Introduction
Gastric cancer (GC) is a commonly diagnosed cancer
with a low survival rate, resulting in its status as one of the
leading causes of cancer-related death. Although surgical tumor
removal is highly effective for the treatment of early-stage GC
(1), many cases are diagnosed at an
advanced stage and cancer recurrence after complete resection is
common. Therefore, chemotherapy is an important option for the
treatment of unresectable or metastasized cancers. Recently,
considerable progress has been achieved in chemotherapeutics, and
various cisplatin-based regimens have shown promising anticancer
outcomes in GC. However, the use of anticancer drugs can give rise
to the drug resistance of cancer cells. In many cases, once cancer
cells acquire drug resistance to one type of drug, they tend to
show cross-resistance to an array of drugs with different
structures and cellular targets. Thus, the acquisition of drug
resistance is a serious obstacle to the success of
chemotherapy.
The development of drug resistance often results
from reduced drug accumulation due to the overexpression of
membrane efflux pumps, such as the canalicular multispecific
organic anion transporter or P-type adenosine triphosphatase
(ATP7B) (2,3). To date, various efforts have been made
to reverse drug resistance with inhibitors to these membrane pumps
(4), but the inhibitors have not
proven clinically useful without side effects. Furthermore,
chemotherapeutic drug resistance is a complex phenomenon, which
also stems from changes in multiple systems including the
upregulation of survival pathways, such as AKT activation and
ERK-dependent MKP-1 induction (2,5,6), and
the suppression of induced death pathways by defects in the
apoptotic pathway or the enhancement of the antioxidant system and
the DNA repair pathway (7,8). Therefore, various strategies to
overcome resistance by targeting these signaling pathways are being
currently evaluated in the field of cancer research.
The hydrophilic bile acid, ursodeoxycholic acid
(UDCA), has been regarded as a suppressor of gastrointestinal
tumors. In addition, UDCA was implicated in the prevention of
colonic cancer through cell cycle arrest and the suppression of
oncogenic factors including Ras and COX-2 (9,10). In
our previous study, we also showed that UDCA performs a suppressive
role in tumor progression in gastric carcinoma cells by inducing
apoptosis (11,12). However, the role of UDCA in
drug-resistant cells is unclear. Our aim in the present study was
to understand the underlying mechanisms involved in the effects of
UDCA on a cisplatin-resistant sub-cell line of SNU601 gastric
cancer cells to aid future progress toward the circumvention of
chemoresistance.
Materials and methods
Cell culture and dosing
The SNU601 human gastric cancer cell line was
obtained from the Korea Cell Line Bank, and the cisplatin-resistant
SNU601 cells (SNU601/R) were a gift from Professor C.H. Choi
(Department of Pharmacology, Chosun University, Korea). The cells
were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% (v/v) fetal bovine serum, and 1% PS at 37°C
in a 5% CO2 atmosphere. Drug treatment of the cells was
performed by adding 500–1000 µM UDCA (ICN Biomedicals, Santa
Ana, CA, USA), 10–50 µM oxaliplatin (L-OHP; Boryung
Pharmaceutical, Korea), 3–10 µM etoposide (Sigma-Aldrich),
or 2–10 ng/ml recombinant human TNF-related apoptosis-inducing
ligand (rhTRAIL), a gift from T.H. Kim (Department of Biochemistry
and Molecular Biology, Chosun University, Korea) in the culture
medium and incubating it for 48 h. Unless specified, drugs were
purchased from Calbiochem (San Diego, CA, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
viability assays
For performance of the MTT assay, cells were plated
in the wells of a 96-well plate at a density of 1×104
cells/well, incubated for 24 h, and then treated with the drugs for
48 h. The MTT solution (0.5 mg/ml) was added to the wells and
incubated at 37°C in a CO2 incubator for 4 h. The plates
were centrifuged at 600 x g for 10 min, and the culture medium was
removed. The cells were solubilized using dimethyl sulfoxide (DMSO)
and the solubilized formazan product was quantified using an
enzyme-linked immunosorbent assay (ELISA) plate reader at 595 nm.
The absorbance of the untreated cells was designated as 100%, and
the cell survival was expressed as a percentage of this value.
Analysis of apoptosis
Treated cells were stained with 1 µg/ml
Hoechst 33342 (HO) for 15 min at room temperature in the dark.
Next, both the floating and attached cells were collected and
centrifuged. The pooled cell pellets were washed with ice-cold
phosphate-buffered saline (PBS), fixed in 3.7% formaldehyde on ice,
washed and resuspended with PBS, and then a fraction of the
suspension was centrifuged in a cytospinner (Shandon, Thermo Fisher
Scientific, Waltham, MA, USA). Slides were prepared, air dried,
mounted in anti-fade solution, and observed under a fluorescence
microscope (DM5000; Leica, Washington, NY, USA) as described
elsewhere. Any condensed/fragmented nuclei were assessed as
apoptotic cells. A total of 500 cells distributed across random
microscopic viewing fields were counted, and the number of
apoptotic cells was expressed as a percentage of the total number
of cells counted.
Immunoblotting
Equal amounts of protein extracts were
electrophoretically separated using 10–12% SDS-PAGE and transferred
to a nitrocellulose membrane using a standard technique. Antibodies
were used to probe for cleaved cysteinyl aspartate-specific
protease-3 (caspase-3; Cell Signaling Technology, Danvers, MA,
USA); poly(ADP-ribose) polymerase (PARP), CD95/Fas, FADD,
cytochrome c, caveolin-1 (Santa Cruz Biotechnology, Santa
Cruz, CA, USA); TRAIL-R1/DR4, TRAIL-R2/DR5 (ProSci, Poway, CA,
USA); ATG5, LC3II (MBL International, Woburn, MA, USA); and c-FLIP
(Alexis Biochemicals, Farmingdale, NY, USA). Anti-α-tubulin
(BioGenex, San Ramon, CA, USA) was used as a loading control.
Signals were acquired using an Image Station 4000MM image analyzer
(Kodak, Rochester, NY, USA).
Caspase-8 activity assays
Caspase-8 activity was assayed using an FADD-like
IL-1β-converting enzyme (FLICE) colorimetric assay kit (BioVision,
Inc., Milpitas, CA, USA), according to the manufacturer's protocol.
Briefly, 200 µg of protein extract in a 50 µl volume
was mixed with the reaction buffer and the IETD-pNA substrate,
incubated for 90 min, and the absorbance at 405 nm was measured.
The fold increase in FLICE activity was determined from comparison
of the results of the treated samples with the untreated
control.
RNA interference (RNAi)
For the RNAi experiment, siRNA of TRAILR-1/DR4,
5′-CUGGAAAGUUCAUCUACUU(dtdt)-3′ (sense) and
5′-AAGUAGAUGAACUUUCCAG(dtdt)-3′ (anti-sense); TRAILR-2/DR5,
5′-CAGACUUGGUGCCCUUUG (dtdt)-3′ (sense) and 5′-UCAAAGGGCACCAAGUCUG
(dtdt)-3′ (antisense); CD95/Fas, 5′-GAGAGUAUUACUA GAG CUU(dtdt)-3′
(sense) and 5′-AAGCUCUAGUAAUACU CUC(dtdt)-3′ (antisense); and
5′-control siRNA, 5′-CCUACGCCACCAAUUUCGU(dtdt)-3′ (sense) and
5′-ACGAAAUUG GUGGCGUAGG(dtdt)-3′ (antisense) were purchased from
Bioneer (Daejeon, Korea). Cells were individually transfected with
siRNA oligonucleotides using an Amaxa Transfection system™ (Basel,
Switzerland) and grown for 24 h prior to drug treatment.
Lipid raft fractionation
Lipid rafts were isolated by sucrose
density-gradient centrifugation. A total of 108 cells
were lysed for 30 min in 1 ml of lysis buffer (1% Brij35 in HEPES
buffer; 25 mM HEPES, 1 mM EDTA, and 150 mM NaCl, pH 6.5)
supplemented with a protease inhibitor cocktail, followed by
homogenization with a glass dounce homogenizer. The homogenates
were mixed with 1 ml of 80% sucrose in HEPES buffer and placed at
the bottom of a centrifuge tube. The samples were then overlaid
with 6.5 ml of 30% sucrose followed by 3 ml of 5% sucrose and
centrifuged at 188,000 × g for 18 h at 4°C. Fractions (1 ml) were
collected from the bottom to the top of the gradient, and rafts
were determined by measurement of the total cholesterol using a
cholesterol assay kit (Wako Diagnostics, Richmond, VA, USA).
Fractions 3 through 5 of the sucrose gradients were pooled and used
as the raft fraction; the rest was designated as the non-raft
fraction.
Statistical analysis
All numerical data are reported as mean ± SE. All
data represent the results of at least 3 independent experiments.
Groups were compared by means of a Student's t-test.
Results
UDCA reduces the viability of the
cisplatin-resistant variant of the gastric cancer cell lines
Cisplatin-resistant human cancer cells have been
reported to have a cross-resistance to various cytotoxic stimuli
(13,14). In agreement with this, the
cisplatin-resistant variant (SNU601/R) derived from the SNU601
human gastric cancer cell line (SNU601/WT) demonstrated a strong
resistance to a wide array of anticancer drugs not only to
cisplatin (15). In this study, we
used SNU601/R cells to evaluate the efficacy of several different
types of anticancer drugs, and found that sensitivity to UDCA
reduced the cell viability, similar to the parental SNU601/WT
cells. However, the other anticancer drugs tested (L-OHP, etoposide
and rhTRAIL) had minimal effects on the viability of the resistant
cells in doses that led to ~50% death in the parental cells
(Fig. 1A). In addition, only
treatment with UDCA decreased the colony-forming ability of the
SNU601/R cells (Fig. 1B).
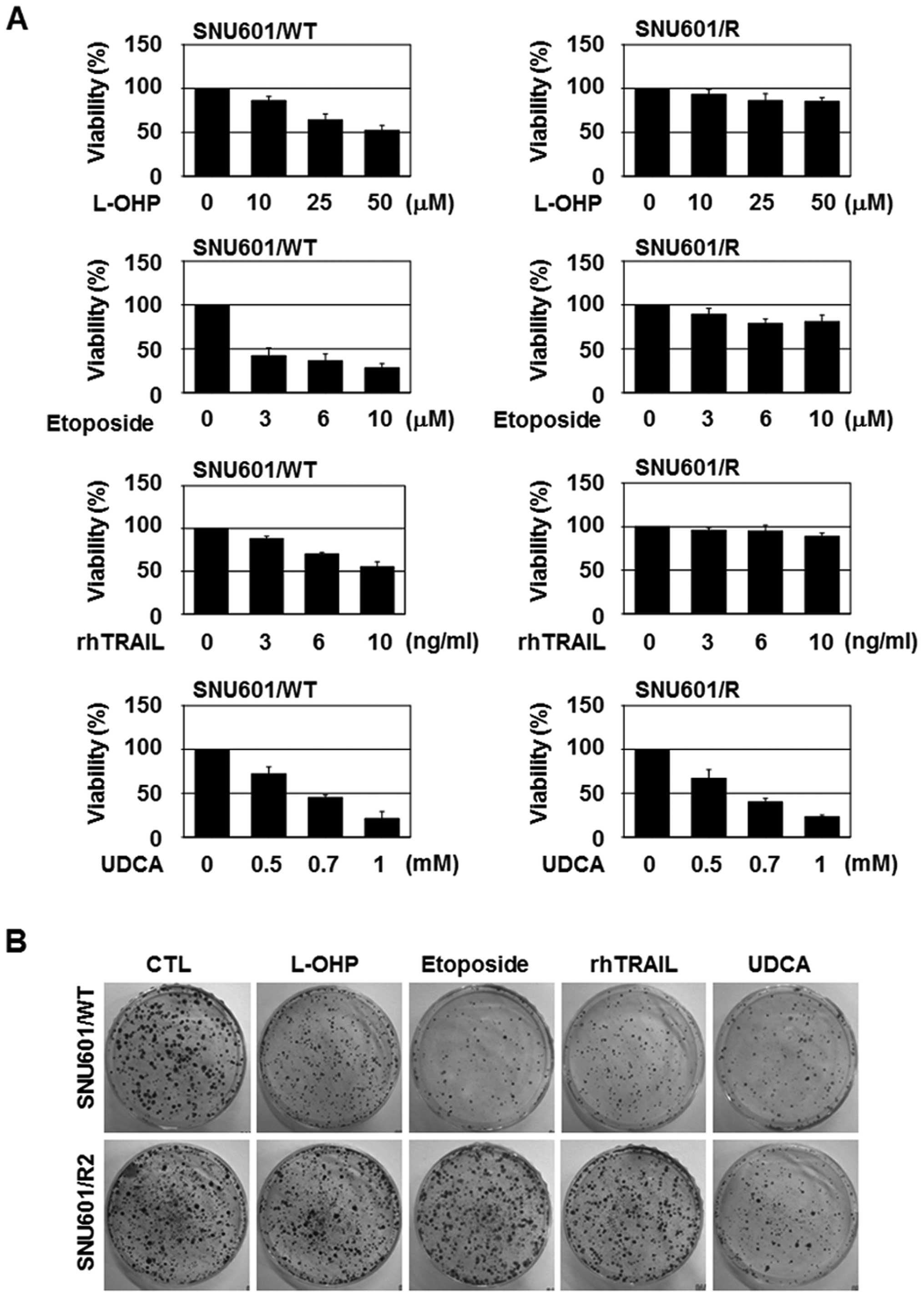 | Figure 1UDCA reduces the viability of a
cisplatin-resistant gastric cancer cell line. (A) SNU601/WT and
SNU601/R cells were exposed to the indicated concentrations of
L-OHP, etoposide, rhTRAIL and UDCA for 48 h, and cell viability was
monitored with MTT assay. (B) SNU601/WT and SNU601/R cells exposed
to 50 µM L-OHP, 10 µM etoposide, 10 ng/ml rhTRAIL and
0.7 mM UDCA for 24 h were trypsinized, and 2,000 cells were
re-seeded and cultured for 2 weeks in a 37°C, 5% CO2
incubator, and colonies were visualized by crystal violet staining.
UDCA, ursodeoxycholic acid; L-OHP, oxaliplatin; rhTRAIL,
recombinant human TNF-related apoptosis-inducing ligand. |
UDCA induces autophagic death in the
SNU601/R cells
As UDCA was the only cytotoxic agent tested to
reduce the viability of the SNU601/R cells, we explored whether the
mechanism involved apoptosis since UDCA was found to induce
apoptosis in the SNU601/WT cells in our previous study (11). Both the parental and the resistant
cells were exposed to L-OHP, etoposide, rhTRAIL and UDCA, and the
resulting apoptotic nuclei were detected by HO staining. Although
UDCA decreased the cell viability of the SNU601/R cells,
UDCA-induced apoptotic body formation was very low in comparison to
the parental cells (Fig. 2A and B).
Other characteristic apoptotic features, namely, the cleavage of
caspase-3, the release of cytochrome c, and the degradation
of PARP, were also blocked in the resistant cells (Fig. 2C). These results indicated that UDCA
did not induce apoptosis in the SNU601/R cells.
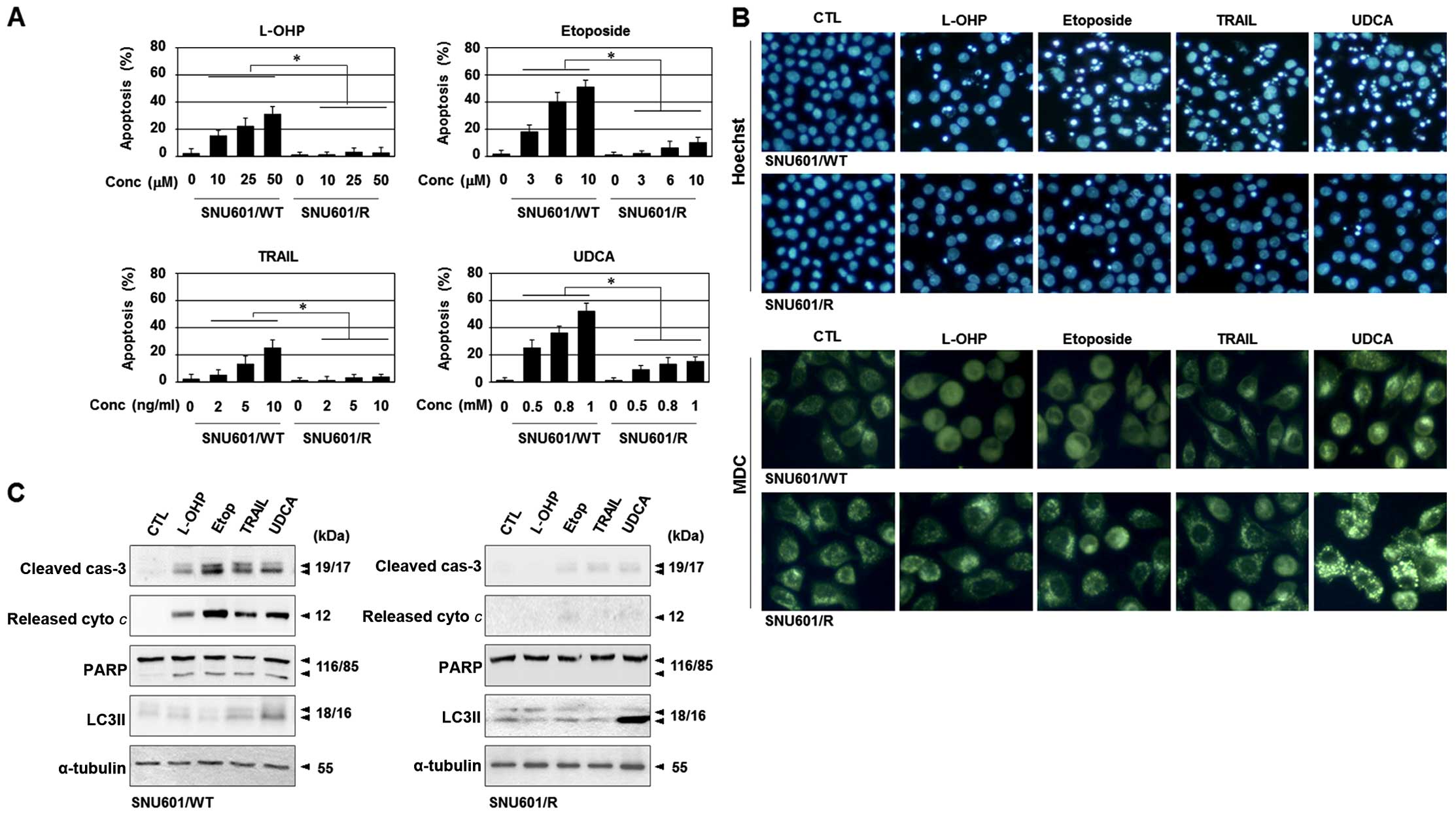 | Figure 2UDCA induces autophagy instead of
apoptosis in the cisplatin-resistant variant of SNU601 cells. (A)
SNU601/WT and SNU601/R cells were exposed to the indicated doses of
L-OHP, etoposide, rhTRAIL and UDCA for 48 h, and were stained by
HO. Apoptotic nuclei were counted under a fluorescence microscope,
and the number of apoptotic cells was expressed as a percentage of
the total number of cells counted. *p<0.01. (B and C)
SNU601/WT and SNU601/R cells were exposed to vehicle, L-OHP,
etoposide, rhTRAIL and UDCA. (B) The treated cells were stained
with HO (upper) and MDC (lower) to visualize apoptotic cells and
autophagosomes, respectively. Stained cells were observed under a
fluorescence microscope (magnification, x200 for HO image and x400
for MDC image), or (C) analyzed by immunoblotting to detect cleaved
caspase, released cytoplasmic cytochrome c, PARP and LC3II.
α-tubulin was used as a loading control. UDCA, ursodeoxycholic
acid; L-OHP, oxaliplatin; rhTRAIL, recombinant human TNF-related
apoptosis-inducing ligand; HO, Hoechst 33342; MDC,
monodansylcadaverine; PARP, poly(ADP-ribose) polymerase. |
Thus, we examined whether other types of cell death
are involved in UDCA-induced cytotoxicity. Upon treatment with
UDCA, the number of autophagic vacuoles was greatly increased in
the SNU601/R cells, as visualized by monodan-sylcadaverine (MDC)
and HO staining (Fig. 2B).
Moreover, treatment with UDCA resulted in a strong elevation in the
level of protein LC3II, an indication of autophagy in the SNU601/R
cells (Fig. 2C). These results
suggest that autophagy occurred in the resistant cells in response
to UDCA.
Autophagy may function as a protective mechanism and
as a cell death mechanism depending on the genetic or signaling
environments. In this study, inhibition of autophagy by
3-methyladenine (3-MA) did not increase UDCA-induced cell toxicity
(data not shown), implying that UDCA-triggered autophagy was not
protective autophagy, but a process of cell death. In contrast,
necrotic features, PI inclusion, or LDH release were not detected
upon UDCA treatment in the SNU601/R cells after 72 h (data not
shown). Therefore, UDCA appears to trigger cell death by autophagy
in SNU601/R cells.
c-FLIP levels are elevated in the
SNU601/R cells
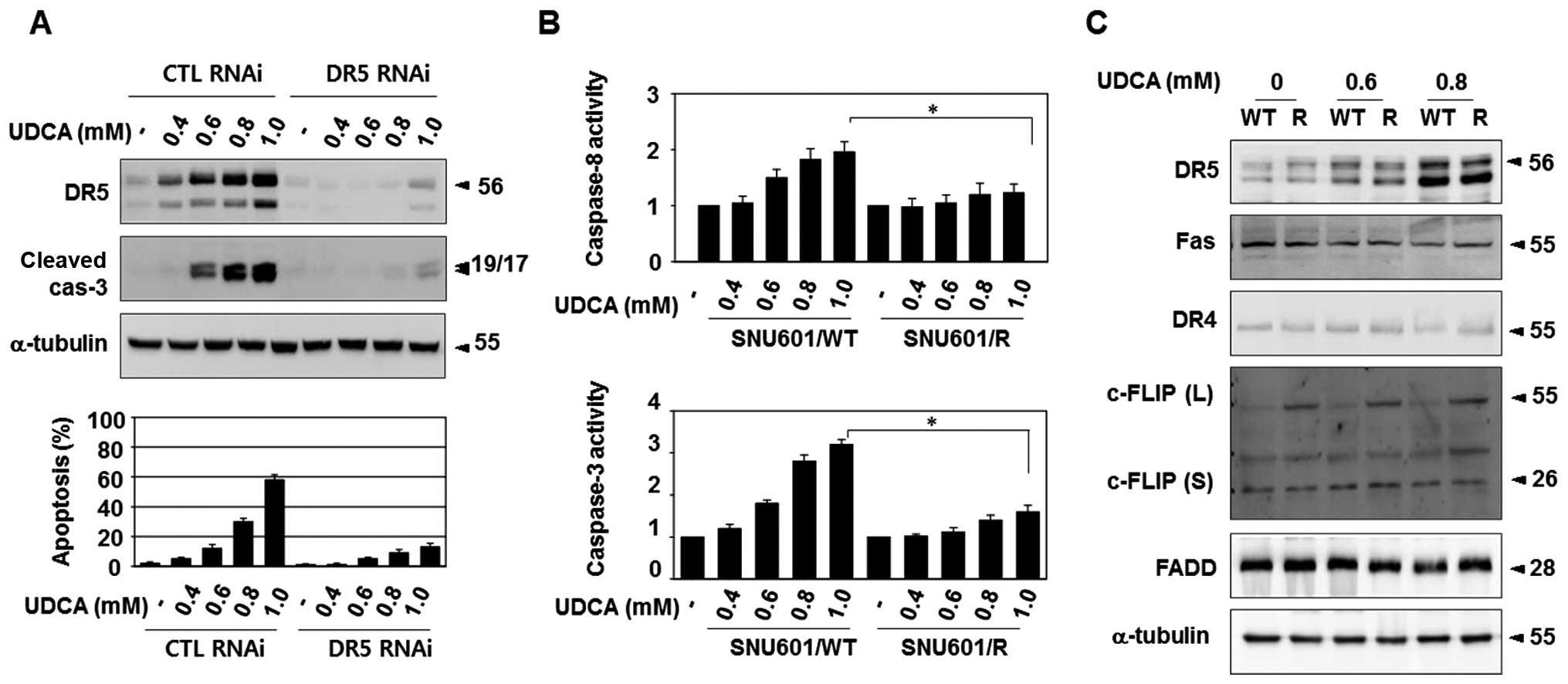
The apoptotic role of TRAIL-R2/DR5 in UDCA-treated
parental SNU601 cells was confirmed via knockdown assays with
TRAIL-R2/DR5-targeting siRNA, resulting in the profound suppression
of caspase-3 activation and formation of apoptotic nuclei (Fig. 3A). In the SNU601/R cells,
UDCA-induced activation of caspase-8, as well as caspase-3, was
severely suppressed compared to the parental cells (Fig. 3B). Thus, we aimed to ascertain
whether the expression or regulation of the components of DISC was
altered in the SNU601/R cells, since DISC is directly linked to
caspase-8 activation.
First, we determined the protein levels of
TRAIL-R2/DR5 and the adaptor protein, FADD, in the resistant cells
with immunoblotting. The basal levels of TRAIL-R2/DR5 and FADD did
not differ between the resistant cells and the parental cells, and
an elevation in the expression of TRAIL-R2/DR5 in the UDCA-treated
cells was also detected in the resistant cells (Fig. 3C).
We also examined protein levels of other death
receptors, including TRAIL-R1/DR4 and CD95/Fas, and DISC inhibitory
factor, c-FLIP, to exclude their possible involvement in the
activation of UDCA-mediated DISC complex. The basal or UDCA-exposed
protein levels of CD95/Fas and TRAIL-R1/DR4 were not altered in the
resistant cells (Fig. 3C). However,
the expression of the long splice variant of cellular
FLICE-inhibitory proteins [c-FLIP(L)] was enhanced in the SNU601/R
cells (Fig. 3C). c-FLIP is a
death-effector domain (DED)-containing protein that is recruited to
DISC and interferes with caspase-8 activation in death receptor
signaling. Therefore, reduction in apoptosis in the resistant cells
in response to various apoptotic stimuli may be due to an increase
in c-FLIP(L) expression, which inhibits DISC activation.
CD95/Fas is involved in the determination
of UDCA-induced cell death mode
Although the elevated expression of c-FLIP may
inhibit TRAIL-R2/DR5-mediated apoptotic signaling, the mechanism by
which autophagic death is triggered by UDCA in the resistant cells
remained unclear. Autophagy is an intracellular degradation system
often triggered by nutrient or energy limitation, which is
regulated by the Atg protein family. Unexpectedly, we found that
the membrane death receptor, CD95/Fas, is associated with the
decision of the mode of cell death in the presence of UDCA, despite
the fact that TRAIL-R2/DR5 acts as a regulator of UDCA-mediated
cell death.
The protein levels of CD95/Fas were observed via
immunoblotting, in the presence and absence of UDCA, and the
expression levels were similar in both cell lines under both
conditions (Figs. 3C and 4A). However, in a raft isolation study, we
found that UDCA increased raft-localized CD95/Fas in the SNU601/WT
cells, but the raft relocalization was not observed in the SNU601/R
cells (Fig. 4B). Translocation of
death receptors into raft areas is one of the essential regulatory
processes of their activity. Therefore, this result implies that
CD95/Fas plays a role in UDCA-treated SNU601/WT, but not in
SNU601/R cells.
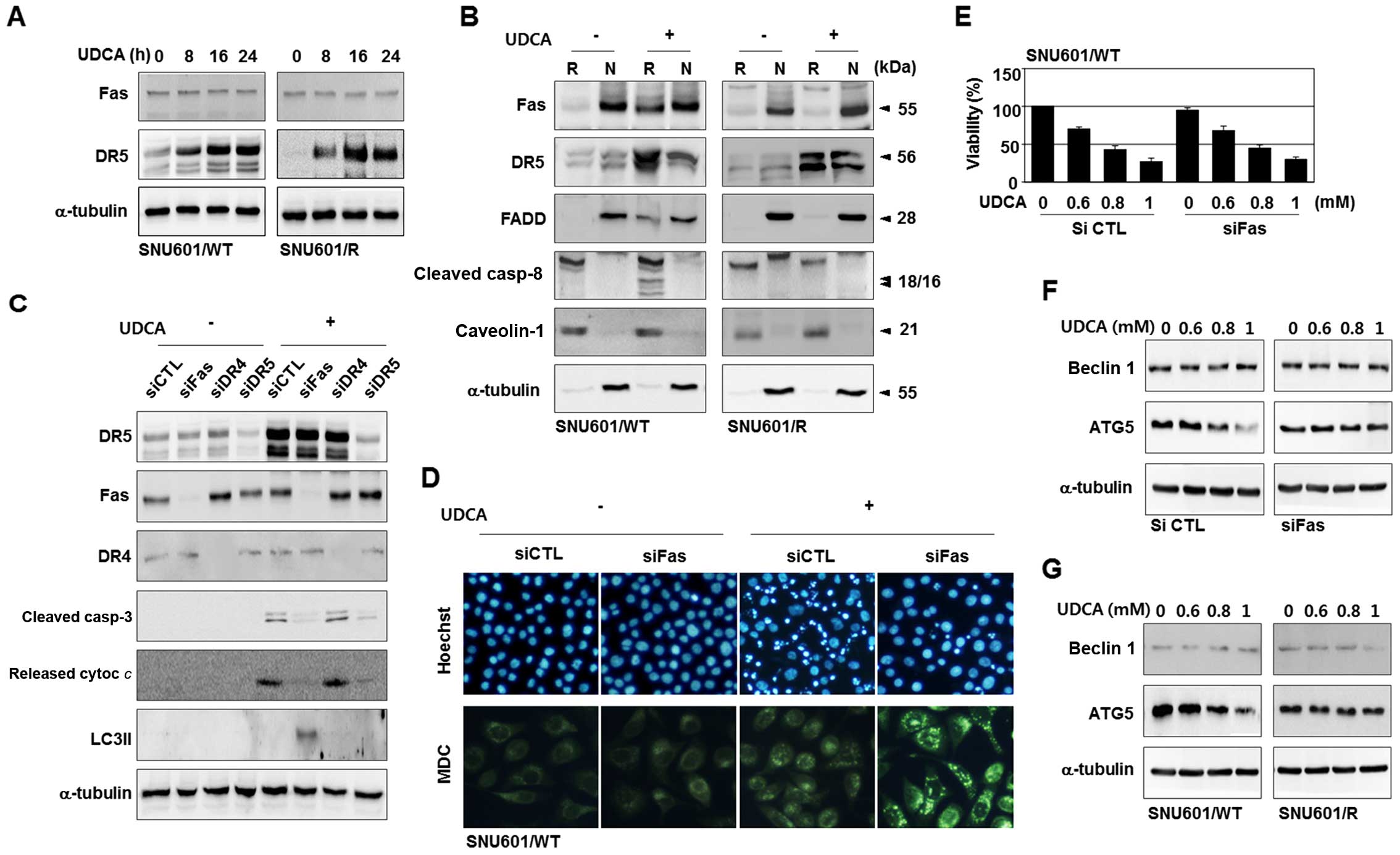 | Figure 4Raft localization of CD95/Fas appears
to be involved in the type of UDCA-induced cell death. (A)
SNU601/WT and SNU601/R cells were treated with UDCA for the
indicated times and analyzed by immunoblotting to detect
TRAIL-R2/DR5 and CD95/Fas. (B) SNU601/WT and SNU601/R cells were
incubated in the absence or presence of UDCA, separated into raft
(R) and non-raft (N) fractions, and analyzed by immunoblotting
using antibodies against CD95/Fas, TRAIL-R2/DR5, FADD, caspase-8,
caveolin-1 and α-tubulin. (C) SNU601/WT cells, transfected with
scrambled control RNA or siRNAs for CD95/Fas, TRAIL-R1/DR4, and
TRAIL-R2/DR5, were exposed to the vehicle control or UDCA and
subjected to immunoblotting. (D-F) SNU601/WT cells, transfected
with control scrambled RNA or CD95/Fas siRNA were exposed to
vehicle or UDCA, and analyzed by (D) HO or MDC staining, (E) MTT
assay, and (F) immunoblotting using antibodies against beclin1 and
ATG5. (G) SNU601/WT and SNU601/R cells were incubated with UDCA and
analyzed by immunoblotting to detect beclin1 and ATG5. UDCA,
ursodeoxycholic acid; HO, Hoechst 33342; MDC,
monodansylcadaverine. |
To understand the role of CD95/Fas further, we
performed interference studies in the parental cells. As shown in
Fig. 4C, silencing of TRAIL-R2/DR5
simply blocked UDCA-induced apoptosis, as detected by the decrease
in cleaved caspase-3 and the release of cytochrome c.
Nevertheless, knockdown of CD95/Fas switched the UDCA-induced death
pattern from apoptosis to autophagy. This was evidenced by a
decrease in apoptotic features, including caspase-3 cleavage,
cytochrome c release, apoptotic nuclei, and the presence of
autophagic markers such as LC3II and autophagosomes (Fig. 4C and D). However, knockdown of
CD95/Fas did not recover cell viability despite suppression of
apoptosis (Fig. 4E), and prevention
of autophagy by 3-MA did not further increase the cell death rate
under the CD95/Fas-silenced condition (data not shown). Hence, the
observed autophagy in the absence of CD95/Fas was also regarded as
a type of cell death, not a protective autophagy, as observed in
the SNU601/R cells.
Finally, we explored whether essential autophagy
regulatory proteins, ATG5 and beclin 1, are involved in the process
of the observed cell death. Expression levels of beclin 1 were
unchanged in response to UDCA, in control or CD95/Fas-silenced
cells; however, ATG5 protein levels were diminished by the presence
of UDCA in the control cells, but not in the CD95/Fas-silenced
cells (Fig. 4F). Furthermore,
UDCA-triggered downregulation of ATG5 was protected in the SNU601/R
cells (Fig. 4G). Taken together,
these results indicate that CD95/Fas assists TRAIL-R2/DR5 in the
induction of apoptosis by inhibiting the autophagic pathway via
downregulation of ATG5, and the loss of CD95/Fas activity results
in the release of autophagic pathway. Therefore, UDCA appears to
induce both apoptosis and autophagic death depending on the
intracellular signaling environment.
Discussion
Apoptosis is a type of programmed cell-suicide in
response to various cytotoxic stimuli, such as anticancer drugs,
and is typically characterized by membrane blebbing, DNA
fragmentation, and apoptotic nuclei. The central components of the
apoptotic machinery are caspases (16,17),
and the caspase cascade is signaled by two main pathways. The first
is an extrinsic pathway, which is initiated by ligation of cell
surface death receptors, leading to DISC formation and caspase-8
activation. The second is an intrinsic mitochondrial pathway,
triggered by cytochrome c release from the mitochondria, and
subsequent caspase-9 activation (16–18).
These extrinsic and intrinsic pathways are often linked and they
work cooperatively to promote apoptosis. Together caspase-8 and -9
activate caspase-3, the main executioner of the apoptotic pathway
that leads to cleavage of cellular proteins, such as
poly(ADP-ribose) polymerase (16,18).
However, cancer cells that survive cytotoxic stimuli often have
defects in apoptosis from the alteration of signaling molecules in
the extrinsic and intrinsic pathways, such as surface death
receptors, p53 and Bcl-2 family members (19–21).
Likewise, the signaling defects in the main apoptotic pathway cause
cross-resistance to various anticancer agents that act through this
pathway.
In this case, stimulation of other types of cell
death rather than apoptosis could be a strategy to circumvent
apoptotic aberrations. To date, necrosis and autophagic death have
been identified as distinct types of cell death. Necrosis is a
strong cellular destructive process; however, induction of necrosis
for cancer therapeutics requires a careful approach as a
necrosis-induced massive inflammatory response promotes tumor
growth or angiogenesis by releasing intracellular cytokines, such
as HMGB1 (22–27).
Autophagy is another type of self-degradative
process involved in the basal turnover of cellular components in
response to nutrient starvation or organelle damage, and it is
controlled by autophagy-related ATG proteins, including beclin 1
and ATG5. This process is characterized by sequestration of
intracellular organelles or portions of the cytoplasm in
autophagosomes, which are degraded after fusion with lysosomes for
subsequent recycling. This process primarily acts as a pro-survival
mechanism to overcome a stressful condition, but it ultimately
commits the cell to death under prolonged or severely stressful
environments (28). This autophagic
death is suggested to be a form of tumor-suppressive cell death, as
shown by the use of tumor-suppressors such as beclin 1 and
DAP-kinase in autophagic pathways. Therefore, autophagic death is
expected to be a suitable option for therapeutic treatment of
cancers that harbor defects in apoptotic signaling.
In this study, we showed that cisplatin-resistant
cancer cells were insensitive to various anticancer agents
including a platinum-based drug (L-OHP), a topoisomerase inhibitor
(etoposide) and a specific ligand to surface death receptor
(rhTRAIL). Thus, SNU601/R cells may have defects in their apoptotic
machinery. However, UDCA was able to reduce the cell viability and
markedly elevate autophagic death, but not apoptosis, in the
drug-resistant cells. These results suggest that autophagic death
is responsible for the decrease in viability in the resistant
cells, despite the fact that UDCA acted as an apoptotic inducer in
the parental, non-resistant cells.
Next, we investigated how UDCA could induce
autophagic death in drug-resistant cells. Previously, we showed
that UDCA elevated the protein levels of TRAIL-R2/DR5, which plays
a critical role in UDCA-induced apoptosis; however,
TRAIL-R2/DR5-mediated apoptosis requires the activity of CD95/Fas.
In the parental cells, translocation of CD95/Fas into membrane
rafts was detected in the UDCA-treated cells, although CD95/Fas
levels were not increased. In addition, knockdown of CD95/Fas in
the parental cells significantly blocked UDCA-induced apoptotic
characteristics, such as caspase cleavage and cytochrome c
release, but, in contrast, encouraged autophagic features,
including higher LC3II levels and the presence of autophagic
vacuoles. Furthermore, UDCA treatment decreased ATG5 levels in the
parental cells, whereas the silencing of CD95/Fas protected these
protein levels. Nevertheless, the knockdown of CD95/Fas did not
affect the cell viability, while knockdown of TRAIL-R2/DR5
partially restored cell viability, in the UDCA-treated cells. These
results suggest that CD95/Fas contributes to the induction of
apoptosis by blocking autophagic processes via inhibition of ATG5
protein levels. In this context, the absence of CD95/Fas
redistribution in the resistant cells appears to be linked to a
release in autophagic signaling, which results in autophagic death.
In support of our hypothesis, ATG5 levels were not reduced in the
UDCA-treated resistant cells. We also found that the resistant
cells had an elevated level of c-FLIP(L), indicating that the
repression of DISC activity is one reason for the multi-drug
resistance of the resistant cells. Therefore, stimulation of
autophagic signaling, which does not require DISC formation, could
be an efficient bypass mechanism to overcome the resistance.
Our results revealed that at least two surface death
receptors, TRAIL-R2/DR5 and CD95/Fas, were stimulated by UDCA, and
cooperation of different death receptors can modulate the mode of
cell death, thereby taking advantage of the intracellular signaling
environment to stimulate various types of death. In fact, several
studies have suggested that several molecular nodes of crosstalk
interconnect autophagy and apoptosis, and autophagy can be
triggered as an evasive system to avoid apoptosis (29,30).
Although further studies are required to clarify the exact
mechanism by which UDCA-mediated CD95/Fas signaling is regulated,
our results demonstrated that UDCA can stimulate both apoptotic and
autophagic death, and may provide at least a part of the
explanation of why the resistant cells are sensitive to UDCA. This
finding may provide a possible strategy to improve chemotherapeutic
efficacy in apoptosis-inducing drug-resistant cancers.
Acknowledgments
This research was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(NRF-2011–0014540), and through the Research Center for Resistant
Cells (R13-2003-009). We thank Professor Cheol-Hee Choi for the
cisplatin-resistant SNU601 cells, Professor Tae-Hyoung Kim for the
kind gift of rhTRAIL, and Ms. Jeong-Eun Choi and Dr Hong-Quan Duong
for their excellent technical assistance.
References
|
1
|
Brozovic A, Fritz G, Christmann M,
Zisowsky J, Jaehde U, Osmak M and Kaina B: Long-term activation of
SAPK/JNK, p38 kinase and fas-L expression by cisplatin is
attenuated in human carcinoma cells that acquired drug resistance.
Int J Cancer. 112:974–985. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Komatsu M, Sumizawa T, Mutoh M, Chen ZS,
Terada K, Furukawa T, Yang XL, Gao H, Miura N, Sugiyama T, et al:
Copper-transporting P-type adenosine triphosphatase (ATP7B) is
associated with cisplatin resistance. Cancer Res. 60:1312–1316.
2000.PubMed/NCBI
|
|
3
|
Taniguchi K, Wada M, Kohno K, Nakamura T,
Kawabe T, Kawakami M, Kagotani K, Okumura K, Akiyama S and Kuwano
M: A human canalicular multispecific organic anion transporter
(cMOAT) gene is overexpressed in cisplatin-resistant human cancer
cell lines with decreased drug accumulation. Cancer Res.
56:4124–4129. 1996.PubMed/NCBI
|
|
4
|
Zhu HJ, Wang JS, Guo QL, Jiang Y and Liu
GQ: Reversal of P-glycoprotein mediated multidrug resistance in
K562 cell line by a novel synthetic calmodulin inhibitor, E6. Biol
Pharm Bull. 28:1974–1978. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCubrey JA, Steelman LS, Abrams SL, Lee
JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA,
D'Assoro AB, et al: Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT
pathways in malignant transformation and drug resistance. Adv
Enzyme Regul. 46:249–279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang J, Zhou JY and Wu GS: ERK-dependent
MKP-1-mediated cisplatin resistance in human ovarian cancer cells.
Cancer Res. 67:11933–11941. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi CH, Kim HS, Kweon OS, Lee TB, You HJ,
Rha HS, Jeong JH, Lim DY, Min YD, Kim MS, et al: Reactive oxygen
species-specific mechanisms of drug resistance in
paraquat-resistant acute myelogenous leukemia sublines. Mol Cells.
10:38–46. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu Z, Chen ZP, Malapetsa A, Alaoui-Jamali
M, Bergeron J, Monks A, Myers TG, Mohr G, Sausville EA, Scudiero
DA, et al: DNA repair protein levels vis-à-vis anticancer drug
resistance in the human tumor cell lines of the National Cancer
Institute drug screening program. Anticancer Drugs. 13:511–519.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alberts DS, Martínez ME, Hess LM, Einspahr
JG, Green SB, Bhattacharyya AK, Guillen J, Krutzsch M, Batta AK,
Salen G, et al: Phoenix and Tucson Gastroenterologist Networks:
Phase III trial of ursodeoxycholic acid to prevent colorectal
adenoma recurrence. J Natl Cancer Inst. 97:846–853. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Khare S, Cerda S, Wali RK, von Lintig FC,
Tretiakova M, Joseph L, Stoiber D, Cohen G, Nimmagadda K, Hart J,
et al: Ursodeoxycholic acid inhibits Ras mutations, wild-type Ras
activation, and cyclooxygenase-2 expression in colon cancer. Cancer
Res. 63:3517–3523. 2003.PubMed/NCBI
|
|
11
|
Lim SC, Duong HQ, Choi JE, Lee TB, Kang
JH, Oh SH and Han SI: Lipid raft-dependent death receptor 5 (DR5)
expression and activation are critical for ursodeoxycholic
acid-induced apoptosis in gastric cancer cells. Carcinogenesis.
32:723–731. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lim SC, Duong HQ, Parajuli KR and Han SI:
Pro-apoptotic role of the MEK/ERK pathway in ursodeoxycholic
acid-induced apoptosis in SNU601 gastric cancer cells. Oncol Rep.
28:1429–1434. 2012.PubMed/NCBI
|
|
13
|
Iwasaki I, Sugiyama H, Kanazawa S and
Hemmi H: Establishment of cisplatin-resistant variants of human
neuroblastoma cell lines, TGW and GOTO, and their drug
cross-resistance profiles. Cancer Chemother Pharmacol. 49:438–444.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun CL and Chao CC: Cross-resistance to
death ligand-induced apoptosis in cisplatin-selected HeLa cells
associated with overexpression of DDB2 and subsequent induction of
cFLIP. Mol Pharmacol. 67:1307–1314. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu H, Choi SM, An CS, Min YD, Kim KC, Kim
KJ and Choi CH: Concentration-dependent collateral sensitivity of
cisplatin-resistant gastric cancer cell sublines. Biochem Biophys
Res Commun. 328:618–622. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martinou JC and Green DR: Breaking the
mitochondrial barrier. Nat Rev Mol Cell Biol. 2:63–67. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsujimoto Y: Cell death regulation by the
Bcl-2 protein family in the mitochondria. J Cell Physiol.
195:158–167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zeuner A, Pedini F, Signore M, Ruscio G,
Messina C, Tafuri A, Girelli G, Peschle C and De Maria R: Increased
death receptor resistance and FLIPshort expression in polycythemia
vera erythroid precursor cells. Blood. 107:3495–3502. 2006.
View Article : Google Scholar
|
|
20
|
Müllauer L, Gruber P, Sebinger D, Buch J,
Wohlfart S and Chott A: Mutations in apoptosis genes: A
pathogenetic factor for human disease. Mutat Res. 488:211–231.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perego P, Giarola M, Righetti SC, Supino
R, Caserini C, Delia D, Pierotti MA, Miyashita T, Reed JC and
Zunino F: Association between cisplatin resistance and mutation of
p53 gene and reduced bax expression in ovarian carcinoma cell
systems. Cancer Res. 56:556–562. 1996.PubMed/NCBI
|
|
22
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hippert MM, O'Toole PS and Thorburn A:
Autophagy in cancer: Good, bad, or both? Cancer Res. 66:9349–9351.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Syntichaki P and Tavernarakis N: The
biochemistry of neuronal necrosis: Rogue biology? Nat Rev Neurosci.
4:672–684. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vakkila J and Lotze MT: Inflammation and
necrosis promote tumour growth. Nat Rev Immunol. 4:641–648. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zong WX and Thompson CB: Necrotic death as
a cell fate. Genes Dev. 20:1–15. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bernales S, McDonald KL and Walter P:
Autophagy counterbalances endoplasmic reticulum expansion during
the unfolded protein response. PLoS Biol. 4:e4232006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moretti L, Cha YI, Niermann KJ and Lu B:
Switch between apoptosis and autophagy: Radiation-induced
endoplasmic reticulum stress? Cell Cycle. 6:793–798. 2007.
View Article : Google Scholar : PubMed/NCBI
|