Introduction
Cancer is the second leading cause of mortality in
the United States and some European countries, such as Finland and
Scotland (1–3). The knowledge of molecular genetic
mechanisms underlying tumorigenesis has increased since the
discovery of the TP53 tumor suppressor gene (4–9).
Tumor suppressor genes normally help prevent unrestrained cellular
growth and promote DNA repair and cell cycle checkpoint
activation.
SAM and SH3 domain containing 1 (SASH1), a novel
tumor suppressor gene mapped on chromosome 6q24.3, is possibly
involved in the tumorigenesis of breast and other solid tumors. It
is a member of the SH3 domain-containing expressed in lymphocytes
(SLY1) gene family that encodes signal adaptor proteins composed of
several protein-protein interaction domains. The other members of
this family are expressed mainly in haematopoietic cells, whereas
SASH1 has demonstrated an ubiquitous expression. It is
downregulated in the majority (74%) of breast tumors in comparison
with corresponding normal breast epithelial tissues. Moreover, the
expression levels of SASH1 are strongly and significantly reduced
in colon cancer of UICC stage II, III and IV, as well as in liver
metastases. Additionally, Martini et al demonstrated that
SASH1 plays a crucial role in tumor formation by regulating the
adhesiveness and migratory behavior of cancer cells (10). Furthermore, Chen et al
indicated that in the A549 lung cancer cells of the pcDNA3.1-SASH1
transfected group, cell viability, proliferation and migration were
significantly reduced compared to the control cells, while a cell
cycle arrest was observed in G1 (11). However, the specific mechanism by
which SASH1 influences these biological behaviors is yet to be
determined. Moreover, to date, no studies have been reported on the
biological role of SASH1 in melanoma cells.
In order to examine the function of the SASH1 gene
in melanoma, we transfected the SASH1 gene into the A-375 melanoma
cells. Subsequently, we examined the changes in the relevant
biological characteristics of the A-375 cell line and preliminarily
analyzed the significance of the SASH1 gene in melanoma. We also
investigated the correlation between SASH1 expression and the
associated molecular changes in the cell cycle to provide further
insight into the potential use of SASH1 for the targeted therapy of
melanoma. The tumor suppressive effects of SASH1 occured due to the
accumulation of cells at the G2/M stage of the cell cycle.
Materials and methods
Construction of recombinant plasmids
The wild-type full-length DNA coding the human SASH1
gene was synthesized by Clontech Laboratory Incorporation and
cloned into the pEGFP-C3 vector (Clontech, Palo Alto, CA, USA) to
construct pEGFP-C3-SASH1, N-terminally tagged with green
fluorescence protein (GFP).
Cell culture and transfection
The cells were cultured and maintained in DMEM high
glucose medium (Gibco BRL, Grand Island, NY, USA), containing 10%
Front FBS (Front Biomedicals, Dunedin, New Zealand), penicillin
(100 U/ml) and streptomycin (100 U/ml), and were incubated at 37°C
in a humidified incubator under an atmosphere of 5% CO2.
The cells transfected with pEGFP-C3 were used as the negative
control. The cells transfected with pEGFP-C3-SASH1 [named A-375
(SASH1)] or pEGFP-C3 [named A-375 (EGFP)] were examined under a
fluorescence microscope (Olympus, Tokyo, Japan) and a corresponding
single cell clone was selected with neomycin. After culture
propagation for certain periods of time, SASH1 gene expression was
measured by western blot analysis.
Western blot analysis
Cells were lysed with cell lysis buffer for western
blot analysis and immunoprecipitation (IP) (Beyotime Institute of
Biotechnology, Jiangsu, China), containing protease inhibitors
(Pierce Biotechnology, Rockford, IL, USA). GAPDH levels were used
to normalize loading. SASH1 antibody was purchased from Bethyl
laboratories Inc. (Montgomery, MA, USA) and the primary GAPDH
antibody was purchased from Proteintech Group (Chicago, IL,
USA).
Cell proliferation assay
Proliferation assay was carried out to assess cell
viability in the A-375 (SASH1), A-375 (EGFP) and A-375 cell lines.
Cells were seeded on 24-well plates and each experiment was
performed in triplicate. After 48 h, the cells were washed with PBS
and assessed using a 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) (AppliChem, Darmstadt, Germany)
assay.
Cell apoptosis assay
To investigate apoptotic body, cells were seeded on
a 24-well plate and each assay was repeated 3 times. After 72 h,
cells were washed with PBS and stained with
4,6-diamidino-2-phenylindole (DAPI) according to the Genmed nuclear
staining kit (Genmed Scientifics Inc., Shanghai, China). Stained
cells were photographed under a Leica fluorescence convert
microscope (Leica Camera AG, Mannheim, Germany).
To examine the rate of apoptosis, cells from each
group (dish, 6 cm in diameter) were gently resuspended in 195 μl of
Annexin V-FITC binding buffer. Subsequently, 5 μl of Annexin V-FITC
were added followed by gentle mixing. The cells were then incubated
for 10 min at room temperature in the dark and subsequently
centrifuged at 1,000 × g for 5 min. The supernatant was discarded,
and 190 μl of Annexin V-FITC binding buffer was added to gently
resuspend the cells. Finally, 10 μl of PI staining solution were
added and gently mixed with the cells. The cells were then placed
on ice and analyzed immediately by flow cytometry.
Cell cycle analysis
Cell cycle analysis was carried out by flow
cytometry (FCM). Briefly, A-375 (SASH1), A-375 (EGFP) and A-375
cell lines were seeded in 10–cm dishes and each experiment was
performed in 4 repeats. The experiments were carried out with the
Genmed cell cycle analysis kit (Genmed Scientifics Inc.). Briefly,
after 72 h, the cells were digested with trypsin after washing
twice with PBS and fixed in ethanol for 16 h at 4°C. The samples
were concentrated by removing the ethanol and treated with Genmed
staining solution for 45 min at 37°C in the dark. Cell cycle
distribution was determined using a flow cytometer (Beckman
Coulter, Miami, FL, USA) and 10,000 cells were analyzed with Expo32
ADC Analysis software.
RNA extraction
RNA was extracted using the Aqua-Spin RNA Isolation
Mini kit (Watson Biotechnologies, Shanghai, China). The
concentration and quality of the isolated DNA and RNA were measured
with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies,
Montchanin, DE, USA).
Real-time PCR
First-strand cDNA was synthesized using PrimeScript
RT reagent kit according to the manufacturer’s instructions
(Takara, Ostu, Shiga, Japan). The CCNB1 (cyclin B), CDK1 (Cdc2),
TP53 (p53) and WEE1 genes were co-amplified with a fragment of the
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene, which served
as the internal standard. Q-PCR was conducted by the SYBR Premix Ex
Taq kit (Takara, Ostu, Shiga, Japan) on the ABI 7900HT Fast
Real-Time PCR System (Life Technologies, Carlsbad, CA, USA). The
primer pairs are shown in Table I
and the cycling conditions of 40 cycles of PCR were as follows:
95°C/5 sec, 55°C/30 sec and 72°C/30 sec. Each sample was run in 4
repeats and all the PCR data were analyzed with the ABI 7900HT
system software 2.3 version.
 | Table IPrimers used for real-time PCR. |
Table I
Primers used for real-time PCR.
| Genes | Primers | Sequences
(5′-3′) | Length (bp) |
|---|
| TP53 | p53 sense |
TCTTTGAACCCTTGCTTGC | 132 |
| p53 antisense |
TCCTACTCCCCATCCTCCTC | |
| CDK1 | Cdc2 sense |
TGTCCGCAACAGGGAAGAAC | 140 |
| Cdc2 antisense |
CGAAAGCCAAGATAAGCAACTC | |
| CCNB1 | CCNB1 sense |
GTAAGCCAAGTCATGGAGAATC | 105 |
| CCNB1 antisense |
GCAGCAATCACAAGAAGAAAC | |
| WEE1 | WEE1 sense |
TGTTACACCAGCCTTTCCAG | 168 |
| WEE1 antisense |
ATGAGTCTTTTAGCATGTCCCT | |
| GAPDH | GAPDH sense |
CAAGAAGGTGGTGAAGCAGG | 116 |
| GAPDH antisense |
CGTCAAAGGTGGAGGAGTGG | |
Statistical analysis
Real-time PCR results were compared using one-way
ANOVA analysis among 3 groups. All p-values were two-sided, and
p<0.05 was considered to indicate a statistically significant
difference. SPSS software version 15.0 (SPSS Inc., Chicago, IL,
USA) was used for all statistical analyses.
Results
Generation of SASH1-overexpressing
cells
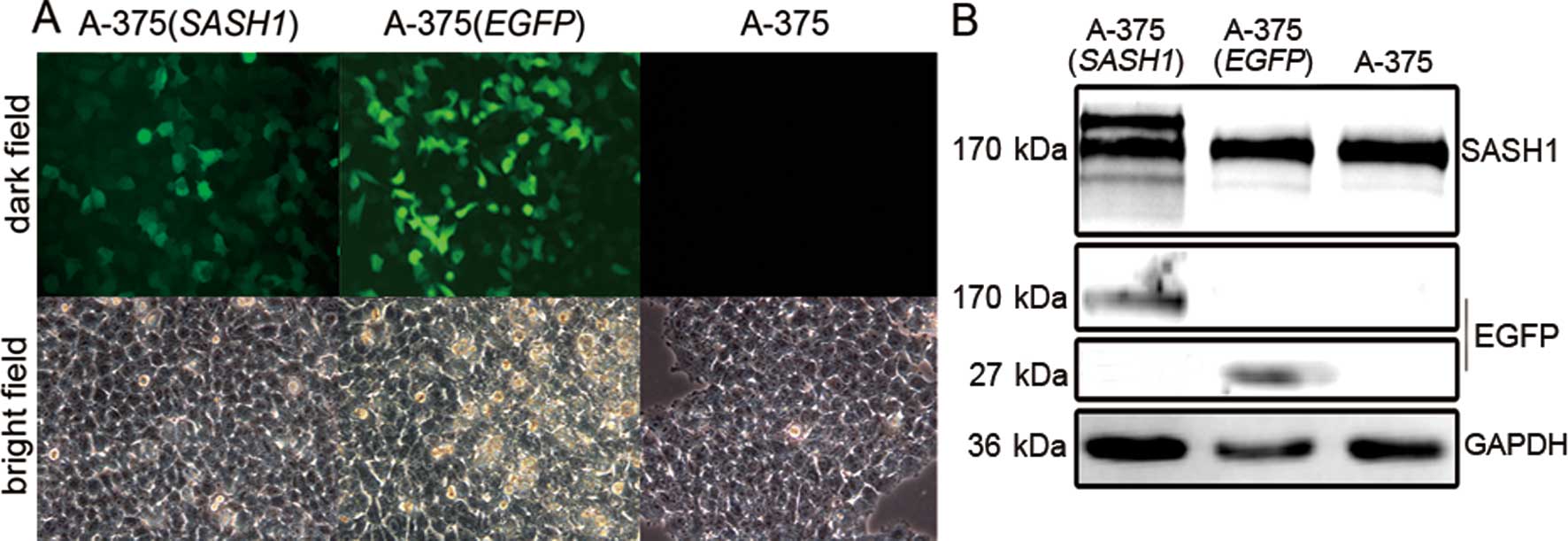
To clarify SASH1 function, a fusion GFP vector
encoding SASH1 was introduced into A-375 cells. GFP was detected by
fluorescence microscopy (Fig. 1A).
GFP was diffusely located in the nucleus and cytoplasm in the A-375
(EGFP) and A-375 (SASH1) cells. The stable cell line [A-375
(SASH1)] was further confirmed by western blot analysis. As shown
in Fig. 1B, the film blotted by
SASH1 antibody reveals the SASH1 fusion protein, 2 bands displayed
near the size of 170 kDa in the A-375 (SASH1) cells, of which, one
was the fusion protein, while the other original protein.
Furthermore, the film blotted by GFP antibody, at the size of 170
kDa displayed a band. All these data proved that the stable cell
lines A-375 (SASH1) and A-375 (EGFP) were successfully
established.
Effects of SASH1 gene on G2/M phase cell
cycle arrest in A-375 cells
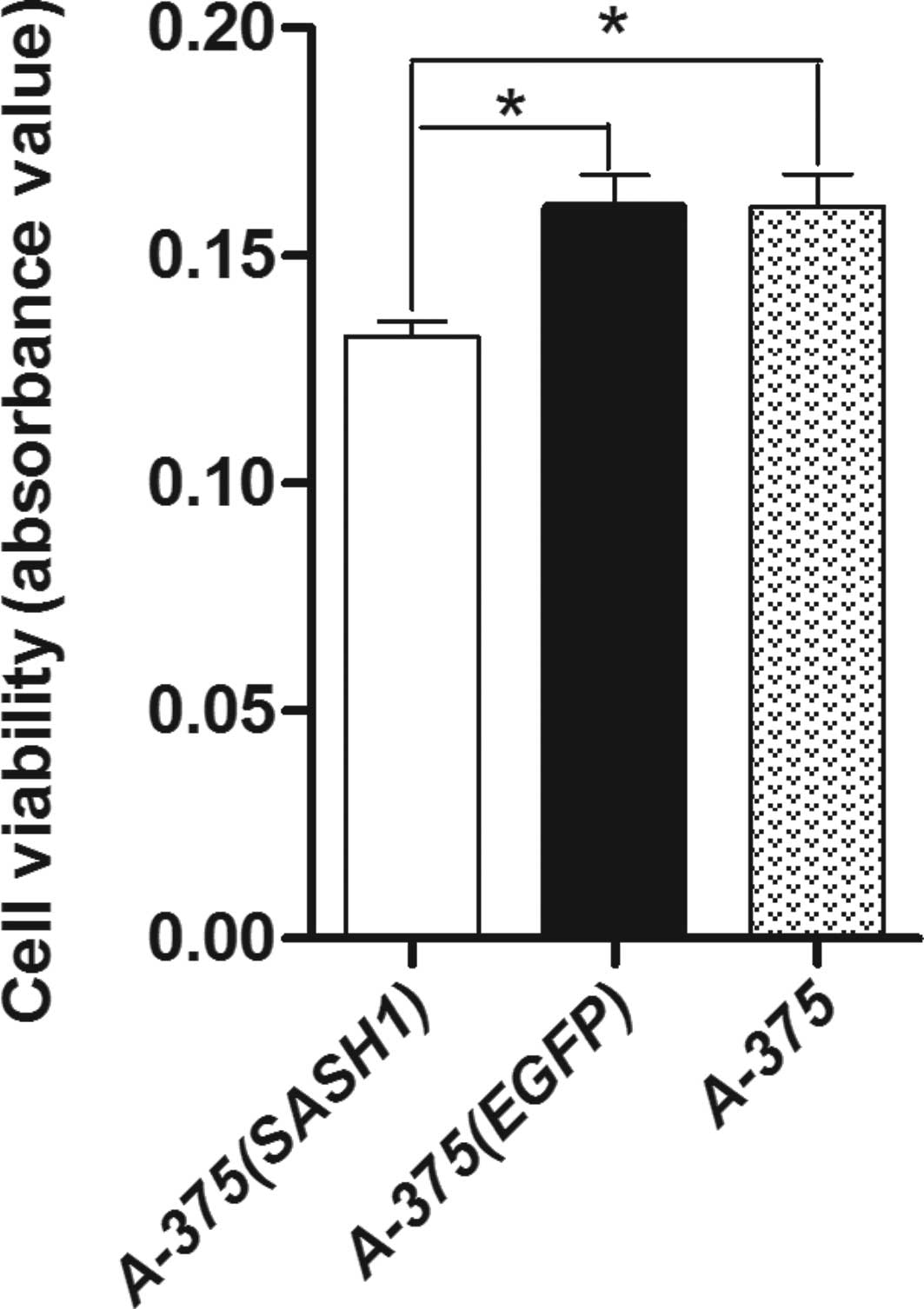
Cell proliferation assay showed that A-375 (SASH1)
cell viability was the lowest among the 3 groups of cells. A-375
(EGFP) cell viability was not significantly diminished compared
with the A-375 cells. Statistical analysis indicated that there was
a significant difference (p<0.001) between the A-375 (SASH1),
A-375 (EGFP) and A-375 cells (Fig.
2). These results suggest that the gene SASH1 inhibits cell
proliferation.
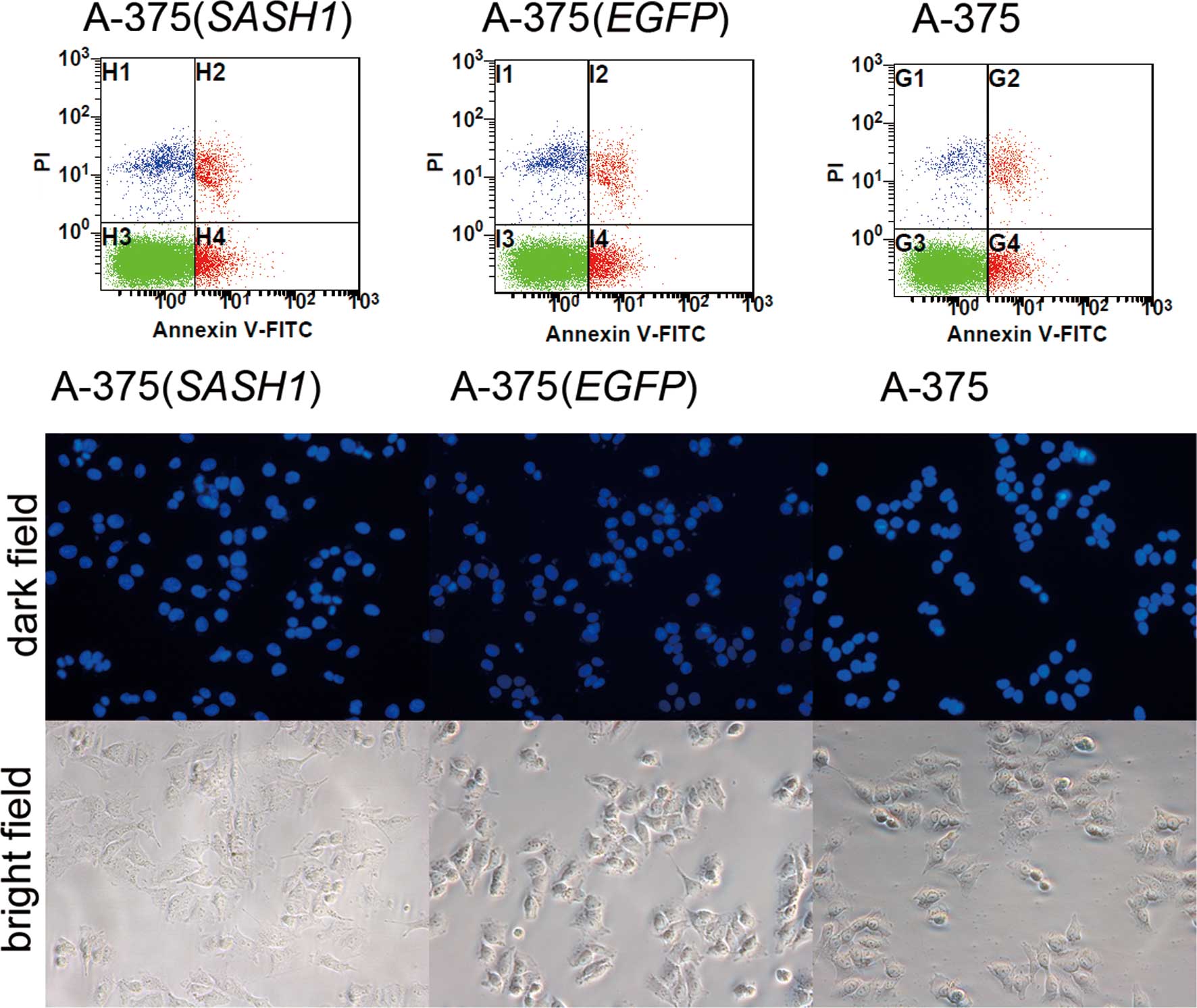
To assess whether cell apoptosis is also involved in
the inhibition of cell proliferation, DAPI staining experiment was
conducted. As shown in Fig. 3, a
slight nuclear fragmentation or chromatin condensation was observed
in the A-375 (SASH1), A-375 (EGFP) and A-375 cells. At the same
time, Annexin V-FITC and PI staining assay also demonstrated that
there was no statistically significant difference in the apoptotic
rate between any 2 groups of the A-375 (SASH1), A-375 (EGFP) and
A-375 cells. Based on these data, it is evident that cell apoptosis
did not participate in the inhibition of cell proliferation.
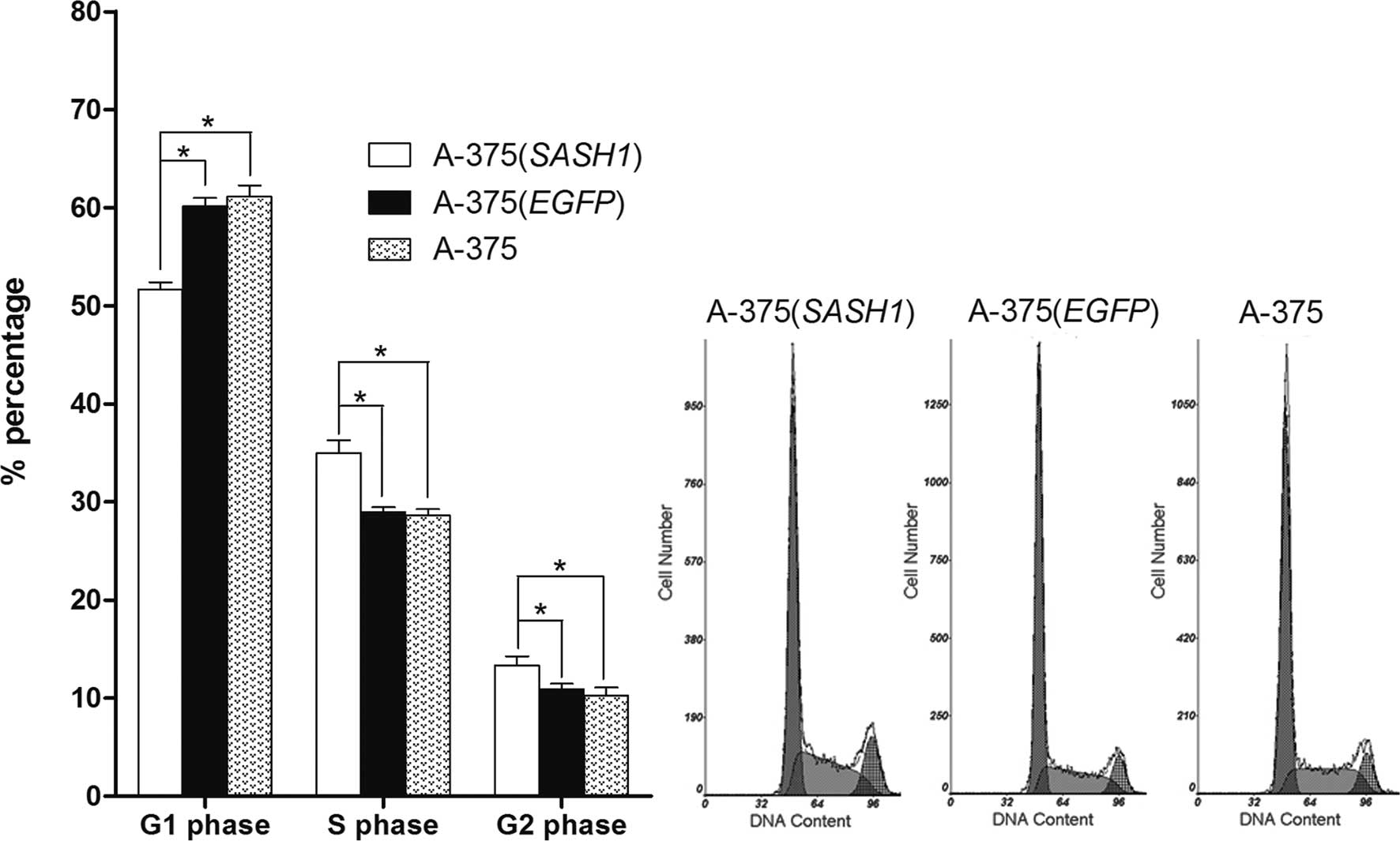
In order to reveal the underlying mechanism behind
the inhibition of cell proliferation, cell cycle assay was
performed. Flow cytometric analysis showed that the number of A-375
(SASH1) cells was significantly increased in the G2/M and S
fractions accompanied by an evident decrease in the number of cells
in G0/G1 compared to the A-375 (EGFP) and A-375 cells. However,
when comparing the A-375 (EGFP) with the A-375 cells, no
statistically significant difference was observed in cell numbers
at the G0/G1, S or G2/M phase (Fig.
4).
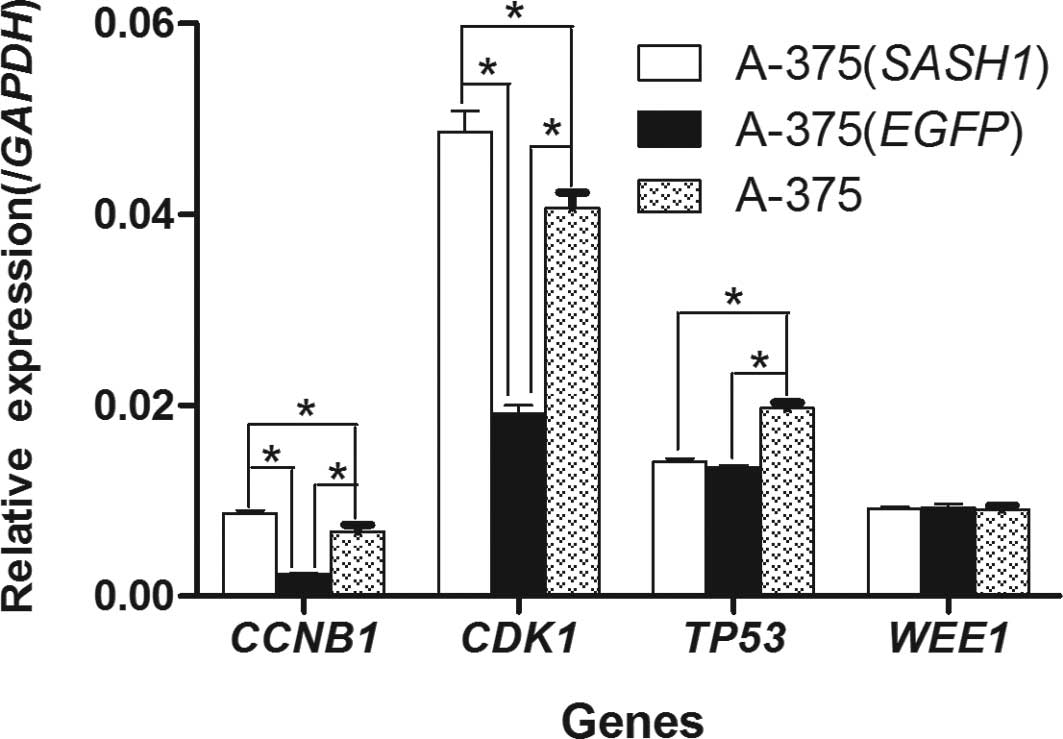
In order to determine the mechanism behind the G2/M
phase cell cycle arrest, the mRNA expression of the CCNB1, CDK1,
TP53 and WEE1 genes was quantified in the A-375 (SASH1), A-375
(EGFP) and A-375 cells by real-time RT-PCR. In the A-375 (SASH1)
cells, the expression of CCNB1 and CDK1 was significantly increased
(p<0.001) as opposed to the A-375 (EGFP) and A-375 cells,
whereas the expression of CCNB1 and CDK1 in the A-375 (EGFP) cells
was evidently (p<0.001) decreased compared to the A-375 cells
(Fig. 5). In particular, compared
to the A-375 (SASH1) cells, the expression of CCNB1 and CDK1 in the
A-375 (EGFP) cells was decreased by 4- and 2.5-fold, respectively.
As for the TP53 gene, its expression in the A-375 (SASH1) and A-375
(EGFP) cells was dramatically lower compared to the A-375 cells
(p<0.001), although no statistically significant difference was
observed between the A-375 (SASH1) and A-375 (EGFP) cells. As for
the WEE1 gene (Fig. 5), no
statistically significant difference was observed when comparing
any any 2 of the 3 groups.
Discussion
The expression of SASH1, a potential tumor
suppressor gene, is either decreased or lost in many types of
cancer (12,13). Based on the characteristics of the
domains within the SASH1 protein, SASH1 may play an important role
in intracellular signaling and transcriptional regulation. Rimkus
et al demonstrated that SASH1 plays a crucial role in
tumorigenesis, development, invasion and metastasis. The decrease
in SASH1 expression correlates with the formation of metachronous
distant metastases (13). In
another study, Martini et al also demonstrated that SASH1
interacts with the actin cytoskeleton and stimulates cell-matrix
adhesion (10). Clinical research
has also documented that the SASH1 mRNA expression level is
significantly reduced in 70% of breast cancer samples (12). Moreover, the SASH1 mRNA expression
level is also significantly reduced in primary lung cancer and
thyroid cancer, while the size of the decrease correlates with the
prognosis and tumor size (13).
Chen et al proved that SASH1 inhibits cell proliferation and
enhances cell apoptosis in the A549 lung cancer cell line (11). All these data suggest that SASH1
plays a crucial role in tumorigenesis, growth and evolution.
However, there are few studies available on the role of SASH1 in
the tumorigenesis and development of melanoma. Therefore, we used
in vitro studies to investigate the role of the SASH1 gene
in A-375 melanoma cells.
An A-375 stable melanoma cell line was established,
overexpressing the SASH1 gene and cell proliferation assay
indicated that the viability of the A-375 (SASH1) cells was
significantly decreased (p<0.05) compared to that of the A-375
(EGFP) and A-375 cells. These results suggest that SASH1 inhibits
cell proliferation. In order to clarify the underlying mechanism
behind the inhibition of cell proliferation, Annexin V-FITC and PI
staining, as well as DAPI staining were performed to assess cell
apoptosis. The results demonstrated that cell apoptosis did not
account for the inhibition of cell proliferation. Subsequent to the
cell cycle analysis conducted, the results proved that the SASH1
gene decreased the percentage of cells in the G1 phase and arrested
cells at the G2/M checkpoint, inducing a significant accumulation
of cells in the S phase. These results suggest that the inhibition
of proliferation by SASH1 occurs by G2/M arrest in the A-375
cells.
The pivotal regulatory step for G2/M transition in
eucaryotes is the activation of the cell division cycle Cdc2/cyclin
B complex (14,15). Cdc2/cyclin B is maintained in an
inactive form during the S and G2 phases by the inhibitory
phosphorylation of the Cdc2 residues, threonine 14 (Thr14) and
tyrosine 15 (Tyr15) (16). The
inhibitory phosphorylation of Cdc2 is modulated by the WEE1 family
protein kinases (human WEE1 and Myt1). The dephosphorylation of the
Thr14 and Tyr15 residues of Cdc2 by Cdc25B (in the cytoplasm) and
Cdc25C (in the nucleus) in the late G2 phase activates the cyclin
B/Cdc2 complex, and the accumulated, active Cdc2/cyclin B1 then
triggers the initiation of mitosis (17). The cyclin B-Cdc2 kinase is the
master regulator of cell cycle progression. The anomalous
regulation of this complex or enhanced expression of cyclin B has
been demonstrated to be associated with several cellular
malfunctions, including G2/M arrest and apoptosis (18,19).
On the basis of G2/M transition, the possible genes
related to G2/M transition were analyzed by real time PCR. Our
results demonstrated that the mRNA expression of cyclin B (CCNB1)
and Cdc2 (CDK1) increased by 4- and 2.5-fold, respectively in the
A-375 (SASH1) cells compared with the A-375 (EGFP) cells. The G2/M
arrest mostly resulted from the inhibition of the expression of
cyclin B or Cdc2 (20,21). However, our findings suggested that
SASH1 enhanced G2/M arrest via the upregulation of cyclin B and
Cdc2. The cyclin B-Cdc2 kinase is usually kept in an inactive state
by the inhibitory phosphorylation of the Cdc2 residues, Thr14 and
Tyr15, until the G2/M transition (17,22).
Thus, we hypothesized that the phosphorylation of Cdc2 may be
responsible for the G2/M arrest. The inhibitory phosphorylation of
Cdc2 is modulated by the WEE1 family protein kinases (human WEE1
and Myt1). The WEE1 gene product is a tyrosine-specific protein
kinase, located in the nucleus and phosphorylates Cdc2 exclusively
at Tyr15 (23). Nevertheless,
there was no difference in the expression of the WEE1 gene in any 2
of the 3 cell lines in the present study. Therefore, we
hypothesized that the crucial role in the phosphorylation of Cdc2
may be played by Myt1. Myt1, as a homologue of the product of the
mik1 gene, is a dual-specificity protein kinase that phosphorylates
Cdc2 at both Thr14 and Tyr15 residues (16). Apart from the inhibitory
phosphorylation of Cdc2, Myt1 seems to influence the normal
shuttling of the Cdc2/cyclin B complex into the nucleus (17). On these grounds, we hypothesized
that the phosphorylation of Cdc2 by Myt1 suppressed its catalytic
activity or stopped the shuttling of the Cdc2/cyclin B complex into
the nucleus, which led to G2/M arrest in the A-375 (SASH1) cells.
At the same time, the overexpression of cyclin B also contributed
to Cdc2 phosphorylation (18).
Nevertheless, additional investigations are required in order to
delineate the influences of SASH1 on Myt1, including promoting its
expression or inhibiting its degradation. Cdc25 family members,
which are dual-specificity phosphatases, have been identified as
positive regulators of Cdc2. To make Cdc2 kinase active and
facilitate the entry of cells from G2 to mitosis, Thr14 and Tyr15
(the 2 residues of Cdc2) are dephosphorylated by active Cdc25C
phosphatase (18). Consequently,
the inactivation or alterations in Cdc25C phosphatase activity may
be another reason for cell cycle arrest in the G2/M phase in the
A-375 (SASH1) cells. Activated Chk1 has been reported to lead to
the inhibitory phosphorylation of Cdc25C on Ser-216, and
subsequently to the inactivation of Cdc25 (24).
Apart from the phosphorylation of Cdc2, the
inhibition of cyclin B-Cdc2 binding may also stop cell entry from
G2 to mitosis (25). In the
present study, cyclin B and Cdc2 were both overexpressed in the
A-375 (SASH1) cells. In view of the above, we assumed that the
SASH1 gene possibly blocked cyclin B1-Cdc2 binding, either directly
or indirectly, leading to G2/M arrest. Although p53 can affect
cyclin B1/Cdc2 activity, resulting in G2/M arrest (26–28),
no statistically significant difference was observed in the TP53
mRNA level between the A-375 (SASH1) and A-375 (EGFP) cells.
Therefore, it did not account for the G2/M arrest. In the present
study, we demonstrate the tumor suppressive effects of SASH1 by
G2/M arrest in A-375 cells. However, additional investigations are
required in order to clarify the exact mechanism involved.
In conclusion, the present study indicates that the
tumor inhibitory effects of SASH1 occur by G2/M arrest in A-375
cells (Fig. 4). Although the exact
mechanism involved remains unclear, our results show that p53 and
WEE1 are not involved in the G2/M arrest and demonstrate that G2/M
arrest correlates with Cdc2 phosphorylation or the inhibition of
cyclin B-Cdc2 binding.
Acknowledgements
The authors are grateful to all the participants in
this study. This study was supported by a grant from the National
Natural Science Foundation of China (No: 90919049), the 973 Program
(2010CB529600, 2007CB947300), the Shanghai Municipal Commission of
Science and Technology Program (09DJ1400601) and the third phase of
the 211 project of the Ministry of Education of China. Sponsors we
not involved with the study design, data collection and analysis,
decision to publish or the preparation of the manuscript.
References
|
1
|
Villanueva A and Llovet JM: Targeted
therapies for hepatocellular carcinoma. Gastroenterology.
140:1410–1426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jousilahti P, Salomaa V, Kuulasmaa K,
Niemela M and Vartiainen E: Total and cause specific mortality
among participants and non-participants of population based health
surveys: a comprehensive follow up of 54 372 Finnish men and women.
J Epidemiol Community Health. 59:310–315. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gray L, Batty GD, Craig P, et al: Cohort
profile: the Scottish health surveys cohort: linkage of study
participants to routinely collected records for mortality, hospital
discharge, cancer and offspring birth characteristics in three
nationwide studies. Int J Epidemiol. 39:345–350. 2010. View Article : Google Scholar
|
|
4
|
Veeck J, Niederacher D, An H, et al:
Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer
is associated with unfavourable prognosis. Oncogene. 25:3479–3488.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Beaumont T and Leadbeater M: Treatment and
care of patients with metastatic breast cancer. Nurs Stand.
25:49–56. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai C and Gu W: p53 post-translational
modification: deregulated in tumorigenesis. Trends Mol Med.
16:528–536. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zegarra-Moro OL, Schmidt LJ, Huang H and
Tindall DJ: Disruption of androgen receptor function inhibits
proliferation of androgen-refractory prostate cancer cells. Cancer
Res. 62:1008–1013. 2002.PubMed/NCBI
|
|
8
|
Zhang H, Pan Y, Zheng L, et al: FOXO1
inhibits Runx2 transcriptional activity and prostate cancer cell
migration and invasion. Cancer Res. 71:3257–3267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao Y, Kong X, Li X, et al: Metadherin
mediates lipopolysaccharide-induced migration and invasion of
breast cancer cells. PLoS One. 6:e293632011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Martini M, Gnann A, Scheikl D, Holzmann B
and Janssen KP: The candidate tumor suppressor SASH1 interacts with
the actin cytoskeleton and stimulates cell-matrix adhesion. Int J
Biochem Cell Biol. 43:1630–1640. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen EG, Chen Y, Dong LL and Zhang JS:
Effects of SASH1 on lung cancer cell proliferation, apoptosis, and
invasion in vitro. Tumour Biol. April 10–2012.(E-pub ahead of
print).
|
|
12
|
Zeller C, Hinzmann B, Seitz S, et al:
SASH1: a candidate tumor suppressor gene on chromosome 6q24.3 is
downregulated in breast cancer. Oncogene. 22:2972–2983. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rimkus C, Martini M, Friederichs J, et al:
Prognostic significance of downregulated expression of the
candidate tumour suppressor gene SASH1 in colon cancer. Br J
Cancer. 95:1419–1423. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Coleman TR and Dunphy WG: Cdc2 regulatory
factors. Curr Opin Cell Biol. 6:877–882. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morgan DO: Cyclin-dependent kinases:
engines, clocks, and microprocessors. Annu Rev Cell Dev Biol.
13:261–291. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krek W and Nigg EA: Mutations of p34cdc2
phosphorylation sites induce premature mitotic events in HeLa
cells: evidence for a double block to p34cdc2 kinase activation in
vertebrates. EMBO J. 10:3331–3341. 1991.
|
|
17
|
Dai X, Yamasaki K, Yang L, et al:
Keratinocyte G2/M growth arrest by 1,25-dihydroxyvitamin D3 is
caused by Cdc2 phosphorylation through Wee1 and Myt1 regulation. J
Invest Dermatol. 122:1356–1364. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ray G, Dhar G, Van Veldhuizen PJ, et al:
Modulation of cell-cycle regulatory signaling network by
2-methoxyestradiol in prostate cancer cells is mediated through
multiple signal transduction pathways. Biochemistry. 45:3703–3713.
2006. View Article : Google Scholar
|
|
19
|
Liu X, He H, Feng Y, Zhang M, Ren K and
Shao R: Difference of cell cycle arrests induced by lidamycin in
human breast cancer cells. Anticancer Drugs. 17:173–179. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seo HR, Lee DH, Lee HJ, et al: Cyclin G1
overcomes radiation-induced G2 arrest and increases cell death
through transcriptional activation of cyclin B1. Cell Death Differ.
13:1475–1484. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang L, Zhang Y, Pan H, et al: Involvement
of cyclin B1 in progesterone-mediated cell growth inhibition, G2/M
cell cycle arrest, and apoptosis in human endometrial cell. Reprod
Biol Endocrinol. 7:1442009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Passalaris TM, Benanti JA, Gewin L, Kiyono
T and Galloway DA: The G(2) checkpoint is maintained by redundant
pathways. Mol Cell Biol. 19:5872–5881. 1999.PubMed/NCBI
|
|
23
|
McGowan CH and Russell P: Cell cycle
regulation of human WEE1. EMBO J. 14:2166–2175. 1995.PubMed/NCBI
|
|
24
|
Wang XM, Li J, Feng XC, Wang Q, Guan DY
and Shen ZH: Involvement of the role of Chk1 in lithium-induced
G2/M phase cell cycle arrest in hepatocellular carcinoma cells. J
Cell Biochem. 104:1181–1191. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ryu MS, Lee MS, Hong JW, Hahn TR, Moon E
and Lim IK: TIS21/BTG2/PC3 is expressed through PKC-delta pathway
and inhibits binding of cyclin B1-Cdc2 and its activity,
independent of p53 expression. Exp Cell Res. 299:159–170. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paulsen MT, Starks AM, Derheimer FA, et
al: The p53-targeting human phosphatase hCdc14A interacts with the
Cdk1/cyclin B complex and is differentially expressed in human
cancers. Mol Cancer. 5:252006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bunz F, Dutriaux A, Lengauer C, et al:
Requirement for p53 and p21 to sustain G2 arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang PM, Huang WC, Lin YC, et al: Loss of
IKKbeta activity increases p53 stability and p21 expression leading
to cell cycle arrest and apoptosis. J Cell Mol Med. 14:687–698.
2010.PubMed/NCBI
|