Introduction
Inflammatory bowel disease (IBD) is a heterogeneous
disease characterized by Crohn’s disease (CD) and ulcerative
colitis (UC). CD most commonly involves the ileum and colon but can
affect any region of the abominal area. UC often involves the
rectum, and inflammation may extend as far as the cecum in a
contiguous pattern (1). Strong
familial aggregation, twin studies and established genetic
associations have demonstrated the importance of genetics in IBD
pathogenesis (2–4). Currently, >32 susceptibility loci
have been identified for IBD (5–10).
However, all these genetic risk factors only account for ~20% of
the genetic risk (11), suggesting
that other factors, including possible epigenetic factors, are
involved in IBD pathogenesis (12). However, the epigenetic aspect of
IBD has not been systemized.
DNA methylation is an important epigenetic
alteration that is involved in the development and differentiation
of diseases (13). The aberrant
DNA methylation involves promoter CpG island methylation and
silenced genes in cancer such as tumor suppressor genes (13). The involvement of DNA methylation
in various types of cancer and several other human diseases
including IBD has been widely investigated (14). Abnormal DNA methylation has been
observed in UC patients in the estrogen receptor (ER),
p14ARF, and E-cadherin gene and
emerging evidence suggests the involvement of epigenetic factors in
the regulation of gene activity as a factors in the pathogenesis of
IBD (15,16). Additionally, it has been previously
reported that CpG islands in the promoter region of
transcription elongation regulator 1-like (TCERG1L) gene are
highly methylated, not only in colon cancer patient tissues,
particularly those obtained from early stage of colon cancer
patients (17), but also in
patients with UC (18). However,
little is known regarding DNA methylation in patients with CD.
Analysis of DNA methylation is leading to a new
generation of cancer biomarkers (19). It was previously demonstrated that
abnormally high DNA concentrations can also be detected in the
serum, plasma, and urine of cancer patients (20,21).
Aberrantly methylated DNA sequences occur frequently in tumors and
have been detected in the circulation of cancer patients by
methylation-specific PCR (MSP) (22,23).
Findings of those studies demonstrated that the presence of
aberrantly methylated gene DNA in serum is highly correlated with
the occurrence of various types of cancer as well as inflammatory
disease such as that present in patients with CD. In the present
study, the methylation status of TCERG1L gene in the serum
samples of CD patients was measured. In addition, detection of the
promoter DNA hypermethylation of TCERG1L in the serum
samples of CD patients was examined. The results obtained show that
the methylation status of TCERG1L gene in the serum of CD
patients has the potential to become a risk marker for the
progression of severe disease.
Materials and methods
Patients and samples
Serum DNA samples were obtained from CD patients
according to the guidelines of the IBD Study Group of the Korean
Association for the Study of Intestinal Diseases (KASID). Of the
101 subjects, 62 were male and 39 female, yielding a male:female
ratio of 1.6:1. The median age at diagnosis of CD was 24 years
(range, 12–66). CD was diagnosed on the basis of conventional
clinical, radiologic, endoscopic, and histopathologic criteria
(24–26). Briefly, patients were diagnosed
with CD if they met at least two of the following criteria: i) a
history of abdominal pain, weight loss, malaise, diarrhea, and/or
rectal bleeding; ii) endoscopic findings of mucosal cobblestoning,
linear ulceration, skip areas, or perianal disease; iii) radiologic
findings of stricture, fistula, mucosal cobblestoning, or
ulceration; iv) macroscopic appearance of bowel wall induration,
mesenteric lymphadenopathy, and ‘creeping fat’ on laparotomy; and
v) pathologic findings of transmural inflammation and/or
epithelioid granulomas. At the time of diagnosis, 26 patients
(25.7%) had disease located in the small bowel alone (L1), 12
(11.9%) had disease in the colon alone (L2), and 63 (62.4%) had
disease in the small bowel and colon (L3). Disease behavior at
diagnosis was inflammatory (B1) in 63 patients (62.4%), stricturing
(B2) in 27 (26.7%), and penetrating (B3) in 11 (10.9%). Clinical
characteristics of these patients are shown in Table I.
 | Table ICharacteristics of samples of patient
with CD. |
Table I
Characteristics of samples of patient
with CD.
| Characteristics | Values |
|---|
| Blood samples (total
n=101) |
| Age (years) |
| Median (range) | 24 (12–66) |
| Gender, n (%) |
| Male | 62 (61.3) |
| Female | 39 (38.6) |
| Disease location at
diagnosis, n (%) |
| Small bowel
alone | 26 (25.7) |
| Colon alone | 12 (11.9) |
| Small bowel and
colon | 63 (62.4) |
| Disease behavior at
diagnosis, n (%) |
| Inflammatory
(B1) | 63 (62.4) |
| Stricturing
(B2) | 27 (26.7) |
| Penetrating
(B3) | 11 (10.9) |
| Tissue samples (total
n=7) |
| Age (years) |
| Median | 21.7 |
| Gender, n (%) |
| Male | 4 (57.1) |
| Female | 3 (42.9) |
| Disease location at
diagnosis, n (%) |
| Small bowel
alone | 3 (42.9) |
| Colon alone | 1 (14.2) |
| Small bowel and
colon | 3 (42.9) |
| Disease behavior at
diagnosis, n (%) |
| Inflammatory
(B1) | 4 (57.2) |
| Stricturing
(B2) | 2 (28.6) |
| Penetrating
(B3) | 1 (14.2) |
Cell culture and treatment
Colorectal cell lines (HCT116, RKO, HT29, SW480,
DLD1, COLO 320, Lovo, and Caco-2) were obtained from the American
Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in
an appropriate medium and under conditions described by the ATCC.
Media were obtained from Invitrogen Life Technologies (Carlsbad,
CA, USA), supplemented with 10% fetal bovine serum (Gemini
Bio-Products, West Sacramento, CA, USA) and 1%
penicillin/streptomycin (Invitrogen Life Technologies).
DNA extraction and methylation
analyses
DNA was extracted following a standard
phenol-chloroform extraction protocol. Bisulfite modification of
DNA was performed using the EZ DNA Methylation kit™ (Zymo Research,
Irvine, CA, USA) as per the manufacturer’s instructions. MSP was
carried out in a 25-μl reaction containing 10X MSP buffer, 10 mM
dNTPs, 10 pmol of each of the methylated or unmethylated primers, 1
unit of JumpStart™ REDTaq® DNA polymerase (Sigma, St.
Louis, MO, USA) and 4 μl of bisulfite-treated DNA. Amplification
cycles were as follows: 1 cycle of 95°C for 5 min followed by 35
cycles of 95°C for 30 sec, 60°C for 30 sec, 72°C for 30 sec, and a
final extension step of 72°C for 5 min. In vitro-methylated
DNA (IVD) was used as a positive control for MSP. A total of 10 μl
of each amplification reaction was loaded and run on 2% agarose gel
containing GelStar™ Nucleic Acid Gel Stain (Lonza, Basel,
Switzerland) and visualized under ultraviolet illumination. Primers
for MSP analysis were previously reported in the study by Yi et
al (17).
Quantitative methylation-specific PCR
using qPCR
Bisulfite modification of genomic DNA was carried
out using the EZ DNA methylation kit (Zymo Research). For
quantitative real-time analyses, the Maxima SYBR-Green qPCR kit
(Fermentas, Seoul, Korea) was used and the amplification conditions
consisted of an initial 10-min denaturation step at 95°C, followed
by 40 cycles of denaturation at 95°C for 15 sec and annealing and
extension for 30 and 60 sec, respectively. A CFX96 real-time PCR
detection system (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and for quantification the comparative cycle threshold (Ct) method
were used, normalizing the Ct values for the indicated gene to the
Ct values of unmethylated reaction relative to a methylated
reaction sample.
Bisulfite sequencing analysis
Genomic DNA (1 μg) from each sample was converted by
bisulfite using the EZ DNA methylation kit (Zymo Research)
following the manufacturer’s instructions. PCR conditions and
primer sequences are available subsequent to request. The PCR
amplicons were gel-purified and subcloned into a pCRII-TOPO vector
(Invitrogen Life Technologies). Clones were randomly selected and
sequenced on an ABI3730xl DNA analyzer to ascertain the methylation
patterns of each locus. The primers for bisulfite sequencing
analysis have been previously reported by Yi et al (17).
Statistical analysis
Statistical analysis was performed using the STATA
9.2 software package (Stata Corporation, College Station, TX, USA).
Most analyses were conducting using a t-test, while continuous
variables were analyzed using the Mann-Whitney U test. P<0.05
was considered statistically significant.
Results and Discussion
Detection of DNA promoter
hypermethylation of TCERG1L gene in sera of patients with CD
Understanding the causes and molecular mechanisms of
CD and UC, the two forms of idiopathic IBD, is a major challenge in
gastroenterology research. Although significant effort has been
made to identify genetic and environmental factors that may
increase the risk of IBD, little is known IBD-specific factors
(8,27). Mounting evidence supports the
theory that IBD is caused by a complex interplay between genetic
predispositions of various genes, combined with an abnormal
interaction with environmental factors. Thus, it appears that
epigenetic factors significantly contribute to the pathogenesis of
disease.
Recently, studies have focused on two separate DNA
hypermethylation biomarker candidate genes in patients with colon
cancer and UC (17,18). In the present study, the hypothesis
that extremely sensitive DNA methylation marker candidates in colon
cancer are capable of detecting high-risk inflammatory diseases
such as UC and CD in patients, led to DNA methylation analysis in
the serum samples of CD patients using TCERG1L gene promoter
region.
The TCERG1L gene which is located on
chromosome 10, has exhibited frequent cancer-specific methylation
in microarray-based approaches (28). TCERG1L is potentially
involved in the elongation-related factors in HeLa nuclear extracts
(29), however, this remains to be
elucidated. CpG islands of the TCERG1L gene promoter region
are known to be frequently methylated in the early stage of colon
cancer (17), thus genomic DNA
from the sera of patients with CD (n=101) was extracted. First, we
assessed the methylation level of TCERG1L gene promoter
region in 101 serum samples of patients with CD by MSP analysis.
Conventional MSP analysis was performed successfully in the
majority of samples. Fig. 1 shows
the frequency of hypermethylation for the TCERG1L gene
promoter region in the sera of patients with CD, and 58 (57%) of
the 101 samples were found to be methylated. We also tested
FBN2 gene in the serum samples of patients with CD which is
known to be frequently methylated in the early stages of colon
cancer. However, no methylation was detected in the samples tested
in the present study (data not shown). Previously, we demonstrated
that the DNA hypermethylation of TCERG1L and FBN2 was
detected in UC patient tissue samples (18). However, in the present study, we
only detected the DNA methylation of the TCERG1L gene in
serum samples of patients with CD. The results suggest that
TCERG1L gene is extremely sensitive as a methylation marker
in the detection of early stages of cancer as well as inflammatory
diseases such as IBD. In addition, it was suggested that there may
be differences in DNA methylation signatures at the gene level
between UC and CD patients. Therefore, genome-wide methylation
analyses are necessary to identify differential methylation levels
between UC and CD in future.
TCERG1L methylation analysis in patients
with CD
TCERG1L was previously reported to be
completely methylated (17).
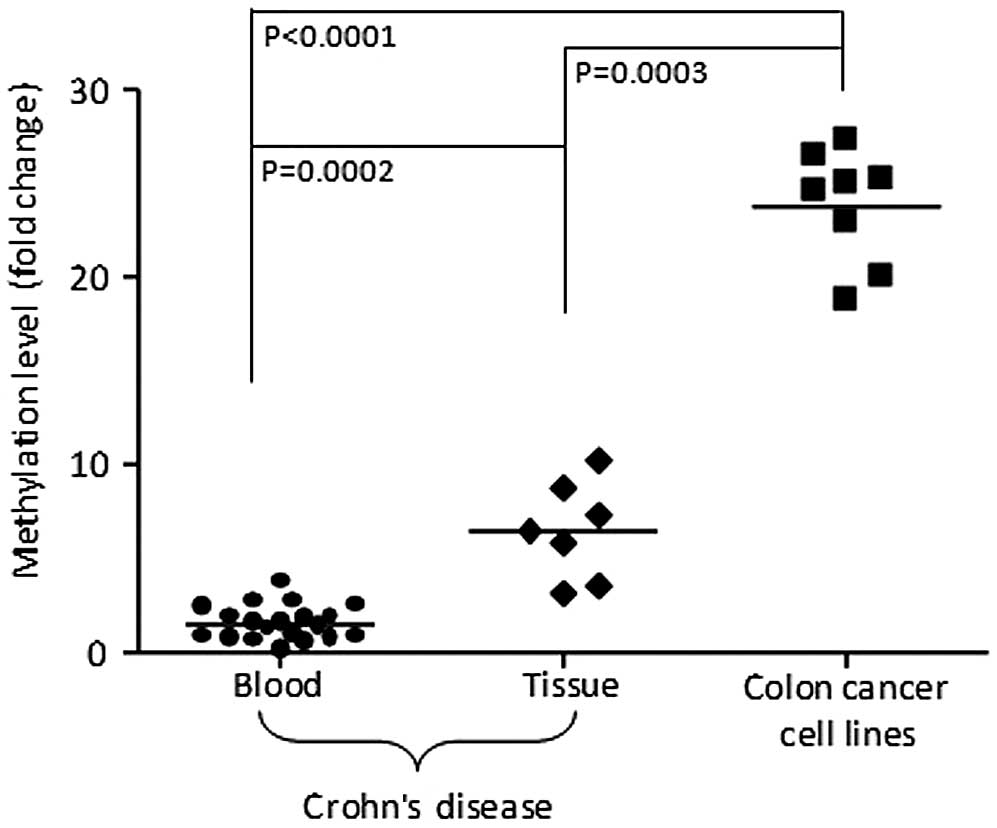
Fig. 2 shows the relative
TCERG1L hypermethylation status in the serum samples (n=20)
and tissue (n=7) of patients with CD compared with colon cancer
cell lines (n=8) using qMSP. In terms of methylation
quantification, the methylation level of TCERG1L gene in
colon cancer cell lines was relatively higher than that of patients
with CD, thus the methylation level in cancer is denser than that
in inflammatory disease. We also compared the methylation level of
TCERG1L between tissue from biopsy and blood samples. The
methylation level in tissue samples with CD was 2- to 4-fold higher
than that in blood samples. We detected TCERG1L methylation
in blood and tissue samples of patients with CD by conventional
MSP, however, quantification of the methylation level distinguished
between cancer and CD. P-values were statistically significant
between CD and colon cancer cell lines (blood, P<0.0001; tissue,
P=0.0003) as well as blood and tissue with CD (P=0.0002). Our data
suggest that DNA hypermethylation of TCERG1L gene is
sensitive enough to detect patient blood or tissue samples with CD.
Limited clinical information of the CD patients in the present
study was obtained. Thus, additional studies are required to
investigate the correlation between clinical data of CD patients
such as duration and methylation level.
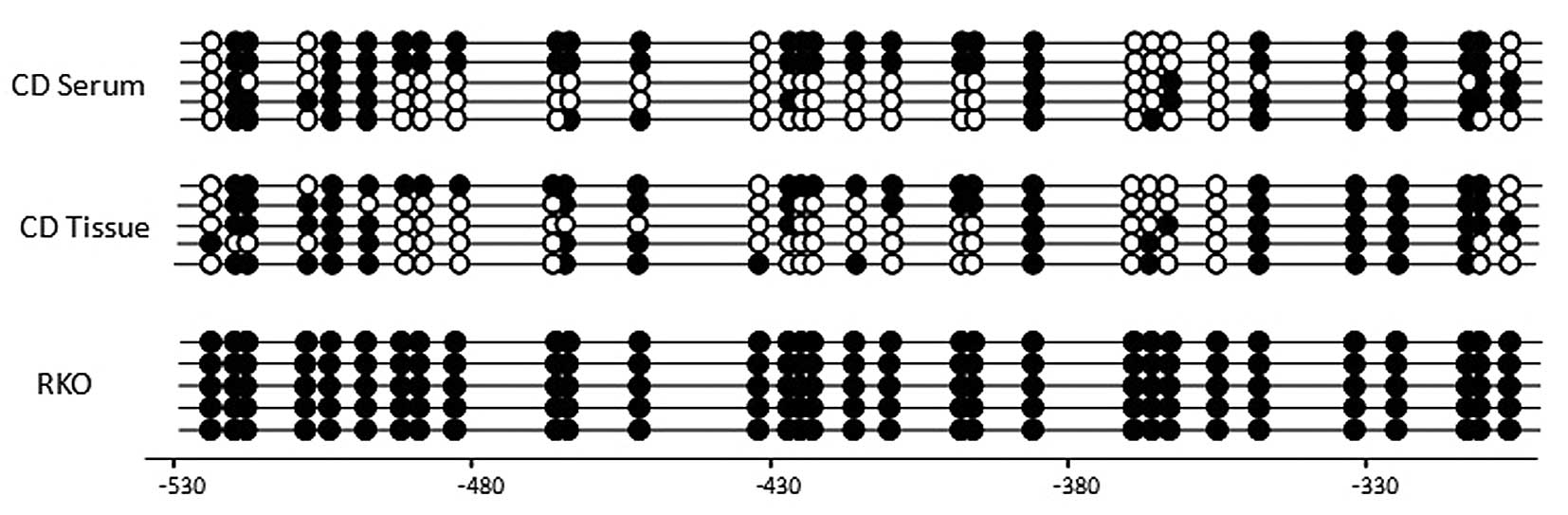
The methylation pattern in the CpG islands of the
promoter region of TCERG1L gene by bisulfite sequencing
analysis was also confirmed (Fig.
3). The bisulfate sequencing region amplified has 31 CpG sites.
The TCERG1L gene was previously reported to have a dense CpG
methylation pattern in CRC tumors (17). In this study, we found that
TCERG1L gene showed a dense CpG methylation pattern in the
blood (patient #11) and tissue (patient #3) of CD patients at 51%
(methylation site per CpG site) compared with complete methylation
(100%) in RKO colon cancer cells. Our results suggest that
TCERG1L gene is densely methylated in CD patients, and the
DNA methylation of TCERG1L is sensitive enough to detect
inflammatory diseases such as CD. However, certain limitations of
the present study should be considered. A lack of control samples
with CD did not yield specificity with TCERG1L gene
methylation. Additionally, there was a lack of clinical information
for CD samples including disease duration in the present study;
thus we were not able to compare results of various analyses
associated with DNA methylation of TCERG1L. Therefore,
additional studies are needed to define the use of DNA methylation
markers with clinical data of CD patients, including control
samples.
In conclusion, we assessed the promoter DNA
methylation pattern of TCERG1L gene in the blood samples of
CD patients. We were able to detect TCERG1L gene promoter
methylation in 57% of patient blood samples with CD by conventional
MSP analysis. The DNA methylation status of TCERG1L in CD
patient tissue and blood samples was also confirmed by bisulfite
sequencing analysis. A comparison of the quantitative methylation
levels among CD patient tissue, blood and colon cancer cell lines
was conducted. Results showed the methylation level of
TCERG1L in colon cancer cell lines to be significantly
higher than that in CD patient blood and tissue samples.
Additionally, results of this study have demonstrated that
methylation of TCERG1L is sensitive enough to detect
inflammatory disease in tissue and blood samples of patients with
CD. Thus, sensitive methylation markers may be useful in the
detection of inflammatory diseases which have the potential risk of
progression of severe disease in CD patients.
Acknowledgements
This study was supported by the National R&D
program (50596-2014) through the Dongnam Institute of Radiological
and Medical Sciences (DIRAMS) funded by the Korean Ministry of
Education, Science and Technology. This study was also supported by
a fund (2013-E63004-00) by Research of Korea Centers for Disease
Control and Prevention.
References
|
1
|
Podolsky DK: Inflammatory bowel disease. N
Engl J Med. 347:417–429. 2002. View Article : Google Scholar
|
|
2
|
Halfvarson J, Bodin L, Tysk C, Lindberg E
and Järnerot G: Inflammatory bowel disease in a Swedish twin
cohort: a long-term follow-up of concordance and clinical
characteristics. Gastroenterology. 124:1767–1773. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thompson NP, Driscoll R, Pounder RE and
Wakefield AJ: Genetics versus environment in inflammatory bowel
disease: results of a British twin study. BMJ. 312:95–96. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tysk C, Lindberg E, Järnerot G and
Flodérus-Myrhed B: Ulcerative colitis and Crohn’s disease in an
unselected population of monozygotic and dizygotic twins. A study
of heritability and the influence of smoking. Gut. 29:990–996.
1988.
|
|
5
|
Yamazaki K, McGovern D, Ragoussis J,
Paolucci M, Butler H, Jewell D, Cardon L, Takazoe M, Tanaka T,
Ichimori T, Saito S, Sekine A, et al: Single nucleotide
polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease.
Hum Mol Genet. 14:3499–3506. 2005.
|
|
6
|
Duerr RH, Taylor KD, Brant SR, Rioux JD,
Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M,
Griffiths A, Dassopoulos T, Bitton A, et al: A genome-wide
association study identifies IL23R as an inflammatory bowel
disease gene. Science. 314:1461–1463. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hampe J, Franke A, Rosenstiel P, Till A,
Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J,
Günther S, Prescott NJ, et al: A genome-wide association scan of
nonsynonymous SNPs identifies a susceptibility variant for Crohn
disease in ATG16L1. Nat Genet. 39:207–211. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rioux JD, Xavier RJ, Taylor KD, Silverberg
MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW,
Shugart YY, Griffiths AM, et al: Genome-wide association study
identifies new susceptibility loci for Crohn disease and implicates
autophagy in disease pathogenesis. Nat Genet. 39:596–604. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wellcome Trust Case Control Consortium.
Genome-wide association study of 14,000 cases of seven common
diseases and 3,000 shared controls. Nature. 447:661–678. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Parkes M, Barrett JC, Prescott NJ,
Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER,
Cummings FR, Soars D, Drummond H, Lees CW, et al: Sequence variants
in the autophagy gene IRGM and multiple other replicating
loci contribute to Crohn’s disease susceptibility. Nat Genet.
39:830–832. 2007.PubMed/NCBI
|
|
11
|
Barrett JC, Hansoul S, Nicolae DL, Cho JH,
Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM,
Bitton A, Dassopoulos T, et al: Genome-wide association defines
more than 30 distinct susceptibility loci for Crohn’s disease. Nat
Genet. 40:955–962. 2008.PubMed/NCBI
|
|
12
|
Petronis A and Petroniene R: Epigenetics
of inflammatory bowel disease. Gut. 47:302–306. 2000. View Article : Google Scholar
|
|
13
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maeda O, Ando T, Watanabe O, Ishiguro K,
Ohmiya N, Niwa Y and Goto H: DNA hypermethylation in colorectal
neoplasms and inflammatory bowel disease: a mini review.
Inflammopharmacology. 14:204–206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tahara T, Shibata T, Nakamura M, Yamashita
H, Yoshioka D, Okubo M, Maruyama N, Kamano T, Kamiya Y, Nakagawa Y,
Fujita H, Nagasaka M, et al: Effect of MDR1 gene promoter
methylation in patients with ulcerative colitis. Int J Mol Med.
23:521–527. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yi JM, Dhir M, Guzzetta AA,
Iacobuzio-Donahue CA, Heo K, Yang KM, Suzuki H, Toyota M, Kim HM
and Ahuja N: DNA methylation biomarker candidates for early
detection of colon cancer. Tumour Biol. 33:363–372. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim TO, Park J, Kang MJ, Lee SH, Jee SR,
Ryu DY, Yang K and Yi JM: DNA hypermethylation of a selective gene
panel as a risk marker for colon cancer in patients with ulcerative
colitis. Int J Mol Med. 31:1255–1261. 2013.PubMed/NCBI
|
|
19
|
Laird PW: The power and the promise of DNA
methylation markers. Nat Rev Cancer. 3:253–266. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Widschwendter M and Menon U: Circulating
methylated DNA: a new generation of tumor markers. Clin Cancer Res.
12:7205–7208. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wallner M, Herbst A, Behrens A, Crispin A,
Stieber P, Göke B, Lamerz R and Kolligs FT: Methylation of serum
DNA is an independent prognostic marker in colorectal cancer. Clin
Cancer Res. 12:7347–7352. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Esteller M, Sanchez-Cespedes M, Rosell R,
Sidransky D, Baylin SB and Herman JG: Detection of aberrant
promoter hypermethylation of tumor suppressor genes in serum DNA
from non-small cell lung cancer patients. Cancer Res. 59:67–70.
1999.PubMed/NCBI
|
|
23
|
deVos T, Tetzner R, Model F, Weiss G,
Schuster M, Distler J, Steiger KV, Grützmann R, Pilarsky C,
Habermann JK, Fleshner PR, Oubre BM, et al: Circulating methylated
SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clin
Chem. 55:1337–1346. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang SK, Yun S, Kim JH, Park JY, Kim HY,
Kim YH, Chang DK, Kim JS, Song IS, Park JB, Park ER, Kim KJ, et al:
Epidemiology of inflammatory bowel disease in the Songpa-Kangdong
district, Seoul, Korea, 1986–2005: a KASID study. Inflamm Bowel
Dis. 14:542–549. 2008.
|
|
25
|
Loftus EV Jr, Silverstein MD, Sandborn WJ,
Tremaine WJ, Harmsen WS and Zinsmeister AR: Crohn’s disease in
Olmsted County, Minnesota, 1940–1993: incidence, prevalence, and
survival. Gastroenterology. 114:1161–1168. 1998.
|
|
26
|
Lee YJ, Yang SK, Byeon JS, Myung SJ, Chang
HS, Hong SS, Kim KJ, Lee GH, Jung HY, Hong WS, Kim JH, Min YI, et
al: Analysis of colonoscopic findings in the differential diagnosis
between intestinal tuberculosis and Crohn’s disease. Endoscopy.
38:592–597. 2006.
|
|
27
|
Brentnall TA, Crispin DA, Rabinovitch PS,
Haggitt RC, Rubin CE, Stevens AC and Burmer GC: Mutations in the
p53 gene: an early marker of neoplastic progression in ulcerative
colitis. Gastroenterology. 107:369–378. 1994.PubMed/NCBI
|
|
28
|
Schuebel KE, Chen W, Cope L, Glöckner SC,
Suzuki H, Yi JM, Chan TA, Van Neste L, Van Criekinge W, van den
Bosch S, van Engeland M, Ting AH, et al: Comparing the DNA
hypermethylome with gene mutations in human colorectal cancer. PLoS
Genet. 3:1709–1723. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sánchez-Alvarez M, Goldstrohm AC,
Garcia-Blanco MA and Suñé C: Human transcription elongation factor
CA150 localizes to splicing factor-rich nuclear speckles and
assembles transcription and splicing components into complexes
through its amino and carboxyl regions. Mol Cell Biol.
26:4998–5014. 2006.
|