Introduction
Breast cancer is one of the most common malignant
diseases in women worldwide and its metastasis to distant organs is
the leading cause of mortality in these patients. The metastatic
sites from primary breast cancer are usually the brain, liver,
lungs and bone tissue (1,2), whereas distant metastases to other
organs, including the uterus, kidney or spleen, are relatively
rare. When patients are diagnosed with breast cancer, ~10–15% of
them develop an aggressive phenotype, and distant metastasis occurs
within 3 years. However, it is not unusual that metastases at
distant sites may appear ≥10 years following the initial diagnosis
(3). Therefore, patients with breast
cancer are at risk of developing lethal metastasis throughout their
entire lifetime.
Epithelial-mesenchymal transition (EMT) is
considered to be closely associated with the invasion and migration
of tumor cells, and it is characterized by a cellular phenotypic
transformation involving acquisition of mesenchymal characteristics
and loss of epithelial characteristics (4–7). The
epithelial and mesenchymal phenotypes are distinct cellular states;
cells are able to transition between each state (8). Additionally, EMT has been regarded as a
reversible process that may be prevented under certain
physiological and pathological conditions. Epithelial cadherin
(E-cadherin) is an important epithelial cell adhesion molecule, and
a decrease in the level of E-cadherin is one of the landmark
features of EMT. The other important phenomenon associated with EMT
is the upregulation of mesenchymal biomarkers such as vimentin and
N-cadherin (9,10). Clinical studies have revealed that
breast cancer, when accompanied by a low expression of E-cadherin
and robust expression of vimentin or N-cadherin, usually exhibits
an aggressive tumor phenotype and a high rate of metastasis
(7,11).
Certain transcription factors, such as zinc finger
protein SNAI2 (SLUG), zinc finger protein SNAI1 (Snail),
twist-related protein 1 (Twist) and zinc finger E-box-binding
homeobox 1 (Zeb1), have been implicated in EMT regulation. Among
them, the transcription factor SLUG inhibits the expression of
E-cadherin by binding to the E-box site on the E-cadherin promoter.
Upregulation of SLUG results in a decrease in the levels of
E-cadherin (12,13), which subsequently leads to an
attenuation of intercellular adhesion and enhanced cell motility
properties. Furthermore, SLUG is able to promote the expression of
vimentin, consequently inducing EMT-like changes (14). Therefore, SLUG has an important role
in promoting the EMT process.
Histone deacetylase inhibitors (HDACIs) are a class
of anti-tumor drugs that exhibit potent anti-tumor activity
(15,16). As a representative of the classical
HDACIs, trichostatin A (TSA) suppresses the activity of histone
deacetylases (HDACs) in a non-competitive and reversible manner.
Previously published studies have revealed that TSA reverses EMT in
non-tumor cells, including human renal tubular epithelial cells,
retinal pigment epithelium cells and hepatocytes (17–19).
Furthermore, a previously published study by our research group
identified that EMT was prevented by TSA in SW480 and PC3 cells
(20). Given these data, we
hypothesized that TSA-induced EMT reversal effects may also occur
in breast cancer cells. Therefore, in the present study,
TSA-mediated changes in EMT-associated biomarkers, including
E-cadherin, vimentin and the transcription factor SLUG, were
investigated, and TSA-induced alterations in the invasive and
migratory abilities of MCF-7 breast cancer cells were
determined.
Materials and methods
Cell culture and cytotoxicity
test
The human breast cancer cell line MCF-7 was obtained
from the Cell Bank of Type Culture Collection of Chinese Academy of
Sciences. The cells were cultured in RPMI-1640 medium (Invitrogen;
Thermo Fisher Scientific, Inc.) under an atmosphere of 37°C
humidified air containing 5% CO2 supplemented with 1%
penicillin/streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.) and 10% fetal bovine serum (HyClone; GE Healthcare Life
Sciences). Cytotoxicity of TSA (Sigma-Aldrich; Merck KGaA) on MCF-7
cells was detected using an MTS assay (Promega Corporation),
according to the manufacturer's protocol. In brief, MCF-7 cells
were plated into 96-well culture plates (5,000 cells in 200 µl per
well) overnight and subsequently treated with 50, 100, 200, 400 or
800 nM TSA for 24 or 48 h. Cells were rinsed with PBS to remove
unattached cells and incubated with 20% MTS reagent in serum-free
medium for 3 h at 37°C. The absorbance of the formazan dye was
measured at 490 nm using a microplate reader (Bio-Rad Laboratories,
Inc.), and the optical density (OD) at 490 nm (OD490) was directly
proportional to the proportion of viable cells. Cell viability (%
of control)=(OD490 of treatment group-OD490 of blank group)/(OD490
of control group-OD490 of blank group) ×100%.
Transwell invasion and migration
assay
Invasion and migration assays were performed as
follows: Boyden chambers equipped with 8-µm polycarbonate filters
were coated with Matrigel™ matrix (BD Biosciences) at 37°C and
dried overnight under sterile conditions for the invasion assay,
whereas Matrigel™ matrix-free polycarbonate filters were used for
the migration assay. Subsequently, MCF-7 cells suspended in 300 µl
RPMI-1640 medium containing 1% FBS were seeded in the upper chamber
at a density of 1×105/well, whereas 600 µl RPMI-1640
medium with 10% FBS (chemotactic agent) was added to the lower
chamber. Cells were incubated with or without 100 nM TSA at 37°C
for 48 h. For the invasion assay, following gentle removal of the
Matrigel that had been coated on the upper side of the filter,
cells adhering to the underside of the filter were fixed with 4%
paraformaldehyde at room temperature for 20 min, and subsequently
stained with DAPI (10 ug/ml) at room temperature for 10 min. For
the migration assay, cells that had passed through the pores of the
filter and fallen into the lower chamber were fixed with 4%
paraformaldehyde at room temperature for 20 min, and subsequently
stained with 0.5% hematoxylin at room temperature for 15 min. An
upright fluorescent microscope (Nikon Corporation) was used for
counting cells (5 fields/chamber) at a magnification of ×100. Each
invasion and migration experiment was repeated at least 3
times.
Wound healing assay
MCF-7 cells (4×105) were seeded in each
well of 6-well plates and were cultured in RPMI-1640 medium
containing 10% FBS until growth was confluent. A defined scratch
was made with a 200-µl pipette tip (cat. no. 94052320; Thermo
Fisher Scientific, Inc.), generating a cell-free area of ~0.5 mm in
width. Cellular debris was gently removed by washing with culture
medium. Subsequently, cells were cultured in serum-free RPMI-1640
with or without 100 nM TSA for 24 or 48 h. Images of the scraped
area were captured using an inverted microscope (Olympus
Corporation) at magnification, ×100. The percentages of wound
closure were subsequently calculated by the following equation:
Wound closure %=[1-(wound area at Tt/wound area at
T0)], where Tt is the time after wounding and
T0 is the time immediately following wounding. A total
of 3 independent experiments were performed.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Briefly, after 48 h incubation with or without 100
nM TSA, MCF-7 cells were washed twice with ice-clod PBS, and total
mRNA was extracted using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). First-strand cDNA was generated by
reverse transcription from 500 ng total RNA using a PrimeScript™ RT
reagent Kit (Takara Bio, Inc.) and the cDNA was synthesized at 37°C
for 15 min. The relative gene expression of target and reference
genes was assessed using a LightCycler® 480 system
(Roche Applied Science) using validated primers for E-cadherin,
vimentin, SLUG, GAPDH and SYBR Premix Ex Taq™ (Takara Bio, Inc.)
for detection. The thermocycling conditions were as follows: 10 min
at 95°C, followed by 40 cycles of denaturation at 95°C for 15 sec,
annealing at 60°C for 30 sec and extension at 72°C for 30 sec. The
ratio between the target gene and GAPDH was used to calculate the
relative quantitation. The relative expression of the amplification
products was analyzed using the 2−ΔΔCq method (21). Data are presented as the mean ±
standard deviation from 3 independent experiments. Primer sequences
used in the RT-qPCR were as follows: E-cadherin forward,
5′-TACACTGCCCAGGAGCCAGA-3′; E-cadherin reverse,
5′-TGGCACCAGTGTCCGGATTA-3′; vimentin forward,
5′-TGAGTACCGGAGACAGGTGCAG-3′; vimentin reverse,
5′-TAGCAGCTTCAACGGCAAAGTTC-3′; SLUG forward,
5′-TTCGGACCCACACATTACCT-3′; SLUG reverse,
5′-GCAGTGAGGGCAAGAAAAAG-3′; GAPDH forward,
5′-GCACCGTCAAGGCTGAGAAC-3′; and GAPDH reverse,
5′-TGGTGAAGACGCCAGTGGA-3′.
Western blot analysis
Briefly, MCF-7 cells were incubated at 37°C for 48 h
and treated with or without 100 nM TSA. Subsequently, cells were
washed 3 times with ice-cold PBS and lysed on ice in cell lysis
buffer and the cell debris was removed by centrifugation at 12,000
× g at 4°C for 2 min. Total protein was quantified using a
bicinchoninic acid assay kit (cat. no. P0010; Beyotime Institute of
Biotechnology) and samples containing equal amounts of protein (60
µg/lane) were separated by 10% SDS-PAGE, and subsequently
transferred onto PVDF membranes. The membranes were blocked with 5%
non-fat milk for 2 h at room temperature, and subsequently
incubated with the primary antibodies against β-actin (1:1,000;
cat. no., sc-8432; Santa Cruz Biotechnology, Inc.), E-cadherin
(1:1,000; cat. no., sc-71008; Santa Cruz Biotechnology, Inc.),
vimentin (1:1,000; cat. no., sc-80975; Santa Cruz Biotechnology,
Inc.), SLUG (1:1,000; cat. no., sc-166476; Santa Cruz
Biotechnology, Inc.) overnight at 4°C. Following incubation with
the appropriate horseradish peroxidase-conjugated secondary
antibodies (1:5,000; cat. no., sc-2031; Santa Cruz Biotechnology,
Inc.) for 2 h at room temperature, specific immune complexes were
detected with chemiluminescence reagent (Western Lightning™,
Chemiluminescence Reagent Plus, PerkinElmer, Inc.). The detection
of β-actin was used as a loading control (22). Protein expression was quantified
using ImageJ software (version 1.46, National Institutes of
Health).
Confocal microscopy for
E-cadherin
MCF-7 cells (3×105) were seeded in a
35-mm glass bottom dish. When grown to ~70% confluence, cells were
stimulated with or without 100 nM TSA for 48 h. Following fixation
in 4% paraformaldehyde at 37°C for 30 min the cells were blocked
with 10% normal goat serum (cat. no. 31873, Thermo Fisher
Scientific, Inc.) at 37°C for 30 min. Cells were then incubated
with an antibody against E-cadherin (1:100; cat. no., sc-71008;
Santa Cruz Biotechnology, Inc.) for 1 h at 37°C. Subsequently,
slides were washed three times with PBS and incubated with Alexa
Fluor 488-conjugated secondary antibodies (1:1,000; Invitrogen;
Thermo Fisher Scientific, Inc.) for 45 min at 37°C. Following an
additional wash step with PBS, cells were stained with 10 µg/ml
DAPI (Invitrogen; Thermo Fisher Scientific, Inc.) at room
temperature for 10 min for the visualization of cell nuclei. The
expression of E-cadherin on cell membranes was detected using
confocal laser scanning microscopy (LSM710; Zeiss GmbH) at
magnification, ×400.
Overexpression of SLUG
MCF-7 cells were seeded into a 6-well plate
(2×105 cells/well) and cultured at 37°C until the
following day. In serum-free medium, cells were transfected with 2
µg control vector pcDNA-3.1 (Generay Biotech Co., Ltd) or 2 µg
pcDNA-SLUG, encoding the human SLUG gene (Generay Biotech Co., Ltd)
mixed with Lipofectamine™ 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). The serum-free medium was then replaced with
complete culture medium 6 h later and the transfection efficiency
was evaluated by RT-qPCR and western blot analysis, as
aforementioned.
SLUG small interfering RNA (siRNA)
transfections
MCF-7 cells were seeded in 6-well plates at a
density of 3×105 cells/well and grown for 24 h. Cell
transfections were performed using Lipofectamine™ RNAi MAX reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. A validated negative control
oligonucleotide
(5′-GCAACGUACAGUGGUUCAA-3′/5′-UUGAACCACUGUACGUUGC-3′, Guangzhou
RiboBio Co., Ltd.) and the siRNA oligonucleotides targeting SLUG
(5′-GCAUUUGCAGACAGGUCAA-3′/5′-UUGAACUGUCUGCAAAUGC-3′, Guangzhou
RiboBio Co., Ltd, Guangzhou, China) were used for transfection. The
final siRNA oligonucleotide concentration was 20 pM and the
transfection efficiency was evaluated by western blot analysis.
Following 48 h transfection, cells were collected for subsequent
assays.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism v.5.0 software (GraphPad Software Inc.). A Student's
t-test was used to analyze differences between two groups. One-way
analysis of variance followed by Tukey's post-hoc test was used to
analyze differences among multiple groups. Data are presented as
the mean ± standard deviation from at least 3 independent
experiments. P<0.05 was considered to indicate a statistically
significant difference.
Results
TSA attenuates invasion and migration
of MCF-7 cells
Transwell invasion and migration assays were
performed to investigate the invasive and migratory abilities of
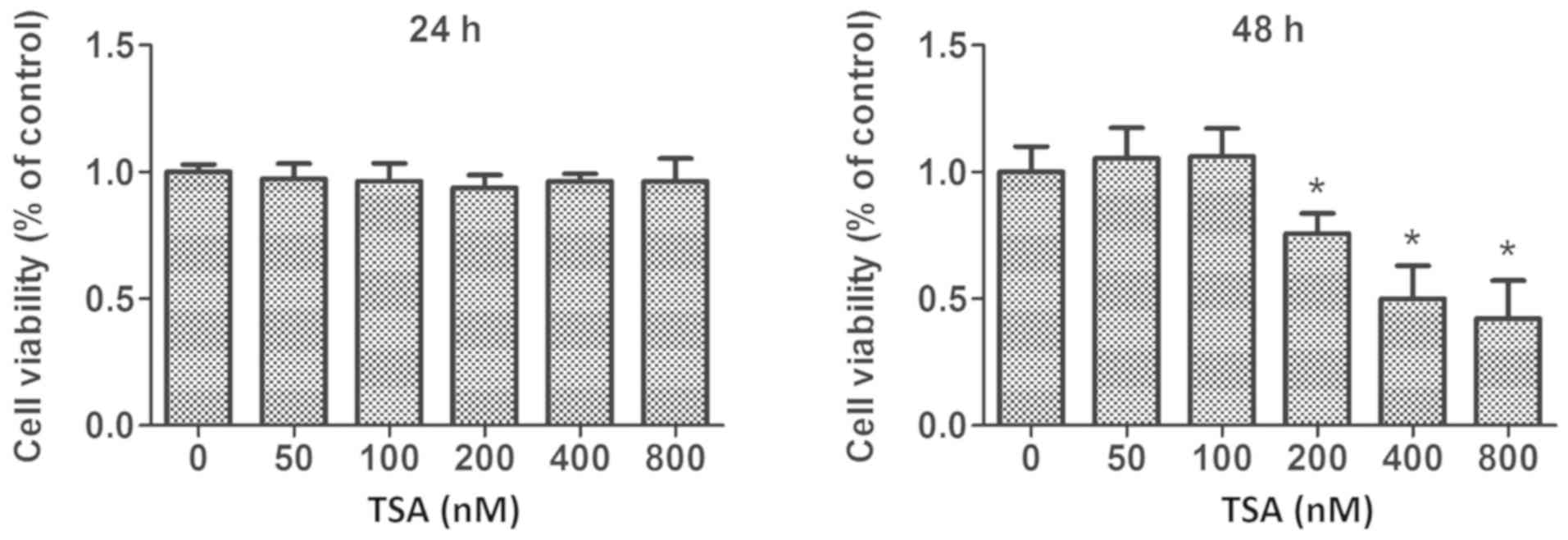
MCF-7 cells. Furthermore, TSA-mediated anti-proliferative effects
were also determined by MTS assay. These experiments revealed that
treatment with 100 nM TSA elicited no significant inhibitory
activity on the proliferation of MCF-7 cells within a 48 h period
(Fig. 1). However, following
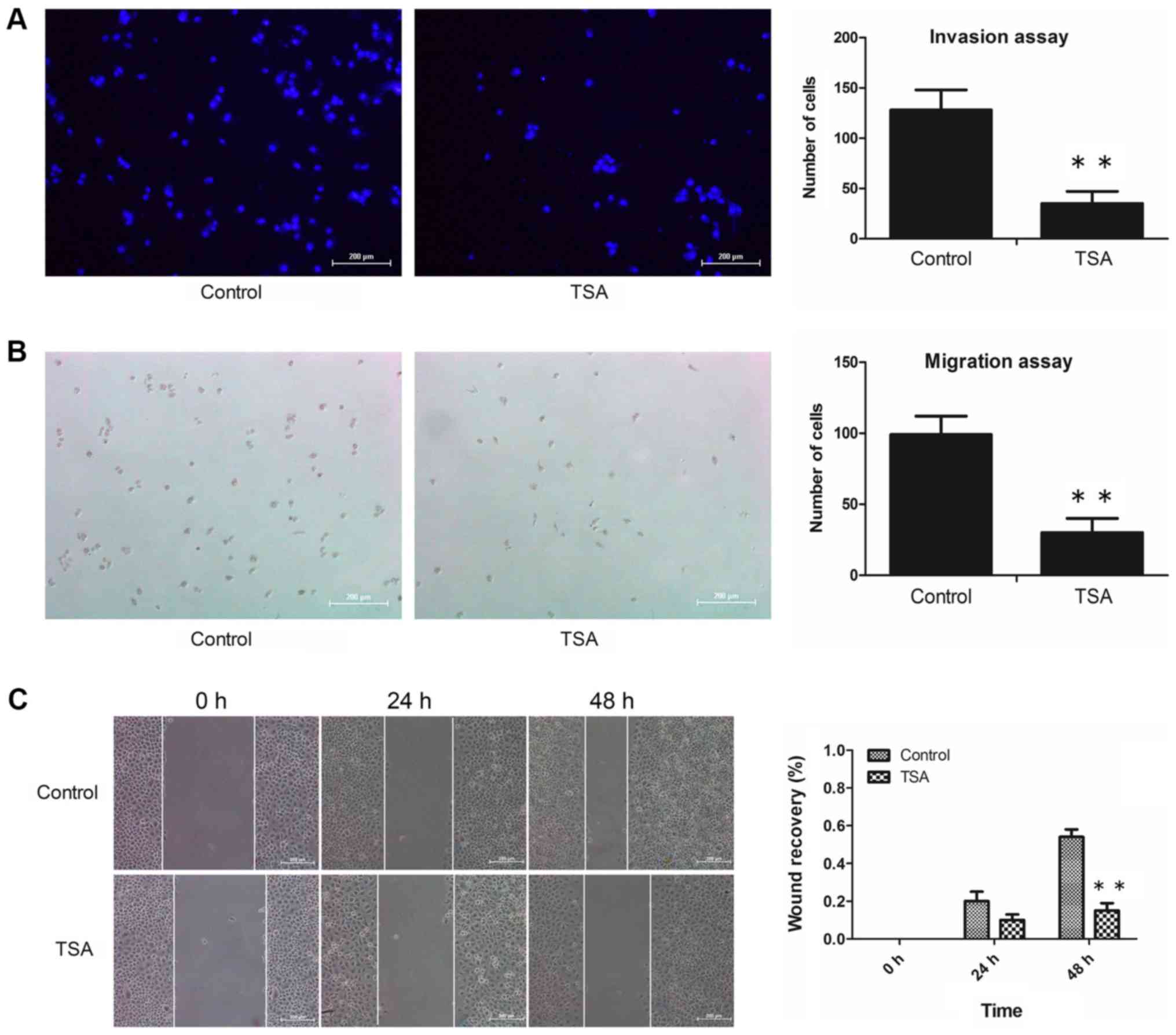
treatment with 100 nM TSA for 48 h, the numbers of invasive cells
adhering to the underside of the filter (Fig. 2A) and migrated cells in the lower
chamber (Fig. 2B) had decreased
markedly compared with the control. Additionally, the results of
the wound healing assay revealed that treatment with TSA caused a
decrease in the migratory capacity of the MCF-7 cells (Fig. 2C). These results suggested that the
decreased invasive and migration capabilities of the cells
following TSA treatment may have resulted from reversion of EMT,
rather than having been caused by any anti-proliferation effect.
Taken together, we concluded that the invasive and migratory
abilities of MCF-7 cells were attenuated by TSA.
TSA reverses EMT in MCF-7 cells
As aforementioned, the epithelial and mesenchymal
phenotypes are distinct cellular states, and cells are capable of
transitioning between them. The expression levels of EMT-associated
biomarkers and transcription factors reflect the nature of the cell
phenotype. Therefore, in MCF-7 cells, changes in TSA-induced mRNA
and protein expression levels of E-cadherin, vimentin and SLUG were
determined. RT-qPCR analysis revealed that TSA led to an
upregulation of E-cadherin, and a downregulation of vimentin and
SLUG mRNA expression levels (Fig.
3A). Similarly, at the protein level, an increased expression
of E-cadherin, and a decrease in the expression levels of vimentin
and SLUG were determined via western blot analysis (Fig. 3B; Table
I.). Furthermore, following treatment with TSA, changes in the
expression level of E-cadherin on the cell membrane were also
detected by confocal microscopy. TSA-induced increases of
E-cadherin was observed visually (Fig.
3C). Taken together, these results suggested that TSA was able
to reverse EMT in MCF-7 cells.
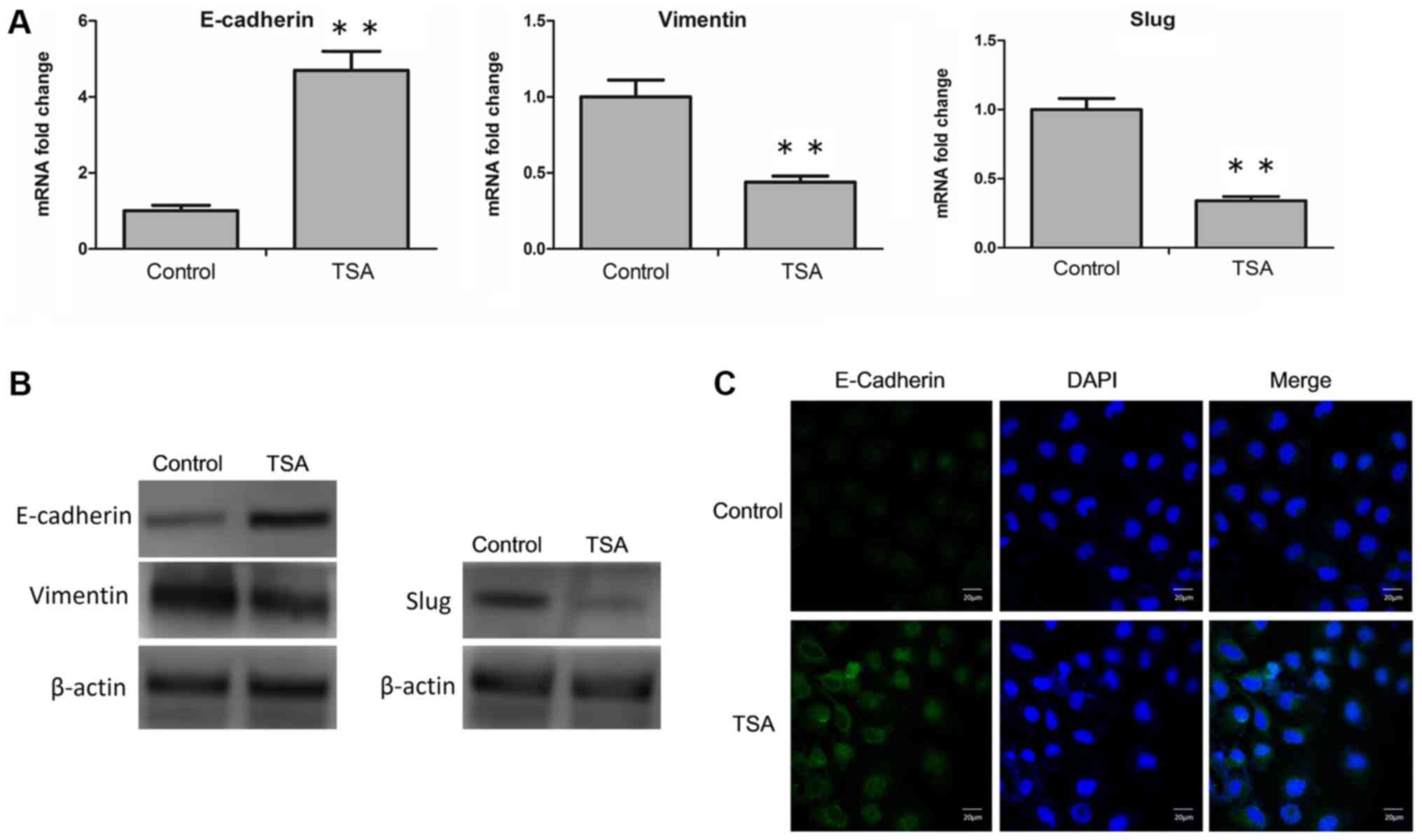 | Figure 3.Treatment with TSA reverses EMT in
MCF-7 cells. (A) MCF-7 cells were treated with or without TSA, and
the mRNA level of E-cadherin, vimentin and SLUG were analyzed by
reverse transcription-quantitative polymerase chain reaction.
**P<0.01 vs. control. (B) MCF-7 cells were incubated with or
without TSA, and subsequently the protein levels of E-cadherin,
vimentin and SLUG were analyzed by western blot analysis. (C) MCF-7
cells were treated with or without TSA, and the cellular location
of E-cadherin (green) was examined by confocal microscopy. Scale
bar, 20 µm. TSA, trichostatin A; EMT, epithelial-mesenchymal
transition; E-cadherin, epithelial cadherin; SLUG, zinc finger
protein SNAI2. |
 | Table I.Protein levels of E-Cadherin,
vimentin and SLUG in MCF7 cells incubated with or without TSA. |
Table I.
Protein levels of E-Cadherin,
vimentin and SLUG in MCF7 cells incubated with or without TSA.
| Group | Control | TSA |
|---|
| E-cadherin/β-actin
expression | 0.70±0.041 |
1.38±0.0097a |
| Vimentin/β-actin
expression | 0.99±0.038 |
0.51±0.028a |
| SLUG/β-actin
expression | 1.02±0.037 |
0.35±0.0095a |
TSA-induced suppression of SLUG is
involved in reversing EMT
SLUG exerts a crucial role in promoting the EMT
process. In MCF-7 cells, upregulation of E-cadherin and
downregulation of vimentin may be due to TSA-mediated suppression
of SLUG. To investigate this, the control vector pcDNA-3.1 and
pcDNA-SLUG were transfected into MCF-7 cells, and changes in the
mRNA and protein levels of SLUG, E-cadherin and vimentin were
examined. The results revealed that E-cadherin was downregulated,
and SLUG and vimentin were upregulated, at the mRNA (Fig. 4A) and protein (Fig. 4B; Table
II) levels. Furthermore, following treatment with 100 nM TSA
for 48 h, these changes were reversed in the SLUG-overexpressing
cells (Fig. 4A and B; Table II). Additionally, the expression
levels of SLUG, E-cadherin and vimentin protein were detected in
SLUG siRNA-transfected MCF-7 cells compared with control
siRNA-transfected MCF-7 cells. The invasive and migratory abilities
of the two groups were also investigated by Transwell invasion and
migration assays. As indicated in Fig.
4C and D, SLUG knockdown led to an upregulation of E-cadherin,
and a downregulation of vimentin, at the protein level (Fig. 4D; Table
III). Compared with the siNC group, cells transfected with SLUG
siRNA exhibited decreased levels of invasion and migration
(Fig. 4C). These results indicated
that TSA-mediated suppression of SLUG was involved in the reversal
of EMT.
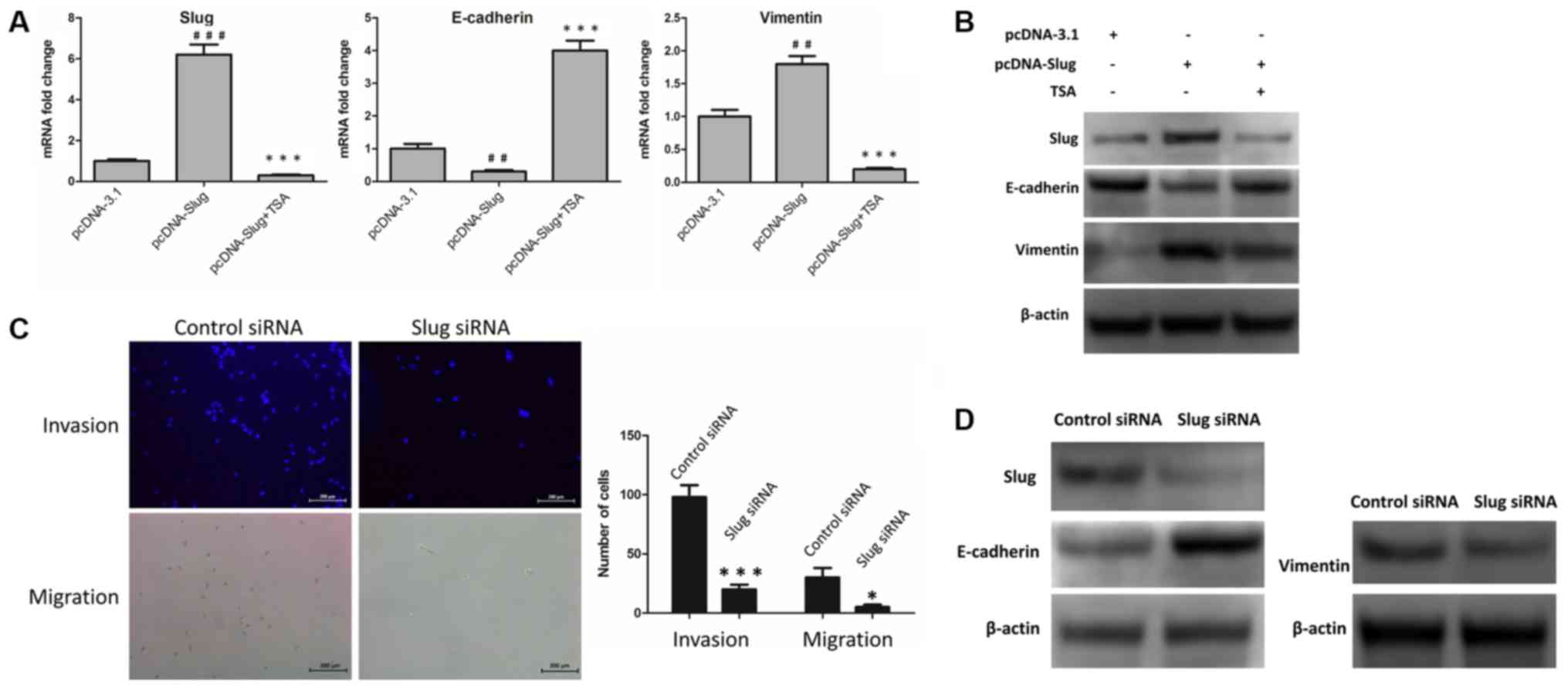 | Figure 4.TSA-mediated suppression of SLUG is
involved in reversing EMT. (A) pcDNA-3.1 and pcDNA-SLUG were
expressed in MCF-7 cells, and cells with pcDNA-SLUG were treated
with or without TSA. Subsequently, the mRNA levels of SLUG,
E-cadherin and vimentin were examined by reverse
transcription-quantitative polymerase chain reaction.
##P<0.01 and ###P<0.001 vs. pcDNA-3.1.
**P<0.01 and ***P<0.001 vs. pcDNA-SLUG. (B) pcDNA-3.1 and
pcDNA-SLUG were expressed in MCF-7 cells, and cells with pcDNA-SLUG
were incubated with or without TSA. Subsequently, the protein
levels of SLUG, E-cadherin and vimentin were examined by western
blot analysis. (C) SLUG siRNA or control siRNA was transfected in
MCF-7 cells, and images were captured of the invasive and migrated
cells. Scale bar=200 µm. *P<0.05 and ***P<0.001 vs. control.
(D) SLUG siRNA or control siRNA were transfected in MCF-7 cells,
and subsequently the expression levels of SLUG, E-cadherin, and
vimentin protein were examined by western blot analysis. TSA,
trichostatin A; EMT, epithelial-mesenchymal transition; SLUG, zinc
finger protein SNAI2; E-cadherin, epithelial cadherin; siRNA, small
interfering RNA. |
| Table II.Protein levels of SLUG, E-Cadherin
and vimentin in cells transfected with pcDNA-3.1 or pcDNA-SLUG and
incubated with or without TSA. |
Table II.
Protein levels of SLUG, E-Cadherin
and vimentin in cells transfected with pcDNA-3.1 or pcDNA-SLUG and
incubated with or without TSA.
|
| pcDNA-3.1 | pcDNA-SLUG | pcDNA-SLUG +
TSA |
|---|
| SLUG/β-actin
expression | 1.54±0.068 |
3.06±0.34b |
1.13±0.076aa |
| E-cadherin/β-actin
expression | 2.08±0.081 |
1.37±0.13b |
1.83±0.07bb |
| Vimentin/β-actin
expression | 0.29±0.019 |
1.20±0.11a |
0.75±0.059bb |
| Table III.Protein levels of SLUG, E-Cadherin
and vimentin in cells transfected with control siRNA or SLUG
siRNA. |
Table III.
Protein levels of SLUG, E-Cadherin
and vimentin in cells transfected with control siRNA or SLUG
siRNA.
|
| Control siRNA | SLUG siRNA |
|---|
| SLUG/β-actin
expression | 1.019±0.037 |
0.28±0.0071a |
| E-cadherin/β-actin
expression | 1.19±0.011 |
2.28±0.034a |
| Vimentin/β-actin
expression | 0.59±0.063 |
0.32±0.083b |
Discussion
It has been well established that E-cadherin and
vimentin are selectively expressed, and perform their specific
functions in epithelial and mesenchymal cellular states,
respectively. E-cadherin is a membrane glycoprotein that normally
binds to another connexin, β-catenin, to maintain the integrity of
the morphology and skeletal structure of epithelial cells. When the
expression of E-cadherin is decreased, the levels of connectivity
between tumor cells decrease, and the dispersed tumor cells become
more motile, which consequently facilitates the processes of tumor
invasion and migration. Vimentin is a structural cytoskeletal
protein that constitutes the intermediate filaments of mesenchymal
cells. High expression of E-cadherin, and the absence of vimentin,
is associated with a low likelihood that cancer cells will be able
to invade and migrate (23,24). In the present study, TSA-mediated
increases in E-cadherin and decreases in vimentin were observed,
and TSA-induced suppression of the invasion and migration
capabilities of the MCF-7 cells was also confirmed, which were
phenomena that suppressed the distant migration of MCF-7 cells.
In addition, the transcription factor SLUG was
downregulated following treatment with TSA. Typically, EMT-inducing
transcription factors are classified into two groups: For example,
SLUG, SNAIL, Krueppel-like factor 8, transcription factor 3 and
Zeb1 repress E-cadherin transcription in a direct manner, whereas
transcription factors such as Twist, Forkhead box protein C2,
transcription factor 4 and Goosecoid repress activity of the
E-cadherin promoter indirectly (25). Importantly, these transcription
factors are involved in the downregulation of E-cadherin.
Furthermore, SLUG is also able to promote the expression of
vimentin, and consequently induce EMT-like changes (14). The results of the present study
indicated that SLUG was able to induce downregulation of
E-cadherin, and upregulation of vimentin in MCF-7 cells, and
TSA-mediated suppression of SLUG was involved in the process of
reversing EMT. In addition, a large number of previously published
studies have suggested that SLUG promotes the initial stages of EMT
in lung, bladder, colorectal, prostate and nasopharyngeal cancer
cells (26–31). Therefore, TSA-mediated repression of
SLUG may be the critical factor with respect to the observed effect
of EMT reversal.
HDACs are involved in various physiological and
pathological regulatory processes, and HDACIs exert potent
EMT-reversal effects on several types of non-tumor cells, and on
tumor cells. TSA-mediated reversal of EMT is closely associated
with the transforming growth factor-β (TGF-β) signaling pathway. It
has reported that HDAC1 and HDAC2 are involved in TGF-β-induced
EMT, and TSA completely inhibits TGF-β-mediated EMT in hepatocytes
and kidney tubular epithelial cells (32,33). In
retinal pigment epithelium cells (34), TSA-mediated inhibition of HDAC
activity was demonstrated to markedly suppress cellular
proliferation and TGF-β-induced EMT. In addition, TSA inhibited
TGF-β2-mediated EMT via regulating not only the canonical Smad
signaling pathway, but also the non-canonical TGF-β/Akt,
mitogen-activated protein kinase and extracellular signal-regulated
kinase 1 and 2 pathways. These studies (32–34) may
partly contribute towards our understanding of the mechanism of
TSA-mediated inhibition of SLUG expression in the present study.
Conversely, our previous study (20)
identified that the amounts of HDAC1 and HDAC2 proteins binding to
the SLUG gene promoter were increased in the TSA-treated SW480 and
PC3 cells. Although few studies have been published on the ability
of HDAC inhibitors to induce an increase in the levels of HDACs
binding to the target gene promoter, a similar mechanism may be
responsible for TSA-induced suppression of SLUG in MCF-7 cells.
However, the underlying mechanism requires further
investigation.
An increasing number of studies have indicated that
EMT not only enhances the invasive and migratory abilities of tumor
cells, but also closely contributes to other of their malignant
characteristics. It was suggested that human mammary epithelial
cells obtained partial stem cell characteristics by inducing EMT
transformation, and the epithelial tumor cells with stem cell
characteristics were identified to express mesenchymal biomarkers
(35). EMT also triggers tumor
immunosuppressive properties, assisting in their immune evasion
(36,37). For example, certain immunosuppressive
cytokines were induced by the EMT-associated transcription factor
Snail, leading to differentiation of regulatory T cells, function
damage in dendritic cells, and tumor resistance to cytotoxic T
cells (36). Furthermore, the
resistance of tumor cells to chemotherapy, radiotherapy and
immunotherapy were also increased by EMT (38–40). In
the presence of EMT, breast and cervical cancer cells were
demonstrated to be highly resistant to paclitaxel (41–43). The
EMT-associated transcription factor SLUG was revealed to be
involved in tumor resistance to radiotherapy and chemotherapy by
antagonizing p53-mediated apoptosis (44). Finally, SLUG inhibition led to an
increase in the radiosensitivity of nasopharyngeal carcinoma
(45). Based on these previous
studies, it may be possible to conclude from the results of the
present study that TSA-induced EMT reversal may have prevented
MCF-7 cells from developing into a more malignant phenotype.
In conclusion, the most significant results of the
present study were that treatment with TSA reversed EMT and
attenuated the invasive and migratory abilities in MCF-7 breast
cancer cells. Subsequent studies will involve the detection of the
invasive and migratory abilities on the condition of E-cadherin
overexpression in the absence of TSA, or blocking of E-cadherin
using neutralizing antibody. The EMT-reversal effect of TSA in
triple-negative breast cancer cells, including MDA-MB-231 and
MDA-MB-435 cells, will also be investigated. Furthermore,
18F-FDG and 18F-FLT positron emission
tomography/computed tomography imaging will be employed to evaluate
TSA-mediated anti-tumor effects in different types of breast cancer
cell lines. However, although these results require further
investigation in vivo, the data from the present study have
provided novel information regarding the chemotherapy of breast
cancer.
Acknowledgements
Not applicable.
Funding
The present study was partially supported by Science
and Technology Department of Sichuan Province (project 2019YJ0574),
Science and Technology Department of Sichuan Province (key project
2017JY0081), and Cadres Health Care of Sichuan Provincial (project
no. 2017-803).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW, XJ and ZC proposed the hypothesis and designed
the majority of the experiments. XW wrote the manuscript. XW, SC
and TS performed the experiments and analyzed the results. HL, DX,
MZ, YY, XL, GZ and XZ assisted in the execution of some
experiments. XJ and ZC were involved in revising the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lee YT: Breast carcinoma: Pattern of
metastasis at autopsy. J Surg Oncol. 23:175–180. 2010. View Article : Google Scholar
|
|
2
|
Weil RJ, Palmieri DC, Bronder JL, Stark AM
and Steeg PS: Breast cancer metastasis to the central nervous
system. Am J Pathol. 167:913–920. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vaidya JS, Keshtgar M and Baum M: Diseases
of the breast. Br J Cancer. 83:1769–1770. 2000. View Article : Google Scholar
|
|
4
|
Behrens J, Mareel MM, Van Roy FM and
Birchmeier W: Dissecting tumor cell invasion: Epithelial cells
acquire invasive properties after the loss of uvomorulin-mediated
cell-cell adhesion. J Cell Biol. 108:2435–2447. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chengsen X, David P, Christo V, Carol X
and Neilson EG: The gatekeeper effect of epithelial-mesenchymal
transition regulates the frequency of breast cancer metastasis.
Cancer Res. 63:3386–3394. 2003.PubMed/NCBI
|
|
6
|
Thompson EW, Newgreen DF and David T:
Carcinoma invasion and metastasis: A role for
epithelial-mesenchymal transition? Cancer Res. 65:5991–5995. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Felipe Lima J, Nofech-Mozes S, Bayani J
and Bartlett JM: EMT in breast carcinoma-a review. J Clin Med.
5(pii): E652016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jean Paul T and Sleeman JP: Complex
networks orchestrate epithelial-mesenchymal transitions. Nat Rev
Mol Cell Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Michael Z and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu F, Gu LN, Shan BE, Geng CZ and Sang
MX: Biomarkers for EMT and MET in breast cancer: An update. Oncol
Lett. 12:4869–4876. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
David S, Socorro María RP, Hardisson D,
Cano A, Moreno-Bueno G and Palacios J: Epithelial-mesenchymal
transition in breast cancer relates to the basal-like phenotype.
Cancer Res. 68:989–997. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yi P, Jing L, Zhang Y, Wang N, Liang H,
Liu Y, Zhang CY, Zen K and Gu H: SLUG-upregulated miR-221 promotes
breast cancer progression through suppressing E-cadherin
expression. Sci Rep. 6:257982016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hajra KM, Chen DY and Fearon ER: The SLUG
zinc-finger protein represses E-cadherin in breast cancer. Cancer
Res. 62:1613–1618. 2002.PubMed/NCBI
|
|
14
|
Vuoriluoto K, Haugen H, Kiviluoto S,
Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB and Ivaska J:
Vimentin regulates EMT induction by SLUG and oncogenic H-Ras and
migration by governing Axl expression in breast cancer. Oncogene.
30:1436–1448. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu S, Ren J, Chen HB, Wang Y, Liu Q, Zhang
R, Jiang SW and Li J: Cytostatic and apoptotic effects of DNMT and
HDAC inhibitors in endometrial cancer cells. Curr Pharm Des.
20:1881–1887. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dowdy SC, Jiang S, Zhou XC, Hou X, Jin F,
Podratz KC and Jiang SW: Histone deacetylase inhibitors and
paclitaxel cause synergistic effects on apoptosis and microtubule
stabilization in papillary serous endometrial cancer cells. Mol
Cancer Ther. 5:2767–2776. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yoshikawa M, Hishikawa KT and Fujita T:
Inhibition of histone deacetylase activity suppresses
epithelial-to-mesenchymal transition induced by TGF-beta1 in human
renal epithelial cells. J Am Soc Nephrol. 18:58–65. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiao W, Chen X, Liu X, Luo L, Ye S and Liu
Y: Trichostatin A, a histone deacetylase inhibitor, suppresses
proliferation and epithelial-mesenchymal transition in retinal
pigment epithelium cells. J Cell Mol Med. 18:646–655. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aki K, Potter JJ, Michael C, Zhen D,
Esteban M and Koteish AA: Histone deacetylase inhibition suppresses
the transforming growth factor beta1-induced
epithelial-to-mesenchymal transition in hepatocytes. Hepatology.
52:1033–1045. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang X, Xu J, Wang H, Wu L, Yuan W, Du J
and Cai S: Trichostatin A, a histone deacetylase inhibitor,
reverses epithelial-mesenchymal transition in colorectal cancer
SW480 and prostate cancer PC3 cells. Biochem Biophys Res Commun.
456:320–326. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hao W, Wang HS, Zhou BH, Li CL, Zhang F,
Wang XF, Zhang G, Bu XZ, Cai SH and Du J: Epithelial-mesenchymal
transition (EMT) induced by TNF-α requires AKT/GSK-3β-mediated
stabilization of snail in colorectal cancer. PLoS One.
8:e566642013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gilles C, Newgreen DF, Sato H and Thompson
EW: Matrix metalloproteases and epithelial-to-mesenchymal
transition. 2005. View Article : Google Scholar
|
|
24
|
Faghihloo E, Akbari A, Adjaminezhad-Fard F
and Mokhtari-Azad T: Transcriptional regulation of E-cadherin and
oncoprotein E7 by valproic acid in HPV positive cell lines. Iran J
Basic Med Sci. 19:601–607. 2016.PubMed/NCBI
|
|
25
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grzegrzolka J, Biala M, Wojtyra P,
Kobierzycki C, Olbromski M, Gomulkiewicz A, Piotrowska A, Rys J,
Podhorska-Okolow M and Dziegiel P: Expression of EMT Markers SLUG
and TWIST in breast cancer. Anticancer Res. 35:3961–3968.
2015.PubMed/NCBI
|
|
27
|
Shih JY and Yang PC: The EMT regulator
SLUG and lung carcinogenesis. Carcinogenesis. 32:1299–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jing Y, Cui D, Guo W, Jiang J, Jiang B, Lu
Y, Zhao W, Wang X, Jiang Q, Han B and Xia S: Activated androgen
receptor promotes bladder cancer metastasis via SLUG mediated
epithelial-mesenchymal transition. Cancer Lett. 348:135–145. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Y, Zhao Z, Xu C, Zhou Z, Zhu Z and You
T: HMGA2 induces transcription factor SLUG expression to promote
epithelial-to-mesenchymal transition and contributes to colon
cancer progression. Cancer Lett. 355:130–140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu YN, Abou-Kheir W, Yin JJ, Fang L,
Hynes P, Casey O, Hu D, Wan Y, Seng V, Sheppard-Tillman H, et al:
Critical and reciprocal regulation of KLF4 and SLUG in transforming
growth factor β-initiated prostate cancer epithelial-mesenchymal
transition. Mol Cell Biol. 32:941–953. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang W, Li X, Zhang W, Li W, Yi M, Yang J,
Zeng Z, Colvin Wanshura LE, McCarthy JB, Fan S, et al:
Oxidored-nitro domain containing protein 1 (NOR1) expression
suppresses SLUG/vimentin but not snail in nasopharyngeal carcinoma:
Inhibition of EMT in vitro and in vivo in mice. Cancer Lett.
348:109–118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lei W, Zhang K, Pan X, Hu Y, Wang D, Yuan
X, Shu G and Song J: Histone deacetylase 1 is required for
transforming growth factor-β1-induced epithelial-mesenchymal
transition. Int J Biochem Cell Biol. 42:1489–1497. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H
and Lee HB: Histone deacetylase-2 is a key regulator of
diabetes-and transforming growth factor-β1-induced renal injury. Am
J Physiol Renal Physiol. 297:F729–F739. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiao W, Chen X, Liu X, Luo L, Ye S and Liu
Y: Trichostatin A, a histone deacetylase inhibitor, suppresses
proliferation and epithelial-mesenchymal transition in retinal
pigment epithelium cells. J Cell Mol Med. 18:646–655. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Knutson KL, Lu H, Stone B, Reiman JM,
Behrens MD, Prosperi CM, Gad EA, Smorlesi A and Disis ML:
Immunoediting of cancers may lead to epithelial to mesenchymal
transition. J Immunol. 177:1526–1533. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ansieau S, Bastid J, Doreau A, Morel AP,
Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S,
et al: Induction of EMT by twist proteins as a collateral effect of
tumor-promoting inactivation of premature senescence. Cancer Cell.
14:79–89. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gal A, Sjoblom T, Fedorova L, Imreh S,
Beug H and Moustakas A: Sustained TGF beta exposure suppresses Smad
and non-Smad signalling in mammary epithelial cells, leading to EMT
and inhibition of growth arrest and apoptosis. Oncogene.
27:1218–1230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yongqing L, Shahenda EN, Darling DS,
Yujiro H and Dean DC: Zeb1 links epithelial-mesenchymal transition
and cellular senescence. Development. 135:579–588. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang AD, Fan F, Camp ER, van Buren G, Liu
W, Somcio R, Gray MJ, Cheng H, Hoff PM and Ellis LM: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12:4147–4153.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q,
Tang F, Chen ZQ, Liu XP and Xu ZD: Twist1-mediated
adriamycin-induced epithelial-mesenchymal transition relates to
multidrug resistance and invasive potential in breast cancer cells.
Clin Cancer Res. 15:2657–2665. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cheng GZ, Joseph C, Qi W, Weizhou Z, Sun
CD and Wang LH: Twist transcriptionally up-regulates AKT2 in breast
cancer cells leading to increased migration, invasion and
resistance to paclitaxel. Cancer Res. 67:1979–1987. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kurrey NK, Jalgaonkar SP, Joglekar AV,
Ghanate AD, Chaskar PD, Doiphode RY and Bapat SA: Snail and Slug
mediate radioresistance and chemoresistance by antagonizing
p53-mediated apoptosis and acquiring a stem-like phenotype in
ovarian cancer cells. Stem Cells. 27:2059–2068. 2010. View Article : Google Scholar
|
|
45
|
Yang H, Zhang G, Che X and Yu S: Slug
inhibition increases radiosensitivity of nasopharyngeal carcinoma
cell line C666-1. Exp Ther Med. 15:3477–3482. 2018.PubMed/NCBI
|