGAAGT |
ATGGTAGCACTGGTGGGAGG | 150 |
| GABARAPL2 |
GTTGACATTGACAAACGGAAGTAC |
CATAGTTAGGCTGGACTGTGGG | 150 |
| HDAC6 |
CATCCGAACTCATACTCCTGTGC |
TAAGACTGTGCTGGGCGTGAT | 136 |
| IRGM |
CTTGCTGCTGCTCATTCTTTG |
CGAGTCTGGAGTTGTTCGTTTC | 133 |
| LAMP1 |
ACAACACGACGGTGACAAGG |
TTCATCCCGAACTGGAAGAGC | 136 |
| MAP1LC3B |
CGCATTTGCCATCACAGTTG |
TAGGAGTCAGGGACCTTCAGC | 94 |
| RAB24 |
CAGAAAGTGGCAGAGGATTACG |
TGACTACCCAAGCCCAGAAAG | 199 |
| RGS19 |
ATCTACACGCTCATGCACCG |
GACAACAACACCTGAAGGGAAC | 175 |
| ULK1 |
CAGCAAAGGCATCATCCACC |
GAAGCCGAAGTCAGCGATC | 115 |
| WIPI1 |
CTACCAACTACCTCCCTACCCAG |
TGTCCACTGGATGACGCAAC | 154 |
Statistical analysis
Each experiment was performed a minimum of three
times. Results are expressed as the mean value ± standard deviation
(SD). Statistical analysis was performed using unpaired Student’s
t-test. The correlations between microarray assays were analyzed
using Spearman’s rank correlation analysis. A P value <0.05 was
considered to indicate a statistically significant result.
Results
Cytotoxicity of chemotherapeutics against
breast cancer cells
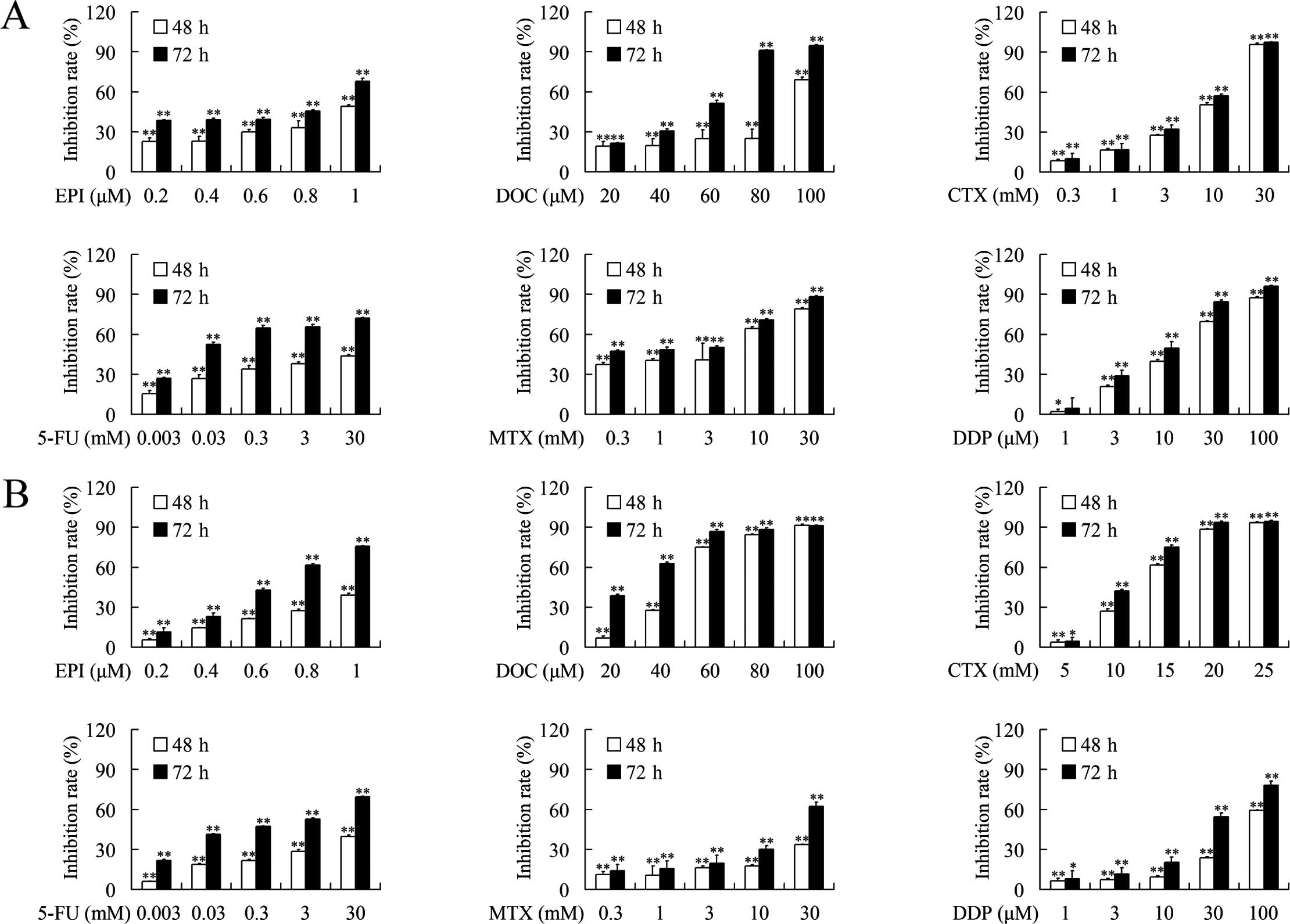
The growth inhibition effects of epirubicin,
docetaxel, MTX, CTX, 5-FU and cisplatin against breast cancer cells
were evaluated using MTT assays. As shown in Fig. 1, epirubicin, docetaxel, MTX, CTX,
5-FU and cisplatin suppressed the growth of MDA-MB-231 and
MDA-MB-453 cells in a dose- and time-dependent manner. The
IC50 values after treatment with each chemotherapeutic
for 48 and 72 h are presented in Table
II.
| Table IIIC50 values of the
chemotherapeutics at 48 and 72 h in the MDA-MB-231 and MDA-MB-453
cells. |
Table II
IC50 values of the
chemotherapeutics at 48 and 72 h in the MDA-MB-231 and MDA-MB-453
cells.
| MDA-MB-231
| MDA-MB-453
|
|---|
| 48 h | 72 h | 48 h | 72 h |
|---|
| EPI | 1.28 µM | 0.60 µM | 2.05 µM | 0.61 µM |
| DOC | 75.35
µM | 45.45
µM | 46.39
µM | 27.38
µM |
| CTX | 5.02 mM | 4.38 mM | 12.04 mM | 10.70 mM |
| 5-FU | 119.33 mM | 89.22
µM | 715.41 mM | 603.47
µM |
| MTX | 2.45 mM | 1.10 mM | 724.39 mM | 17.29 mM |
| DDP | 13.48
µM | 8.61 µM | 225.78
µM | 72.94
µM |
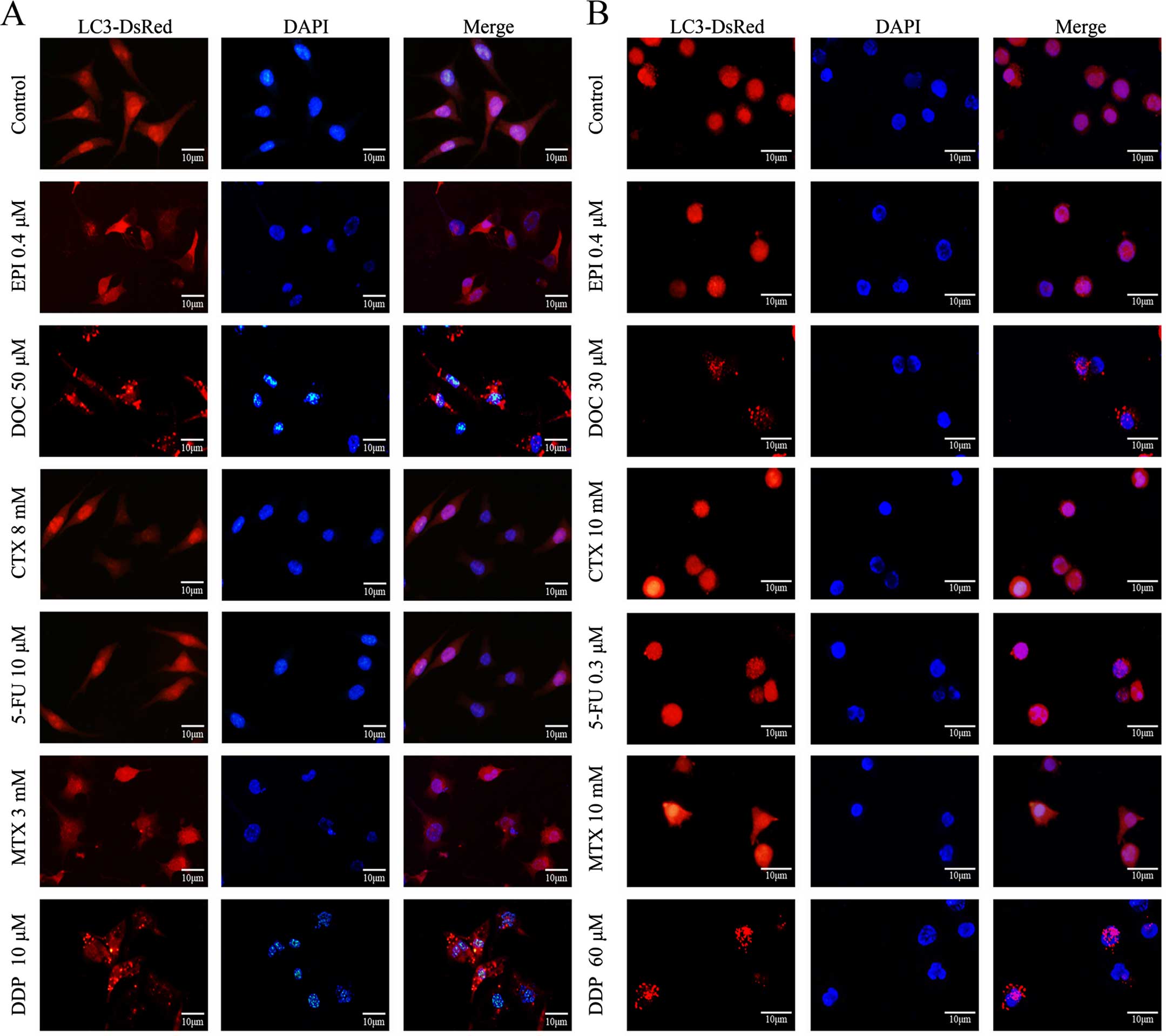
Docetaxel and cisplatin induce autophagy
in breast cancer cells
Using the DsRed-LC3 reporter, increased formation of
LC3 punctate was observed following the treatment with docetaxel or
cisplatin, yet not following other chemotherapeutics, suggesting
docetaxel and cisplatin induced autophagy in the breast cancer
cells (Fig. 2).
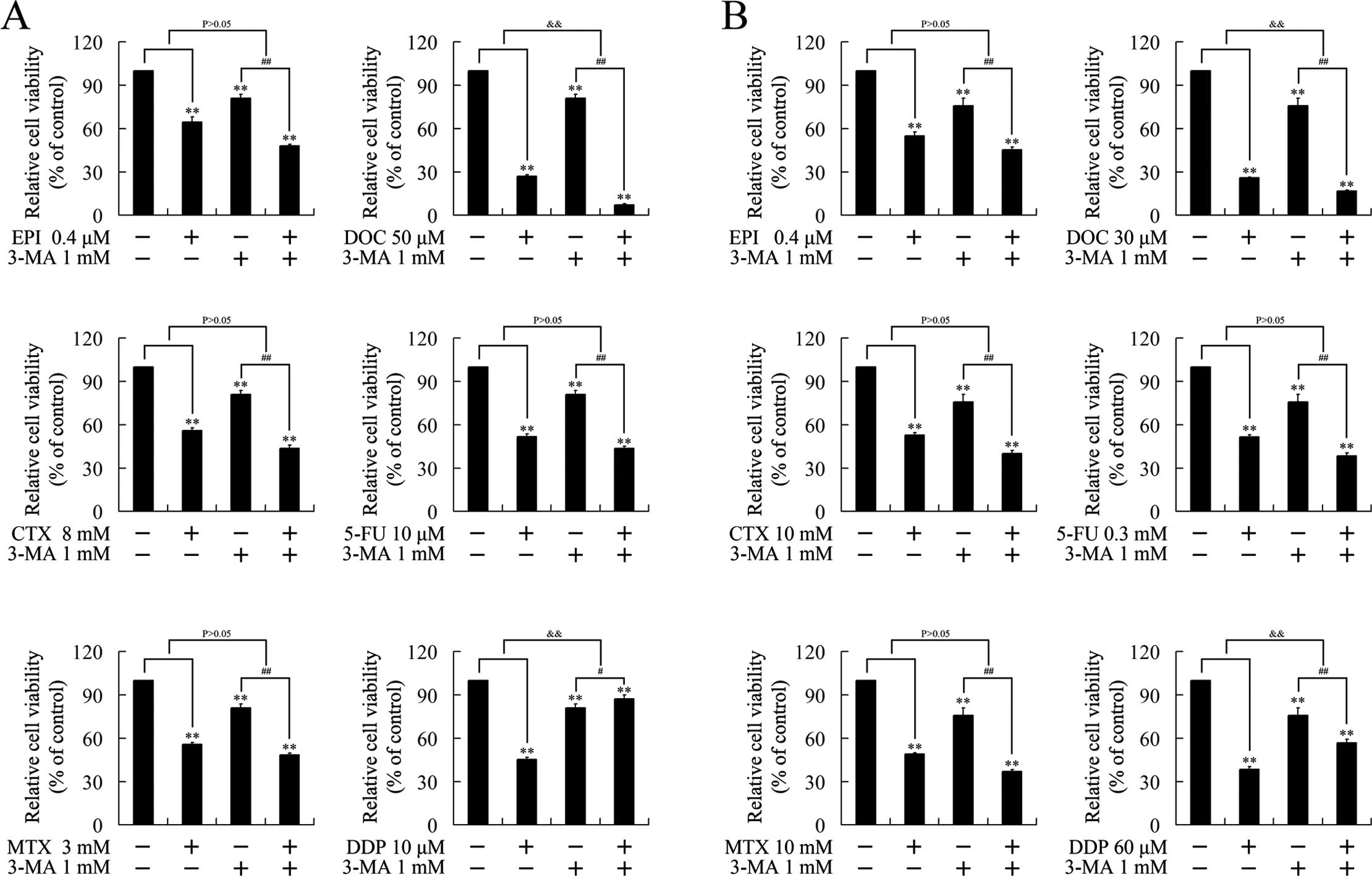
Cisplatin suppresses breast cancer growth
through an autophagy-dependent manner
To verify whether the growth inhibition induced by
chemotherapeutics was executed in an autophagy-dependent manner, we
investigated the effect of 3-MA, a classic autophagy inhibitor, on
the cytotoxicity of the chemotherapeutics. As expected, 3-MA had no
effect on epirubicin, MTX, CTX and 5-FU, consistent with the
undetectable autophagy by these chemotherapeutics. 3-MA attenuated
the cytotoxicity of cisplatin, suggesting that cisplatin suppressed
cell growth in an autophagy-dependent manner. Notably, 3-MA
strengthened the growth inhibition induced by docetaxel, indicating
the autophagy induced by docetaxel impaired its cytotoxicity
(Fig. 3).
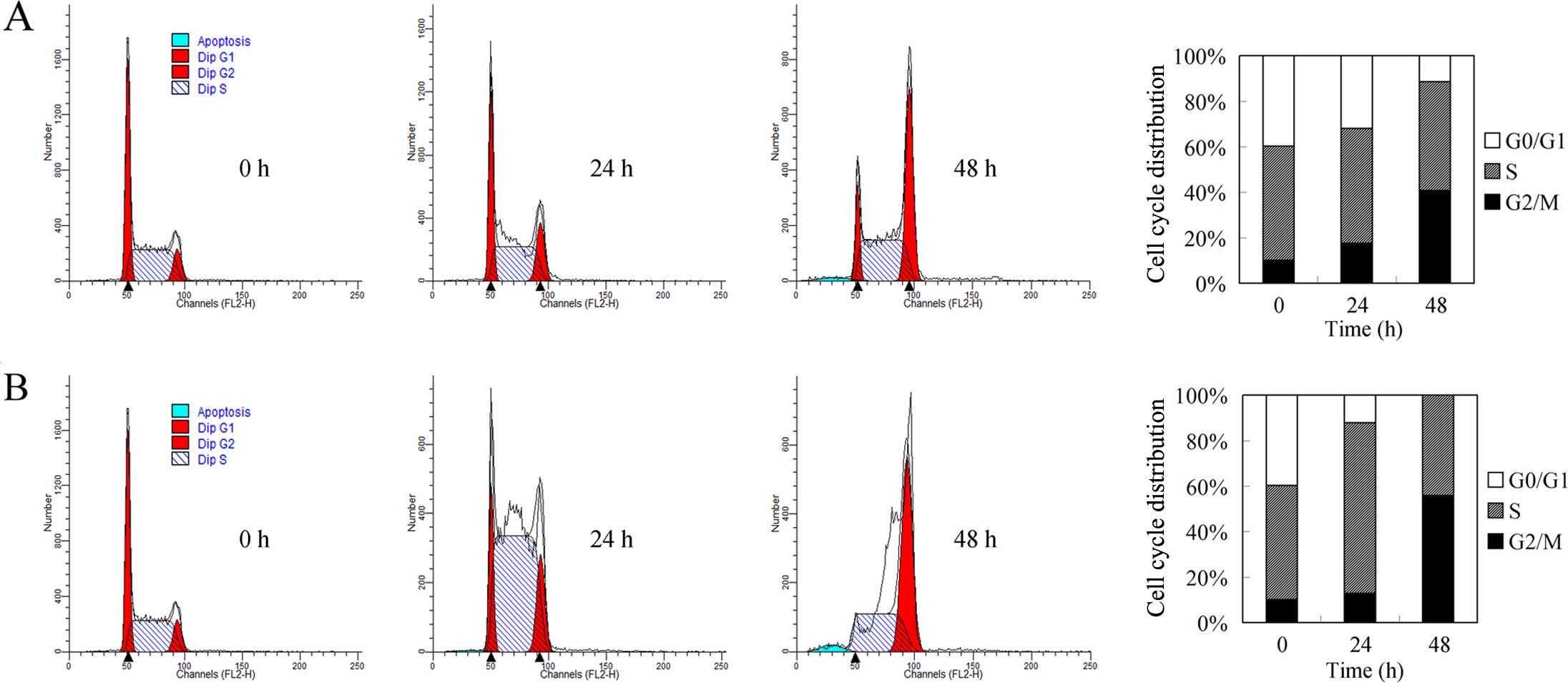
Cell cycle regulation by docetaxel and
cisplatin in breast cancer cells
Cell cycle arrest and autophagy are closely linked
biological processes (11). We
previously reported that autophagy induction results in G2/M cell
cycle arrest. To investigate the mechanism involved in docetaxel
and cisplatin-induced growth repression, we carried out flow
cytometric analysis. As shown in Fig.
4A, docetaxel induced G2/M cell arrest and decreased the
induced accumulation of cells in the S phase (Fig. 4B), consistent with its DNA
crosslinking role (12). However,
with prolonged treatment, an increased G2/M population and
annihilation of the G0/G1 population were noted, suggesting that
mechanisms beyond DNA crosslinking could be involved. Considering
the G2/M population accumulating role of autophagy, we speculated
that autophagic cell death could be involved in the G2/M cell cycle
arrest by cisplatin.
Cisplatin treatment increases the
expression levels of multiple autophagy-related genes
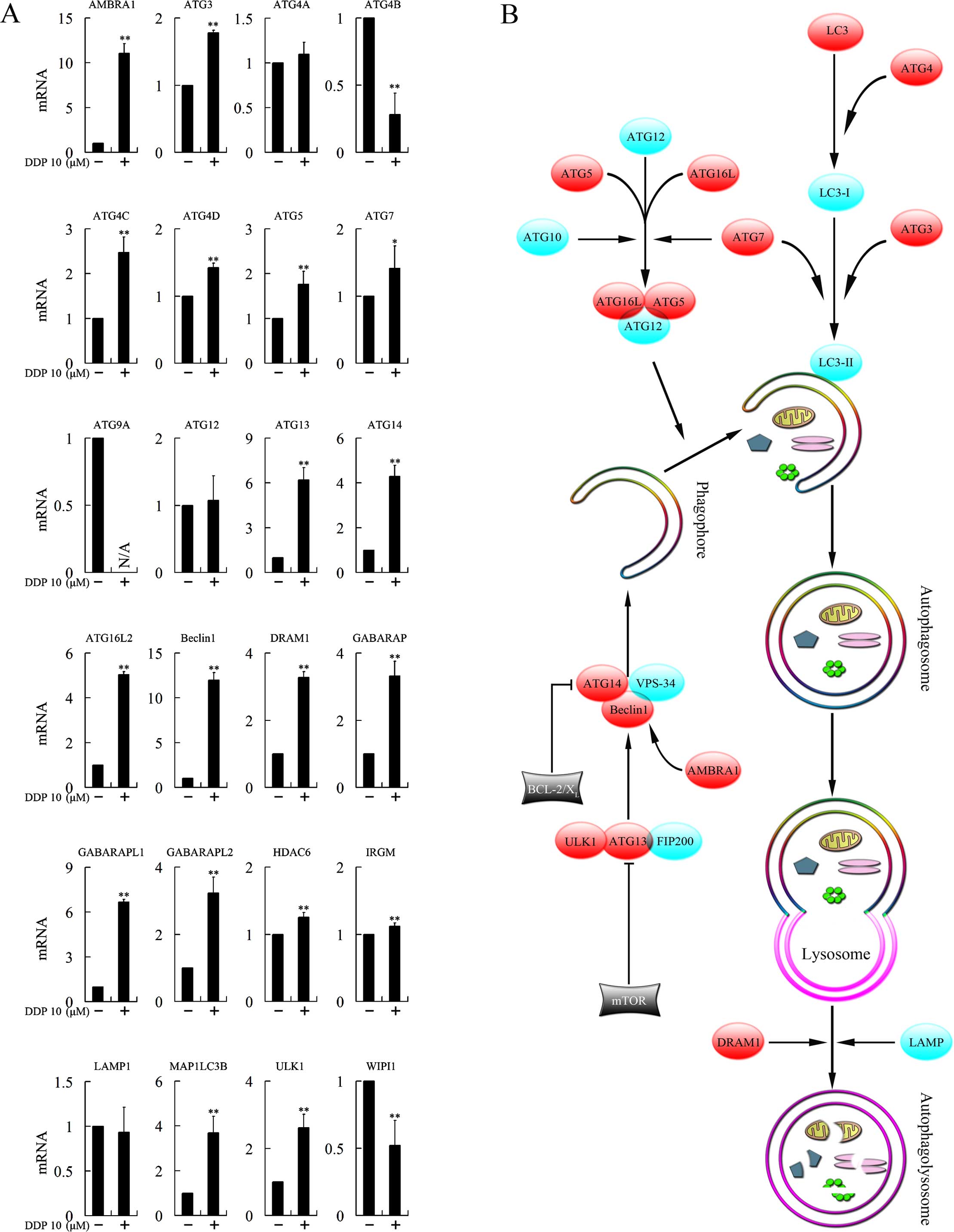
To investigate the mechanism involved in the
autophagy-dependent cytotoxicity of cisplatin, we tested the
expression levels of 29 genes closely associated with autophagy
following treatment with cisplatin. ATG9B, ATG10, ATG16L1, RAB24
and RGS19 were not found to be expressed. Expression of ATG4A,
ATG12 and LAMP1 was not changed. ATG4B, ATG9A and WIPI1 were
downregulated, while AMBRA1, ATG3, ATG4C, ATG4D, ATG5, ATG7, ATG13,
ATG14, ATG16L2, Beclin1, DRAM1, GABARAP, GABARAPL1, GABARAPL2,
HDAC6, IRGM, MAP1LC3B and ULK1 were upregulated, and most of the
upregulations were >1.5-fold (Fig.
5A).
Discussion
Autophagy is a homeostatic and evolutionarily
conserved process that degrades cellular organelles and proteins,
and maintains cellular biosynthesis during nutrient deprivation or
metabolic stress (13). In the
present study, we found that induction of autophagy was involved in
the cytotoxicity of cisplatin. Mechanistically, cisplatin binds to
and causes crosslinking of DNA, which repressed DNA replication and
ultimately triggers apoptosis. Consistent with the repression on
DNA replication, we found that treatment with cisplatin reduced
accumulation of cells at the S phase. Notably, prolonged treatment
increased the population of cells in the G2/M phase, suggesting
that suppression of DNA replication is not the solo mechanism
involved in the cytotoxicity of cisplatin. Considering the close
link between autophagy and G2/M cell cycle arrest (11), we speculated that the autophagic
pathway could be involved in the cisplatin-triggered G2/M cell
cycle arrest and cytotoxicity. Autophagy is a complex process
(Fig. 5B) that is divided into
several distinct phases controlled by the autophagy-related (ATG)
family of proteins (14). Autophagy
begins with the formation of double-membrane vesicles, known as
autophagosomes, that engulf cytoplasmic constituents. The
autophagosome then fuses with lysosomes, where the sequestered
contents undergo degradation and recycling (13). The formation of the autophagosome
could be divided into four stages: induction, nucleation (isolation
of the membrane), elongation and completion of the double-membrane
vesicle. In the present study, we assessed the mRNA expression of a
range of genes closely associated with autophagy following
treatment with cisplatin. Several genes participating in the
formation of the autophagosome was clearly upregulated. These genes
included AMBRA1, ATG3, ATG4C, ATG4D, ATG5, ATG7, ATG13, ATG14,
ATG16L2, Beclin1, DRAM1, GABARAP, GABARAPL1, GABARAPL2, HDAC6,
IRGM, MAP1LC3B and ULK1, suggesting that cisplatin induced
autophagy by affecting multiple steps of autophagosome
formation.
The induction stage of autophagy requires the ULK1
kinase complex, which consists of ULK1 (termed as ATG1 in yeast and
ULK1 in mammals), ATG13 and FIP200 (15). ATG13 binds ULK1 and mediates the
interaction of ULK1 with FIP200. The ULK1 kinase complex is a
critical positive regulator of autophagosome formation. In the
present study, both ULK1 and ATG13 were upregulated after treatment
with cisplatin, suggesting the induction stage of autophagy was
activated.
The vesicle nucleation stage depends on a complex
containing Beclin1 (termed as Atg6 in yeast and Beclin1 in
mammals), VPS34 and ATG14 (16).
Stimulation of this complex generates
phosphatidylinositol-3-phosphate (PI3P), which promotes
autophagosomal membrane nucleation. Decreased expression of Beclin1
has been found in several types of cancers, including non-small
cell lung (17) and breast cancer
(18). Overexpression of Beclin1
represses cell proliferation, whereas knockdown of Beclin1 boosts
cell growth and inhibits apoptosis (17). In our previous study, autophagy
inducer artesunate repressed the growth of breast cancer cells
through overexpression of Beclin1. In the present investigation,
both Beclin1 and ATG14 were found to be overexpressed after
cisplatin treatment. In addition, the activating molecule in
Beclin1-regulated autophagy (AMBRA1), which was found to be a
positive regulator of the Beclin1-dependent programme of autophagy
(19), was overexpressed upon
treatment with cisplatin, suggesting the participation of cisplatin
in the nucleation stage of autophagy.
The elongation stage requires the participation of
two ubiquitin-like protein conjugation systems, ATG12-ATG5 and
LC3-phosphatidylethanolamine (PE). The ATG12-ATG5 conjugation
system is a large multimeric complex, in which ATG12 is conjugated
to ATG5 by ATG7 (an E1-like enzyme) and ATG10 (an E2-like enzyme),
and then forms a complex with ATG16L. In the present study, ATG5
and ATG16L2 were found to be significantly upregulated and
expression of ATG7 was also increased slightly, indicating
promotion of the formation of the ATG12-ATG5 complex.
LC3 (as termed as ATG8) is a credible marker for
autophagosomes. Mammalian LC3 homologs includes MAP1LC3A (LC3A),
MAP1LC3B (LC3B), MAP1LC3C (LC3C), GABARAP, GABARAPL1 and GABARAPL2
(20). During the formation of
autophagosomes, LC3 forms punctate structures within the cytoplasm
that correspond to autophagic vesicles (9). In the LC3-PE conjugation system, LC3
is cleaved by the protease ATG4 to generate the LC3-I form. PE is
conjugated to cleaved LC3 by an ATG7- and ATG3-dependent activation
and transfer cascade to fulfill the conversion of LC3-I (a
cytosolic truncated form) to LC3-II (autophagosomal
membrane-associated, PE-conjugated form) (21). LC3-II binds with newly formed
complete double-membrane autophagosomes and remains on mature
autophagosomes until after fusion with lysosomes into the
autophagolysosomes. Thereby LC3-II is commonly used to monitor
autophagy. In the present study, overexpression of ATG4A, ATG4C,
ATG4D, GABARAP, GABARAPL1, GABARAPL2 and MAPILC3B was observed.
Moreover, damage-regulated modulator of autophagy (DRAM1), a
lysosomal protein that induces autophagy through lysosomal
acidification, fusion of lysosomes with autophagosomes and
clearance of autophagosomes (22),
was shown to be overexpressed as well. Mature autophagosomes then
fuse their external membranes with those from lysosomes to degrade
their cargo and recycle essential biomolecules. Finally, the ATG
protein complexes from matured autophagosomes disintegrate
(23).
The relationship between autophagy and tumorigenesis
has been extensively explored (24,25).
However, whether autophagy is protumorigenic or antitumorigenic is
still contradictory. In fact, autophagy is either cancer-promoting
or cancer-suppressive, depending on the cell type, developmental
stage of cancer and the stimulator (24,25).
On the basis of genetic studies, the tumor-suppressive functions of
autophagy are most apparent during tumor initiation (26). The requirement for autophagy becomes
more apparent in later stages, during which tumor cells cope with
microenvironmental stress (27).
Some environmental cues (starvation, high temperature, low oxygen
and hormonal stimulation) or intracellular stresses (damaged
organelles, accumulation of mutant proteins and microbial invasion)
activate signaling pathways that induce autophagy-promoting cancer
cell survival in these severe environments (28). Inhibition of protumorigenic
autophagy by genetic or pharmacological strategies succeeds in
killing tumor cells and triggering apoptosis in vivo
(13). Furthermore, the combination
of autophagy inhibitors and chemotherapeutics displays synergistic
effects against cancer cells (13).
On the contrary, some effective anticancer agents, such as
rapamycin (29) and nilotinib
(8), have been shown to repress
cancer cell growth through the induction of autophagy. In our
previous study, we confirmed that artesunate, a derivative of the
active component of the Chinese medicinal herb Artemisia
annua L., repressed breast cancer growth through autophagy
induction and exhibited a synergistic anticancer effect with
chemotherapy (5). These
investigations suggest that both inhibition and induction of
autophagy could be potential strategies in cancer treatment
depending on whether the participating autophagy is protumorigenic
or antitumorigenic.
In the present study, both protumorigenic and
antitumorigenic autophagy were observed upon treatment with
chemotherapeutics. Autophagy inhibition strengthened the
cytotoxicity of docetaxel, yet impaired the cytotoxicity of
cisplatin, suggesting the docetaxel-induced autophagy could be
protumorigenic, while the cisplatin-induced autophagy could be
antitumorigenic. In our previous study, we found that artesunate,
an autophagy inducer, did not present a synergistic effect with
docetaxel. This could be explained by the present results, as
docetaxel itself could induce protumorigenic autophagy. Therefore,
therapeutic targeting of the autophagic pathway should take many
factors into consideration, as its chemotherapeutic allies may also
affect autophagy induction and this effect could be either
protumorigenic or antitumorigenic.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81472296, 81101867,
81272542, 81200369 and 81372443), the China International Medical
Foundation (grant no. CIMF-F-H001-057), the Special Foundation of
Clinical Medicine of Jiangsu Provincial Bureau of Science and
Technology (grant no. BL2014039), the Scientific Research Project
of Jiangsu Provincial Bureau of Traditional Chinese Medicine (grant
no. L213236), the Medical Scientific Research Project of Jiangsu
Provincial Bureau of Health (grant no. Z201206), the Special
Foundation of Wu Jieping Medical Foundation for Clinical Scientific
Research (grant nos. 320.6753.1225 and 320.6750.12242), the Science
and Education for Health Foundation of Suzhou for Youth (grant nos.
SWKQ1003 and SWKQ1011), the Science and Technology Project
Foundation of Suzhou (grant nos. SYS201112, SYSD2012137 and
SYS201335), the Science and Technology Foundation of Suzhou
Xiangcheng (grant nos. SZXC2012-70 and XJ201451), and a Project
Founded by the Priority Academic Program Development of Jiangsu
Higher Education Institutions.
Abbreviations:
|
PCD
|
programmed cell death
|
|
DOC
|
docetaxel
|
|
MTX
|
methotrexate
|
|
CTX
|
cyclophosphamide
|
|
5-FU
|
fluorouracil
|
|
DDP
|
cisplatin
|
|
ER
|
estrogen receptor
|
|
ATG protein
|
autophagy-related protein
|
|
3-MA
|
3-methyladenine
|
|
MTT
|
methyl thiazolyl tetrazolium
|
|
DMSO
|
dimethyl sulfoxide
|
|
PI
|
propidium iodide
|
|
SD
|
standard deviation
|
|
PI3P
|
phosphatidylinositol-3-phosphate
|
|
PE
|
phosphatidylethanolamine
|
References
|
1
|
Sasco AJ, Kaaks R and Little RE: Breast
cancer: Occurrence, risk factors and hormone metabolism. Expert Rev
Anticancer Ther. 3:546–562. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
3
|
Tan AA, Mu AK, Kiew LV and Chen Y:
Comparative secretomic and N-glycoproteomic profiling in human
MCF-7 breast cancer and HMEpC normal epithelial cell lines using a
gel-based strategy. Cancer Cell Int. 14:1202014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carlson RW and McCormick B: Update: NCCN
breast cancer Clinical Practice Guidelines. J Natl Compr Canc Netw.
3(Suppl 1): S7–S11. 2005.PubMed/NCBI
|
|
5
|
Chen K, Shou LM, Lin F, Duan WM, Wu MY,
Xie X, Xie YF, Li W and Tao M: Artesunate induces G2/M cell cycle
arrest through autophagy induction in breast cancer cells.
Anticancer Drugs. 25:652–662. 2014.PubMed/NCBI
|
|
6
|
Yu L, McPhee CK, Zheng L, Mardones GA,
Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al: Termination of
autophagy and reformation of lysosomes regulated by mTOR. Nature.
465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li W, Xie L, Chen Z, Zhu Y, Sun Y, Miao Y,
Xu Z and Han X: Cantharidin, a potent and selective PP2A inhibitor,
induces an oxidative stress-independent growth inhibition of
pancreatic cancer cells through G2/M cell-cycle arrest and
apoptosis. Cancer Sci. 101:1226–1233. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu HC, Lin CS, Tai WT, Liu CY, Shiau CW
and Chen KF: Nilotinib induces autophagy in hepatocellular
carcinoma through AMPK activation. J Biol Chem. 288:18249–18259.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen Y, Azad MB and Gibson SB: Methods for
detecting autophagy and determining autophagy-induced cell death.
Can J Physiol Pharmacol. 88:285–295. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sheen JH, Zoncu R, Kim D and Sabatini DM:
Defective regulation of autophagy upon leucine deprivation reveals
a targetable liability of human melanoma cells in vitro and in
vivo. Cancer Cell. 19:613–628. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Capparelli C, Chiavarina B,
Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, Andò S, Howell
A, Martinez-Outschoorn UE, Sotgia F, et al: CDK inhibitors
(p16/p19/p21) induce senescence and autophagy in cancer-associated
fibroblasts, ‘fueling’ tumor growth via paracrine interactions,
without an increase in neoangiogenesis. Cell Cycle. 11:3599–3610.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Plaimee P, Weerapreeyakul N, Barusrux S
and Johns NP: Melatonin potentiates cisplatin-induced apoptosis and
cell cycle arrest in human lung adenocarcinoma cells. Cell Prolif.
48:67–77. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: Therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Carew JS, Kelly KR and Nawrocki ST:
Autophagy as a target for cancer therapy: New developments. Cancer
Manag Res. 4:357–365. 2012.PubMed/NCBI
|
|
15
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
FU LL, Cheng Y and Liu B: Beclin-1:
Autophagic regulator and therapeutic target in cancer. Int J
Biochem Cell Biol. 45:921–924. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang W, Fan H, Zhou Y, Duan P, Zhao G and
Wu G: Knockdown of autophagy-related gene BECLIN1 promotes cell
growth and inhibits apoptosis in the A549 human lung cancer cell
line. Mol Med Rep. 7:1501–1505. 2013.PubMed/NCBI
|
|
18
|
Negri T, Tarantino E, Orsenigo M, Reid JF,
Gariboldi M, Zambetti M, Pierotti MA and Pilotti S: Chromosome band
17q21 in breast cancer: Significant association between beclin
1loss and HER2/NEU amplification. Genes Chromosomes Cancer.
49:901–909. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pattingre S, Espert L, Biard-Piechaczyk M
and Codogno P: Regulation of macroautophagy by mTOR and Beclin 1
complexes. Biochimie. 90:313–323. 2008. View Article : Google Scholar
|
|
20
|
Tanida I, Ueno T and Kominami E: In vitro
assays of lipidation of mammalian Atg8 homologs. Curr Protoc Cell
Biol. 64:11.20:11.20.1–11.20.13. 2014.
|
|
21
|
Ravikumar B, Sarkar S, Davies JE, Futter
M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M,
Korolchuk VI, Lichtenberg M, Luo S, et al: Regulation of mammalian
autophagy in physiology and pathophysiology. Physiol Rev.
90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Crighton D, Wilkinson S, O’Prey J, Syed N,
Smith P, Harrison PR, Gasco M, Garrone O, Crook T and Ryan KM:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar :
|
|
24
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh
H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al:
Promotion of tumorigenesis by heterozygous disruption of the beclin
1 autophagy gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Debnath J: The multifaceted roles of
autophagy in tumors-implications for breast cancer. J Mammary Gland
Biol Neoplasia. 16:173–187. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roy S and Debnath J: Autophagy and
tumorigenesis. Semin Immunopathol. 32:383–396. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bao XX, Xie BS, Li Q, Li XP, Wei LH and
Wang JL: Nifedipine induced autophagy through Beclin1 and mTOR
pathway in endometrial carcinoma cells. Chin Med J. 125:3120–3126.
2012.PubMed/NCBI
|
|
29
|
Zhu BS, Xing CG, Lin F, Fan XQ, Zhao K and
Qin ZH: Blocking NF-kappaB nuclear translocation leads to
p53-related autophagy activation and cell apoptosis. World J
Gastroenterol. 17:478–487. 2011. View Article : Google Scholar : PubMed/NCBI
|