Introduction
Osteoclastogenesis and osteoblastogenesis must be
properly balanced for bone homeostasis to be maintained (1,2), and
abnormal activation of osteoclasts has been identified in a variety
of bone diseases, such as osteoporosis, osseous metastasis and
arthritis, which are associated with pathologic bone resorption.
Some studies (3,4) have been conducted to investigate how
various inflammatory cytokines drive excessive bone loss by
suppressing the anabolic functions of osteoblasts and inducing
receptor activator of nuclear factor (NF)-κB ligand (RANKL)
expression in the osteoblast lineage and stromal cells, thereby
promoting osteoclastogenesis. These studies have demonstrated that
inflammatory cytokines such as tumor necrosis factor alpha (TNF-α)
and interleukin-1 can act in concert with RANKL to augment
signaling by RANK and directly promote osteoclastogenesis, but the
underlying molecular mechanism remains unclear (5,6).
Osteoclast precursors are derived from hematopoietic
stem cells identified in the bone marrow and are the only cells
capable of bone resorption (7).
Thus, these cells are required for both normal bone homeostasis and
pathological bone loss. Their differentiation and activation are
regulated by a variety of hormones and cytokines. In particular,
RANKL and macrophage colony-stimulating factor (M-CSF) were
demonstrated to be essential cytokines for osteoclast
differentiation (8). RANKL is a
member of the TNF superfamily, and through its receptor RANK, RANKL
acts as the primary cytokine mediating osteoclast differentiation
by inducing fusion of preosteoclasts to produce mature
multinucleated cells (9,10). Binding between RANKL and RANK on
preosteoclasts leads to the recruitment of TNF receptor-associated
factor (TRAF) family proteins, such as TRAF2/6, which activate
mitogen-activated protein kinases, as well as NF-κB (11). This signaling activates
transcription factors that are required for osteoclast
differentiation, such as c-Fos, activator protein-1 and nuclear
factor of activated T cells 1 (NFATc1) (12,13)
Stimulation with RANKL selectively induces NFATc1 expression, and
thus, NFATc1 is considered to function as a master switch for
controlling the terminal differentiation of osteoclasts (14).
TNF-α is known to be among the most potent
multifunctional inflammatory cytokines. By inducing macrophage cell
killing activity, TNF-α serves an important role in the host immune
response (15). In addition, TNF-α
has been demonstrated to be involved in bone resorption associated
with inflammatory diseases of bone (16,17).
TNF-α can influence osteoclast precursor differentiation and bone
resorption activity through various processes, one of which is the
induction of RANKL and M-CSF expression within osteogenic cells,
which indirectly promotes the differentiation and function of
osteoclasts (18). Another process
involves the direct induction of preosteoclast fusion and
differentiation, but the specific underlying mechanisms have not
been elucidated (19).
To investigate the molecular mechanism underlying
the positive effects of TNF-α on RANKL-induced osteoclast
differentiation, then the authors examined the activation of the
NF-κB pathway and the expression of RANK during the course of
osteoclastogenesis in bone marrow-derived macrophages (BMMs). The
results of the present study indicated that TNF-α promotes
osteoclast precursor differentiation by working together with RANKL
and upregulating RANK expression.
Materials and methods
Cell culture and reagents
The growth medium for primary mouse BMMs was
α-minimum essential medium (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) and antibiotics (100 U/ml
penicillin and 100 g/ml streptomycin, Gibco; Thermo Fisher
Scientific, Inc.). Recombinant murine M-CSF and recombinant murine
soluble RANKL were purchased from PeproTech, Inc. (Rocky Hill, NJ,
USA). Tartrate-resistant acid phosphatase (TRAP) staining solution
387-A was purchased from Sigma-Aldrich; Merck KGaA (Darmstadt,
Germany), and the Cell Counting Kit-8 (CCK-8), a TRAP assay kit
(cat. no. P0332) and an inhibitor of NF-κB (BAY 11–7082) were
purchased from Beyotime Institute of Biotechnology (Haimen, China).
For reverse transcription-quantitative polymerase chain reaction
(RT-qPCR), TaqMan® gene expression assays were conducted
with Reverse Transcriptase qPCR™ Mastermix No-ROX from
Promega Corporation (Madison, WI, USA). Primary antibodies to RANK
(sc-59981; 1:500), NF-κB (sc-166588; 1:100), and NFATc1 (sc-7294;
1:3,000) were obtained from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA).
Isolation of BMMs and in vitro
induction of osteoclastogenesis
A total of 20 C57BL/6 mice (weighing approximately
18–22 g) were obtained from the Animal Experiment Laboratory of
Daping Hospital (Chongqing, China). All mice were fed ad libitum
with standard rodent chow and had free access to food and water
(Prolab RMH 3000; PMI LabDiet, Richmond, IN, USA). All experiments
involving mice were approved by the Institutional Animal Care and
Use Committee at the Third Military Medical University (Chongqing,
China). Femurs and tibiae were removed from 6-week-old male C57BL/6
mice following sacrificed the mice by cervical dislocation. Bone
marrow cells were collected, treated with red blood cell lysis
buffer (150 mM ammonium chloride, 10 mM potassium bicarbonate and
0.1 mM EDTA, pH 7.4), and then cultured in growth medium containing
M-CSF (10 ng/ml) in 5% CO2 at 37°C. Following incubation
overnight, nonadherent cells were collected, seeded at
5×107 cells per well in 6-well plates, and treated with
M-CSF (30 ng/ml) for a further 48 h. Then the cells that adhered to
the plates were used as BMMs, and cells that continued to be
nonadherent were washed away. Detachin™ (Genlantis, San
Diego, CA, USA) was used for cell detachment, and, upon
resuspension, the obtained cells were seeded on dishes or plates
for induction of osteoclastogenesis. To induce osteoclastic
differentiation, BMMs (1×105 cells/ml) were cultured in
growth media containing 30 ng/ml M-CSF and 50 ng/ml RANKL with or
without TNF-α at various concentrations for 4 days.
Cell viability assay
BMMs were cultured in growth media containing 30
ng/ml M-CSF in the presence or absence of TNF-α (0, 10, 20, 40, 80,
100 or 200 µM) for 4 days. Cell viability was assessed using CCK-8,
according to the manufacturer's instructions. Briefly, the cells
were plated in 96-well plates at 1×104 cells per well
and cultured in growth medium. At the indicated time points, cells
were incubated with CCK-8 solution for 4 h at 37°C. The viability
of cells was assessed by measuring the absorbance at 450 nm using
the Biotek ELX-800 plate reader (Biotek Instruments, Inc.,
Winooski, VT, USA). All experiments were performed in
triplicate.
Measurement of TRAP activity and TRAP
staining
BMMs (1×105 cells/well) were seeded in
wells of 24-well plates and incubated for 24 h prior to treatment
with 10 ng/ml M-CSF for 2 days followed by treatment with 30 ng/ml
M-CSF + 50 ng/ml RANKL in media containing 10% FBS for 4 days.
Following incubation, the cells were fixed in 3.7% formaldehyde,
permeabilized with 0.1% Triton X-100 and incubated with TRAP
staining solution 387-A in the dark at 37°C. Following rinsing the
cells, TRAP-positive multinucleated cells were visualized by
phase-contrast light microscopy (Olympus Corporation, Tokyo,
Japan). Following staining, TRAP-positive multinucleated cells (3
nuclei) were counted manually in six randomly selected visual
fields. TRAP activity was assessed using a TRAP staining kit and
absorbance measurement at 540 nm.
RT-qPCR
Briefly, total RNA was extracted on the indicated
days using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), and purified total RNA was used for cDNA synthesis with
Moloney murine leukemia virus (M-MLV) reverse transcriptase and
oligo(dT) primers (Promega Corporation). The specific primer pairs
are listed in Table I. Nfatc1 and
other mRNAs were measured using the StepOne real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The PCR
program involved an initial step for 20 sec at 95°C, followed by 40
thermal cycles of 3 sec at 95°C and 30 sec at 60°C, and finished
with 15 sec at 95°C, 1 min at 60°C and 15 sec at 95°C. Data were
analyzed according to the comparative cycle threshold method
(20) and were normalized to GAPDH
expression in each sample. The expression of individual genes was
assessed in triplicate samples and repeated each experiment at
least three times.
 | Table I.List of primer sequences. |
Table I.
List of primer sequences.
| Transcript | Primer sequence (5′
to 3′) |
|---|
| RANK | F:
CATGGCAGAGGCGGGAGTAC |
|
| R:
GCCCGCTAGAGATGAACGTG |
| NFATc1 | F:
CCCCGTCCAAGTCAGTTTCTATG |
|
| R:
CGTCCGTGGGTTCTGTCTTTATA |
| NF-κB (P65) | F:
CCCAACACTGCCGAGCTCAAG |
|
| R:
CGCCGTAGCTGCATGGAGAC |
| β-actin | F:
CGCTCCTCGTGCTGTCTTC |
|
| R:
TCTTCTCCATGTCATCCCAGTTG |
Immunostaining
For immunostaining analysis, BMMs were fixed in 4%
paraformaldehyde and permeabilized with 0.1% Triton X-100 for 30
min. Cells were rinsed again with PBS and blocked with 5% bovine
serum albumin, washed three times with PBS prior to incubation with
the primary antibody against RANK (dilution 1:300) at 4°C
overnight. Cells stained with RANK were then visualized following
incubation with a fluorescein isothiocyanate-conjugated secondary
antibody (A0568; 1:500; Beyotime Institute of Biotechnology,
Haimen, China) for 2 h at room temperature and treatment with 50
µg/ml DAPI for nuclear staining. Fluorescence images were obtained
using a fluorescence microscope (Fluoview 400; Olympus
Corporation).
Western blot analysis
Cell lysates were prepared using
radioimmunoprecipitation assay buffer [50 mM Tris-Cl (pH 7.4), 150
mM NaCl, 1% NP-40, 0.25% Na-deoxycholate, 0.1% SDS with 1 mM EDTA
(pH 8.0), 1 mM phenylmethylsulfonyl fluoride, 2 µg/ml aprotinin, 2
µg/ml leupeptin, 4 mM Na3VO4 and 10 mM NaF].
The samples (10–30 µg protein/well) were resolved using SDS-PAGE
(6–10% gels), and proteins were transferred to nitrocellulose
membranes. The membranes were blocked in 5% skim milk and incubated
with antibodies against NFATc1 (1:3,000), RANK (1:500), IκB kinase
α (sc-1643; 1:1,000) and α-tubulin (sc-69970; 1:500) at 4°C
overnight. Next, the membranes were incubated with a horseradish
peroxidase-conjugated secondary antibody rabbit anti-mouse IgG
H&L (ab6728; Abcam, Cambridge, MA, USA) at a dilution of 1:300
for 3 h at 4°C. Protein bands were visualized with an enhanced
chemiluminescence detection kit (Roche Diagnostics, Basel,
Switzerland). Chemiluminescence was detected using a motorized
molecular imaging system (GE Healthcare, USA).
Statistical analysis
Unless otherwise specified, the results are
presented as mean ± standard deviation. Statistical comparisons
were performed using Student's t-tests, and P<0.05 was
considered to indicate a statistically significant difference.
Results
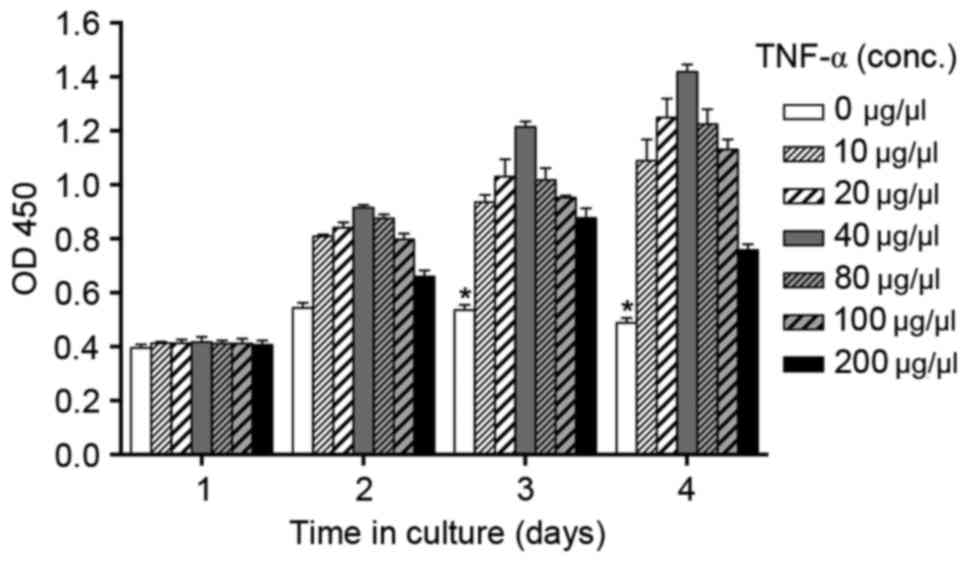
Effect of TNF-α on BMM viability
The authors first investigated whether specific
concentrations of TNF-α may be cytotoxic to BMMs in culture.
Following treatment with TNF-α at concentrations between 0 and 200
µg/µl for 4 days, BMMs remained viable in general (Fig. 1). However, the effect of TNF-α on
BMM viability was indicated to be dose dependent. At concentrations
of ≤40 µg/µl, TNF-α positively affected the viability of BMMs,
inducing BMM proliferation, whereas BMMs treated with higher doses
of TNF-α presented less activity with the CCK-8 assay, although
still more than that of cells not exposed to TNF-α. Therefore, the
authors selected 40 µg/µl TNF-α as the appropriate concentration
for stimulation of BMMs.
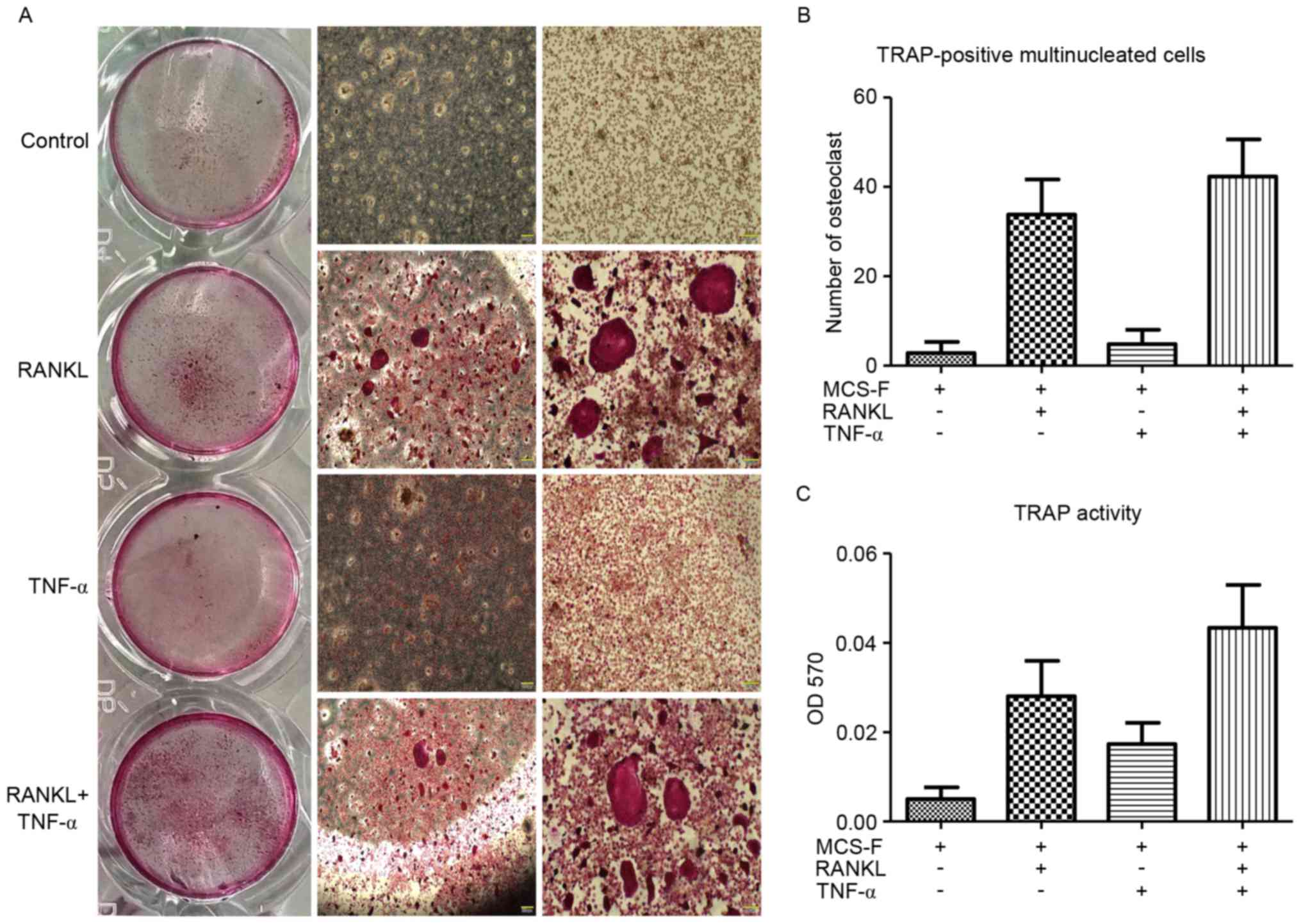
TNF-α promotes osteoclast formation
and fusion from BMMs
The effects of TNF-α on osteoclast formation were
analyzed among BMMs in vitro by treating BMMs with RANKL and
M-CSF in the presence or absence of TNF-α. TNF-α treatment promoted
osteoclast formation as indicated by an increase in the number of
TRAP-positive multinucleated osteoclasts (Fig. 2A). Large TRAP-positive
multinucleated osteoclasts (≥10 nuclei) were clearly observed in
the TNF-α-treated group (Fig. 2B).
In addition, the TRAP activity of BMMs induced by RANKL was further
promoted by TNF-α (Fig. 2C). These
results indicated that TNF-α promotes osteoclast formation and
fusion.
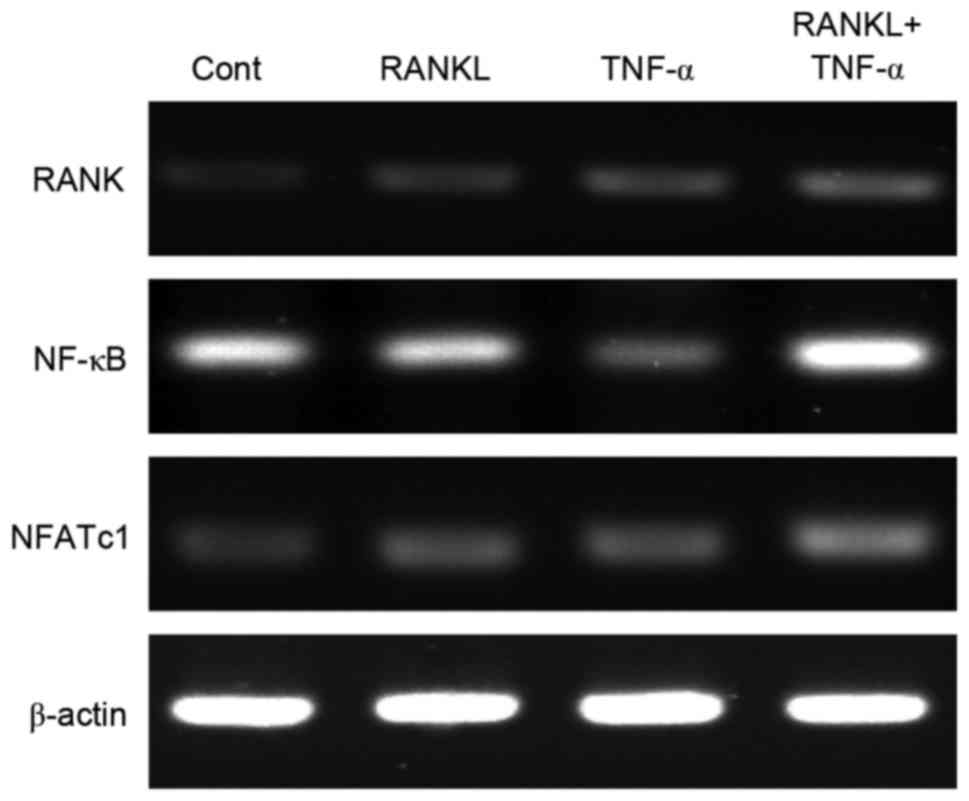
TNF-α and RANKL together upregulate
expression of genes related to osteoclastogenesis
To explore the molecular mechanism underlying the
promotion of osteoclast formation and fusion by TNF-α, the authors
analyzed the mRNA levels of genes related to osteoclast
differentiation, specifically RANK, NFATc1 and NF-κB,
in BMMs exposed to TNF-α, RANKL or both. The mRNA levels of
RANK and NFATc1 were increased by varying degrees by
TNF-α (Fig. 3). Furthermore, the
synergistic actions of TNF-α and RANKL may be reflected by the
activation of NFATc1 signaling, which is essential for
osteoclastogenesis, unlike NF-κB signaling. Notably, together TNF-α
and RANKL induced upregulation of RANK.
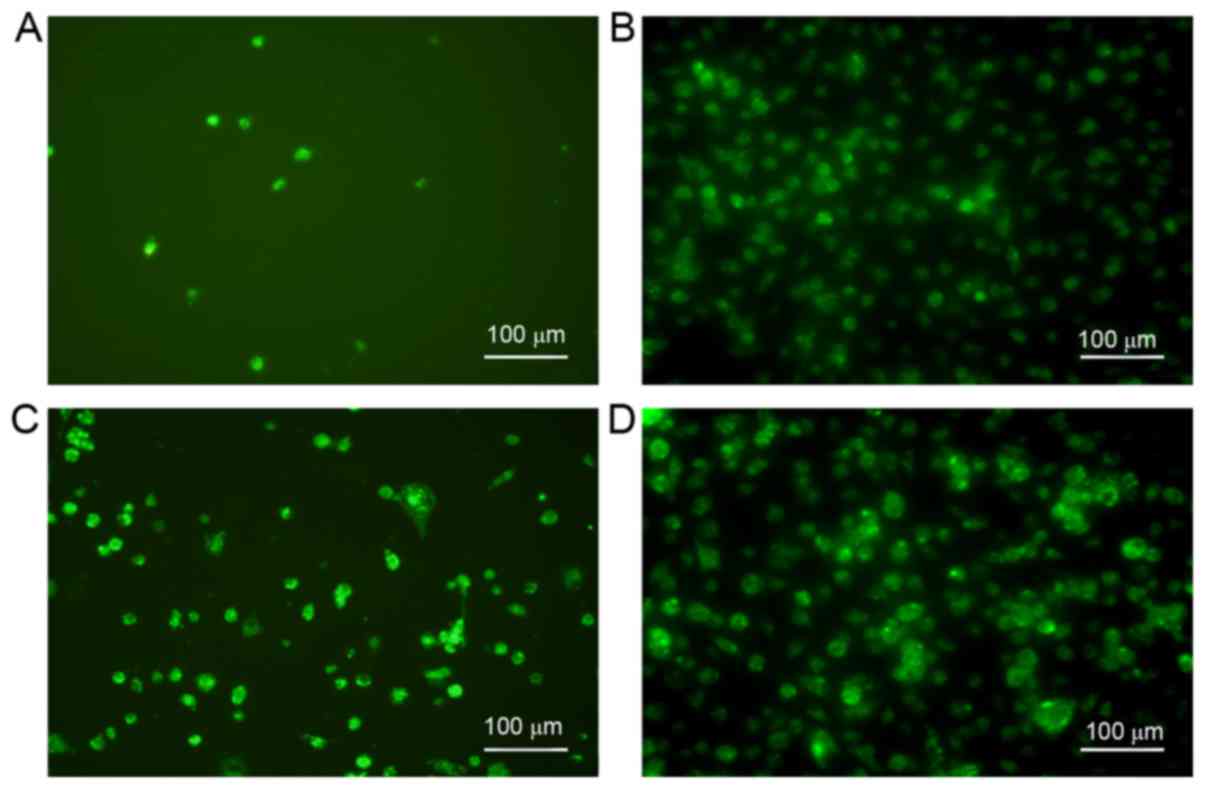
TNF-α and RANKL increase expression of
RANK in BMMs
To confirm that TNF-α and RANKL together increase
the expression of RANK, RANK expression was assessed in BMMs
exposed to TNF-α, RANKL or both, via immunofluorescence. The
results demonstrated that either TNF-α or RANKL may upregulate the
expression of RANK, yet exposure to both cytokines led to even
greater expression of RANK (Fig.
4).
Inhibition of NF-κB signaling prevents
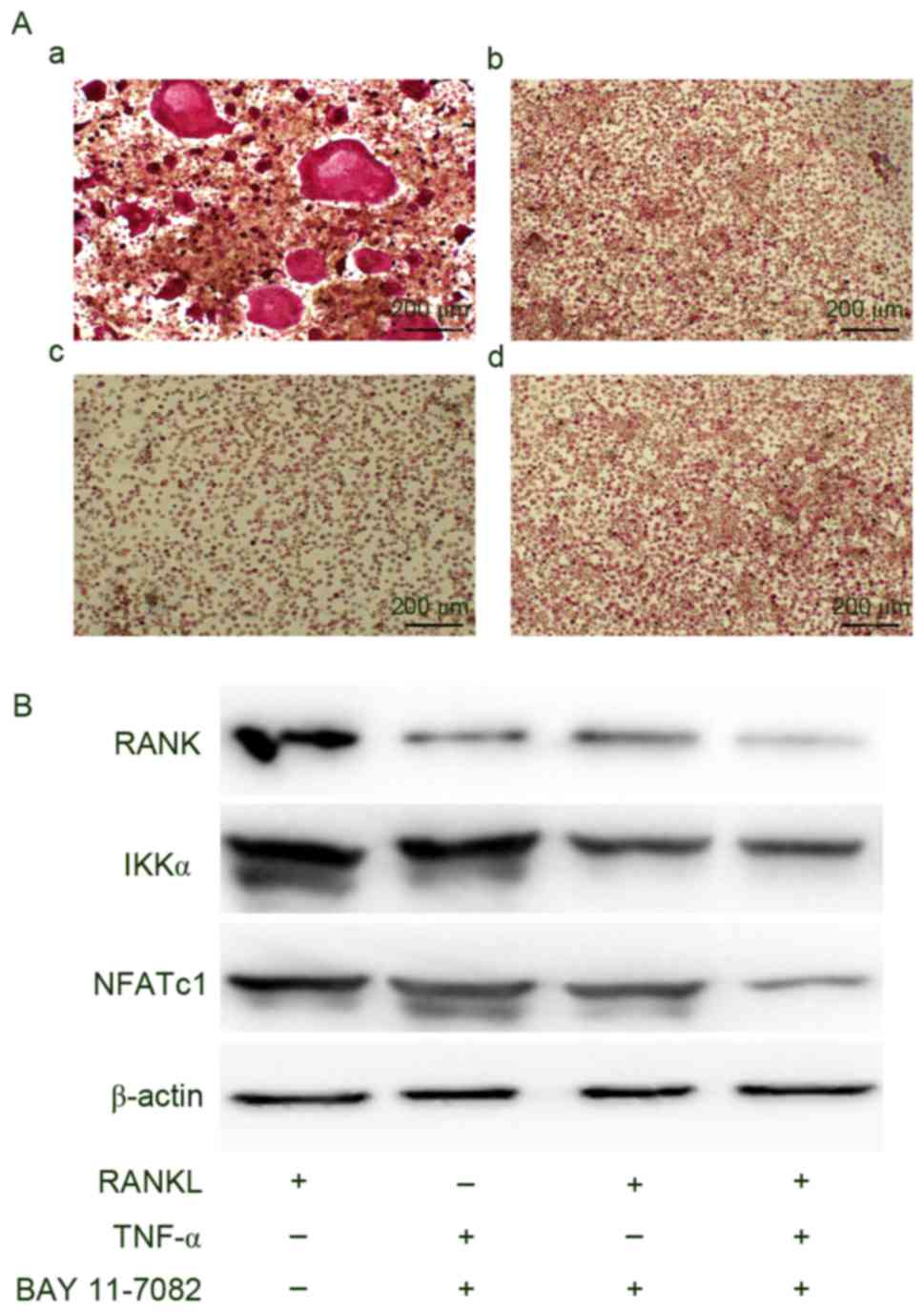
osteoclastogenesis even with stimulation by TNF-α and/or RANKL
Finally, the authors examined whether the NF-κB
signaling pathway participates in osteoclastogenesis among BMMs
using BAY 11–7082, a specific inhibitor of NF-κB. The addition of
BAY 11–7082 to BMMs in culture medium containing TNF-α, RANKL or
both resulted in the suppression of osteoclastogenesis and
downregulation of IKKα, NFATc1 and RANK expression (Fig. 5).
Discussion
Bone undergoes continuous renewal, which requires a
precise balance of bone resorption and formation. Bone resorption
by osteoclasts is known to be largely mediated by inflammatory
cytokines including TNF-α, which has been indicated to modulate a
broad spectrum of responses including inflammation, proliferation,
apoptosis, immunoregulation and antiviral activity at the cellular
level (21). In bone, TNF-α
inhibits extracellular matrix deposition and stimulates matrix
metalloprotease synthesis. Moreover, previous studies have
demonstrated that TNF-α can be used to induce bone resorption both
in vitro and in vivo (22,23).
To better understand the molecular mechanisms underlying the
functions of TNF-α in osteoclast differentiation, the authors
examined the activation of specific signaling pathways and the
expression of RANK and other osteogenic factors during the course
of osteoclastogenesis induced by M-CSF and RANKL with or without
TNF-α.
Initially, the viability of BMMs exposed to various
concentrations of TNF-α was examine in culture and concentrations
of TNF-α that were nontoxic to BMMs were identified. In addition,
greater activity among cells treated with TNF-α at concentrations
of ≤40 µg/µl was observed. However, all BMMs treated with TNF-α
presented greater viability than control cells not treated with
TNF-α. These data suggested that TNF-α can influence on the
viability of BMMs directly and were distributed normally between
the effect and concentration. Following this, whether TNF-α could
stimulate osteoclastic differentiation of BMMs with or without
RANKL was analyzed. The current results demonstrated that TNF-α
could not induce osteoclastogenesis of BMMs without RANKL, but did
enhance osteoclastogenesis when delivered with RANKL, as confirmed
by increased numbers of TRAP-positive multinucleated osteoclasts
and increased TRAP activity. Nevertheless, the osteoclastogenic
effect of TNF-α independent of RANKL has been controversial, with
studies reporting that TNF-α directly promotes osteoclast formation
in vitro in the absence of RANKL (21,24).
In contrast to the present approach, peripheral blood mononuclear
cells (PBMCs) were isolated from whole blood of psoriatic arthritis
patients as the target cells treated with TNF-α, which then led to
the observation of an enhanced osteoclastogenic effect with TNF-α
alone. In contrast, the authors suggested that PBMCs should be seen
as the cells to be pretreated with various cytokines. However,
consistent with the present findings, another study reported that
TNF-α does not induce osteoclastogenesis without RANKL. Moreover,
Li et al (25) demonstrated
that administration of TNF-α to RANK-deficient mice is not able to
induce osteoclastogenesis in vivo. The current results
further support that TNF-α cannot serve as a substitute for RANKL
toward the induction of osteoclastogenesis in physiological
conditions.
Previous studies have indicated that TNF-α increases
the production of osteoclastogenic cytokines, such as M-CSF and
RANKL in osteoblast-like cells (26,27).
Therefore, TNF-α likely promotes the differentiation of osteoclast
precursors, thereby supporting bone resorption. To further
investigate the molecular mechanisms by which TNF-α induces
osteoclastogenesis, the authors evaluated the mRNA expression of
RANK, NFATc1 and NF-κB in BMMs exposed to TNF-α and RANKL
individually or in combination using RT-qPCR. The results indicated
that both TNF-α and RANKL individually upregulated the expression
of RANK and NFATc1, indicating the potential for similar activities
between these cytokines. Moreover, greater increases in the
expression of RANK and NFATc1, as well as increased NF-κB
expression, were observed upon treatment of BMMs with both TNF-α
and RANKL. The increased expression of RANK mRNA substantiated
matching trends in the intensity of immunofluorescent staining of
RANK in BMMs treated with TNF-α and RANKL individually or in
combination.
The present findings suggested that RANKL and TNF-α
function in concert in the osteoclastogenic process. Therefore, the
authors sought to determine whether this coordinated activity is
reflected in a signaling pathway essential for osteoclast
differentiation. When RANKL binds to its receptor RANK on
osteoclasts, activation of the NF-κB signaling pathway is one
critical distal event in osteoclastogenesis (28). To determine whether TNF-α also
activates the same signaling pathways as RANKL in the cytoplasm, a
specific inhibitor of NF-κB (BAY 11–7082) was used to block the
pathway. The results indicated that blocking the NF-κB pathway
inhibited the formation of osteoclasts as well as the upregulation
of RANK, IKKα and NFATc1 typically observed during
osteoclastogenesis. Thus, the NF-κB pathway may represent a target
for interrupting bone absorption caused by chronic
inflammation.
The current results confirm that the relationship
between TNF-α and RANKL in osteoclastogenesis is intricate. Both
factors, individually and in combination, have major effects on
osteoclast progenitors. The current data for the role of TNF-α in
inflammatory osteolysis indicate that TNF-α acts on the stromal
environment to enhance expression of osteoclastogenic factors,
thereby increasing osteoclast differentiation. These results
demonstrate that TNF-α alone cannot induce BMM differentiation into
osteoclasts, but TNF-α may directly stimulate BMM synthesis of RANK
and other osteoclastogenic cytokines, as well as amplify the
effects of RANKL via NF-κB signaling. Therefore, the NF-κB pathway
may represent a potential target for preventing osteoporosis and
arthritis caused by chronic inflammatory factors, such as
TNF-α.
Acknowledgements
The authors gratefully acknowledge all of the
members of the laboratory for sharing reagents and advice.
Funding
The present study was supported by the Ministry of
Science and Technology (grant no. 011cb964701).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZGW and BZ planned the study. GL, FL and XL
conducted the experiments. GL analyzed the data and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experiments involving mice were approved by the
Institutional Animal Care and Use Committee at the Third Military
Medical University (Chongqing, China).
Consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Rodan GA: The development and function of
the skeleton and bone metastases. Cancer. 97:726–732. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tanaka Y, Nakayamada S and Okada Y:
Osteoblasts and osteoclasts in bone remodeling and inflammation.
Curr Drug Targets Inflamm Allergy. 4:325–328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pathak JL, Bravenboer N, Verschueren P,
Lems WF, Luyten FP, Klein-Nulend J and Bakker AD: Inflammatory
factors in the circulation of patients with active rheumatoid
arthritis stimulate osteoclastogenesis via endogenous cytokine
production by osteoblasts. Osteoporos Int. 25:2453–2463. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jules J and Feng X: In vitro investigation
of the roles of the proinflammatory cytokines tumor necrosis
factor-alpha and interleukin-1 in murine osteoclastogenesis.
Methods Mol Biol. 1155:109–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Córdova LA, Trichet V, Escriou V, Rosset
P, Amiaud J, Battaglia S, Charrier C, Berreur M, Brion R, Gouin F,
et al: Inhibition of osteolysis and increase of bone formation
after local administration of siRNA-targeting RANK in a
polyethylene particle-induced osteolysis model. Acta Biomater.
13:150–158. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weitzmann MN: The Role of inflammatory
cytokines, the RANKL/OPG axis, and the immunoskeletal interface in
physiological bone turnover and osteoporosis. Scientifica (Cairo).
2013:1257052013.PubMed/NCBI
|
|
7
|
Gallois A, Lachuer J, Yvert G, Wierinckx
A, Brunet F, Rabourdin-Combe C, Delprat C, Jurdic P and Mazzorana
M: Genome-wide expression analyses establish dendritic cells as a
new osteoclast precursor able to generate bone-resorbing cells more
efficiently than monocytes. J Bone Miner Res. 25:661–672. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Vries TJ, Schoenmaker T, Aerts D,
Grevers LC, Souza PP, Nazmi K, van de Wiel M, Ylstra B, Lent PL,
Leenen PJ and Everts V: M-CSF priming of osteoclast precursors can
cause osteoclastogenesis-insensitivity, which can be prevented and
overcome on bone. J Cell Physiol. 230:210–225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim HJ, Yoon HJ, Kim SY and Yoon YR: A
medium-chain fatty acid, capric acid, inhibits RANKL-induced
osteoclast differentiation via the suppression of NF-kappaB
signaling and blocks cytoskeletal organization and survival in
mature osteoclasts. Mol Cells. 37:598–604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi J, Choi SY, Lee SY, Lee JY, Kim HS,
Lee SY and Lee NK: Caffeine enhances osteoclast differentiation and
maturation through p38 MAP kinase/Mitf and DC-STAMP/CtsK and TRAP
pathway. Cell Signal. 25:1222–1227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lampiasi N, Russo R and Zito F: The
alternative faces of macrophage generate osteoclasts. Biomed Res
Int. 2016:90896102016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ishida N, Hayashi K, Hoshijima M, Ogawa T,
Koga S, Miyatake Y, Kumegawa M, Kimura T and Takeya T: Large scale
gene expression analysis of osteoclastogenesis in vitro and
elucidation of NFAT2 as a key regulator. J Biol Chem.
277:41147–41156. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Johnson RS, Spiegelman BM and Papaioannou
V: Pleiotropic effects of a null mutation in the c-fos
proto-oncogene. Cell. 71:577–586. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takayanagi H, Kim S, Koga T, Nishina H,
Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T and Inoue J:
Induction and activation of the transcription factor NFATc1 (NFAT2)
integrate RANKL signaling in terminal differentiation of
osteoclasts. Dev Cell. 3:889–901. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Danks L and Takayanagi H: Immunology and
bone. J Biochem. 154:29–39. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Almeida M, Han L, Ambrogini E, Weinstein
RS and Manolagas SC: Glucocorticoids and tumor necrosis factor α
increase oxidative stress and suppress Wnt protein signaling in
osteoblasts. J Biol Chem. 286:44326–44335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dong J, Cui X, Jiang Z and Sun J:
MicroRNA-23a modulates tumor necrosis factor-alpha-induced
osteoblasts apoptosis by directly targeting Fas. J Cell Biochem.
114:2738–2745. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garcia-Lopez S, Villanueva R and Meikle
MC: Alterations in the Synthesis of IL-1β, TNF-α, IL-6, and their
downstream targets RANKL and OPG by mouse calvarial osteoblasts
in vitro: Inhibition of bone resorption by cyclic mechanical
strain. Front Endocrinol (Lausanne). 4:1602013.PubMed/NCBI
|
|
19
|
Lin NY, Stefanica A and Distler JH:
Autophagy: A key pathway of TNF-induced inflammatory bone loss.
Autophagy. 9:1253–1255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kitaura H, Kimura K, Ishida M, Kohara H,
Yoshimatsu M and Takano-Yamamoto T: Immunological reaction in
TNF-α-mediated osteoclast formation and bone resorption in
vitro and in vivo. Clin Dev Immunol. 2013:1818492013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mukai T, Ishida S, Ishikawa R, Yoshitaka
T, Kittaka M, Gallant R, Lin YL, Rottapel R, Brotto M,
Reichenberger EJ and Ueki Y: SH3BP2 cherubism mutation potentiates
TNF-α -induced osteoclastogenesis via NFATc1 and TNF-α -mediated
inflammatory bone loss. J Bone Miner Res. 29:2618–2635. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Osta B, Benedetti G and Miossec P:
Classical and paradoxical effects of TNF-alpha on bone homeostasis.
Front Immunol. 5:482014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ritchlin CT, Haas-Smith SA, Li P, Hicks DG
and Schwarz EM: Mechanisms of TNF-alpha- and RANKL-mediated
osteoclastogenesis and bone resorption in psoriatic arthritis. J
Clin Invest. 111:821–831. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li J, Sarosi I, Yan XQ, Morony S,
Capparelli C, Tan HL, McCabe S, Elliott R, Scully S, Van G, et al:
RANK is the intrinsic hematopoietic cell surface receptor that
controls osteoclastogenesis and regulation of bone mass and calcium
metabolism. Proc Natl Acad Sci USA. 97:pp. 1566–1571. 2000;
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu Y, Yang D, Qiu L, Okamura H, Guo J and
Haneji T: Tumor necrosis factor-α induces interleukin-34 expression
through nuclear factor-κB activation in MC3T3-E1 osteoblastic
cells. Mol Med Rep. 10:1371–1376. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiong J, Piemontese M, Thostenson JD,
Weinstein RS, Manolagas SC and O'Brien CA: Osteocyte-derived RANKL
is a critical mediator of the increased bone resorption caused by
dietary calcium deficiency. Bone. 66:146–154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Swarnkar G and Abu-Amer Y: Regulation of
NF-κB signaling in osteoclasts and myeloid progenitors. Methods Mol
Biol. 1280:527–542. 2015. View Article : Google Scholar : PubMed/NCBI
|