In the event of external triggers, cardiac
insufficiency responds to adaptive alterations in both the
structure and function of the heart, commonly referred to as
cardiac remodeling. These alterations include changes in genomic
expression levels, cell morphology and abnormal interstitial
secretion (1,2). Cardiac remodeling is divided into
physiological and pathological types. Physiological cardiac
remodeling is a reversible adaptive reaction that primarily occurs
during growth, exercise and pregnancy (3). Where pathological cardiac remodeling
is an irreversible adaptive response caused by numerous conditions,
including myocardial infarction (MI), ischemia/reperfusion (I/R)
injury, pressure loading, inflammation and oxidative stress
(4,5). Direct manifestations of cardiac
remodeling include myocardial hypertrophy and cardiac fibrosis and
continued poor remodeling can lead to heart failure (6–8).
Thus, determining the mechanisms that lead to cardiac remodeling
and preventing undesirable remodeling is essential.
Sodium-glucose cotransporter type 2 inhibitors
(SGLT2is) are hypoglycemic medications that inhibit SGLT2 in the
renal tubules, decreasing glucose reabsorption, lowering the renal
glucose threshold and initiating glucose excretion in the urine
(9). Compared with other
traditional hypoglycemic drugs, SGLT2is are primarily used for
treating type 2 diabetes but have also been reported to exert
cardiovascular benefits. According to cardiovascular outcome
studies, SGLT2is reduce the incidence of hospitalization due to
heart failure (10–14). The ‘new tetrad’ of cornerstone
heart failure medications has replaced the original ‘golden
triangle’ and now includes beta-blockers, aldosterone receptor
antagonists, renin-angiotensin system inhibitors and SGLT2is
(15,16).
The four SGLT2 inhibitors widely used in clinical
treatment are Empagliflozin (EMPA), Dapagliflozin (DAPA),
Canagliflozin (CANA) and Ertugliflozin; these have been approved by
the US Food and Drug Administration and the European Medicines
Agency (17). EMPA and DAPA are
widely used for heart failure prevention and treatment (15,16).
Existing studies have confirmed the lack of SGLT2 expression in
cardiac tissue, thereby necessitating the investigation of the
myocardial protective effect of SGLT2is (18). It has been suggested that SGLT2is
exerts a diuretic effect via glomerular reabsorption, reducing
blood volume and cardiac load and protecting the heart by reducing
myocardial oxygen consumption. However, as this diuretic effect is
dependent on blood glucose concentration, the cardiac benefit in
non-diabetic patients has not been determined (19). Therefore, the protective effect of
SGLT2is on the myocardium may be exerted directly on the heart.
Several studies have shown that the anti-heart failure effect of
SGLT2is may be mediated by inhibiting or reversing cardiac
remodeling (20–22).
Currently, the molecular mechanisms and signaling
pathways of SGLT2is in cardiac remodeling are being investigated.
The present review provides a foundation and supports the
investigation of novel mechanisms of cardiac remodeling and heart
failure, as well as the development of novel drug targets based on
the function and molecular mechanisms of SGLT2is-mediated
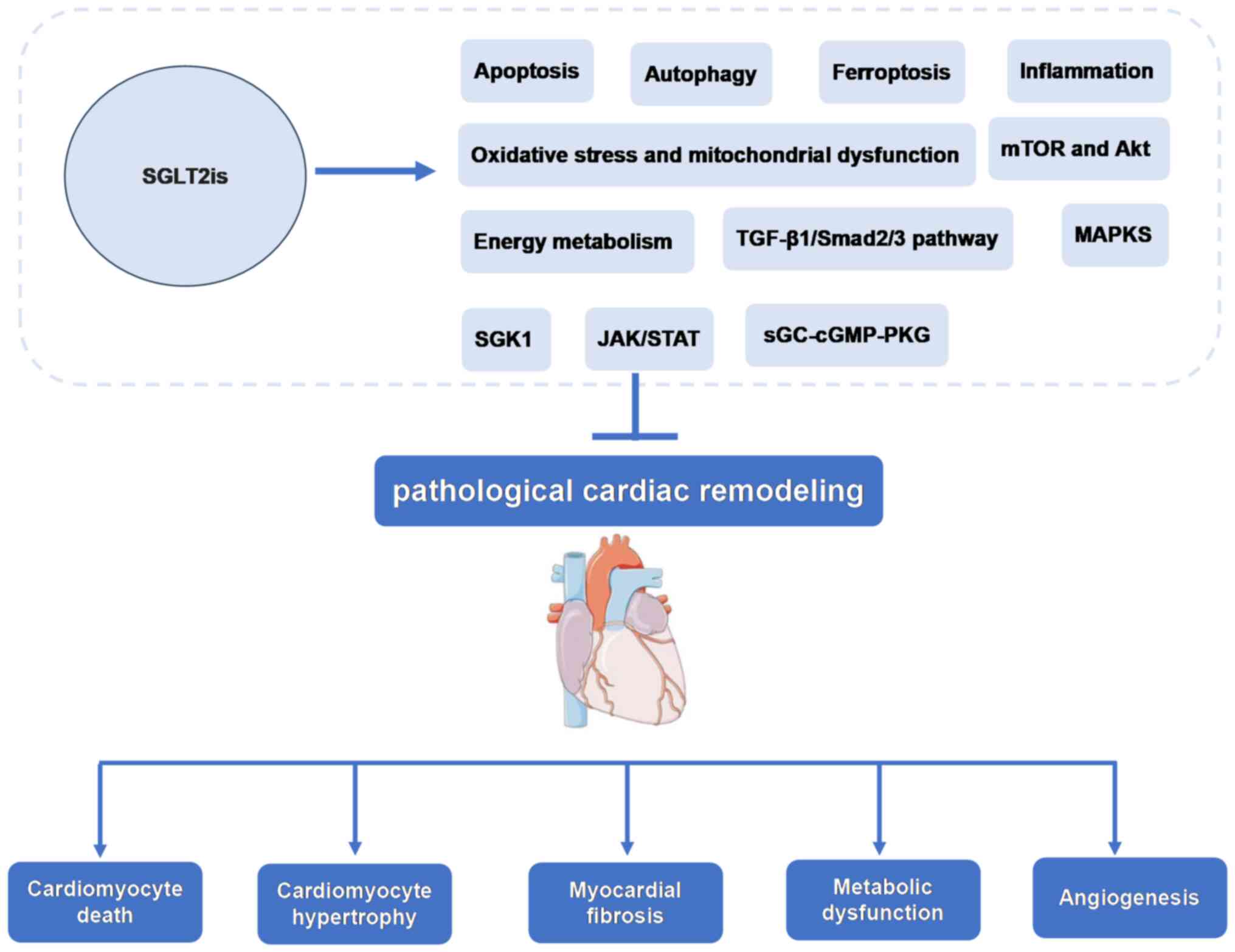
inhibition of pathological cardiac remodeling (Fig. 1).
Pathological cardiac remodeling is often manifested
by changes in the morphology and size of the left ventricle. In
addition, the left ventricular (LV) mass index (LVMI) and LV
ejection fraction (LVEF) are used as evaluation indexes of cardiac
structural function (23,24). Studies have reported that
SGLT2isThe improved cardiac function is mainly manifested as
increased LVEF and decreased LV end-diastolic volume (LVEDV), LV
end-systolic volume (LVESV), left atrial volume index (LAVI) and
LVMI (22,25–35).
However, LV diastolic dysfunction (LVDD) often manifests as altered
LV diastolic filling and is assessed based on the peak mitral E
wave velocity to early mitral or septal annular tissue Doppler
velocity ratio (E/e'), peak mitral E wave velocity to A wave
velocity ratio and LAVI (36).
SGLT2is also improved ejection fraction, LVEDV, LVESV and diastolic
dysfunction in animal models of heart failure (37–46).
Regarding cardiac structure, the LV mass (LVM), LV
wall thickness and LV wall thickness-to-cavity radius can be used
to determine the structural and morphological changes of the LV.
Specifically, increased LVM has been considered a marker of
clinical LV hypertrophy (47).
Several studies have demonstrated that the cardiovascular benefits
of SGLT2is may be achieved through reduced LVM, as it occurs
without a decline in the volume, which reflects the decrease in
ventricular wall thickness (26,29,30,48).
However, the mechanisms of decreasing wall thickness are yet to be
elucidated. In the present review, the effects of SGLT2is on
cardiac structure and function in patients with cardiovascular
disease and animal models were summarized (Tables I and II).
Pathological cardiac remodeling causes hypertrophy
of cardiomyocytes and the proliferation of non-cardiomyocytes in
numerous cardiovascular diseases, including hypertension, diabetic
cardiomyopathy, aortic stenosis, MI, pathological stimulation,
cardiomyocyte hypertrophy and cardiac fibrosis (3). The characteristics of cardiac
hypertrophy are abnormal size and function of myocardial cells,
often manifested as increased ventricular mass, myocardial cell
volume and expression of fetal genes, such as atrial natriuretic
peptide, brain natriuretic peptide and β-myosin heavy chain
(49). Myocardial fibrosis, the
excessive deposition of extracellular matrix, is closely associated
with the severity of myocardial fibrosis. Type I collagen is the
most abundant structural protein (50,51).
Myocardial fibroblasts are the main cellular effectors that lead to
cardiac fibrosis. Pathological stimuli can reduce the number of
cardiomyocytes, which in turn stimulates inflammation. In order to
compensate for the loss of cardiomyocytes, cardiac fibroblasts
proliferate and differentiate into myofibroblasts, leading to scar
formation (52).
Cardiac hypertrophy and cardiac fibrosis are the
major pathological processes in cardiac remodeling and are closely
related to the prognosis of cardiovascular diseases, making them
the primary intervention targets for heart failure (53,54).
Several studies have demonstrated that SGLT2is attenuates or
inhibits cardiomyocyte hypertrophy and cardiac fibrosis by
regulating multiple signaling pathways in numerous models, such as
transverse aortic constriction (TAC), left coronary artery ligation
MI and diabetes (39,40,55–63).
Apoptosis is a type of programmed cell death that
serves a key role in embryonic development and tissue homeostasis
(64). Apoptosis is mediated by
death receptors, also known as extrinsic apoptotic pathways and
mitochondria, also called intrinsic apoptotic pathways, both of
which can activate cysteine-dependent proteases (caspases)
(65). Apoptosis serves a crucial
role in the development of the heart and is associated with the
occurrence and development of numerous cardiovascular diseases.
Studies have reported that apoptosis is a pathological feature of
MI and heart failure, and that the inhibition of apoptosis can
prevent and treat post-MI remodeling and heart failure (66).
Further studies have reported that SGLT2is reduces
cardiac remodeling and improves cardiac function by inhibiting the
apoptosis pathways. EMPA inhibits cardiomyocyte apoptosis and
improves cardiac remodeling in early MI in non-diabetic mice
(67).
In mice with autoimmune myocarditis induced by
α-myosin-heavy chain peptides, CANA markedly reduces the Bax/Bcl-2
ratio and the level of cleaved caspase-3 protein, followed by
inhibition of apoptosis, which was reported to improve myocarditis
(68). In cardiac I/R rats,
DAPA-induced pre-ischemia upregulated the levels of anti-apoptotic
protein Bcl-2 to protect cardiomyocytes from apoptosis, thereby
alleviating cardiac mitochondrial dysfunction by reducing reactive
oxygen species (ROS) production (43). DAPA mediates the cardioprotective
effect in diabetic rats by activating the phosphorylation of Akt,
JAK2 and MAPK signaling cascades, increasing the erythropoietin
levels and reducing apoptosis (69). DAPA also normalizes mitochondrial
fission and reduces cardiomyocyte apoptosis by activating the
phosphoglycerate mutase member 5 (PGAM5)/dynamin-related protein 1
(Drp1) signaling pathway, thereby improving cardiac remodeling
after acute MI (70). However,
this study did not use an agonist for the PGAM5/Drp1 pathway, so
the relationship between DAPA and the PGAM5/Drp1 signaling pathway
could not be assessed.
A recent study reported that in Doxorubicin-induced
cardiac dysfunction, DAPA decreased the cardiac expression of Bax
and cleaved caspase-3, but increased the expression of Bcl-2, as
well as signal transducer and activator of transcription 3 (STAT3)
that was subsequently inhibited by Doxorubicin (71). These findings indicate that DAPA
activates the expression of sirtuin1 (SIRT1), which inhibits the
protein kinase RNA-like endoplasmic reticulum (ER) kinase
(PERK)-eukaryotic translation initiation factor 2α (eIF2α)-C/EBP
homologous protein signaling pathway of the ER stress response in
angiotensin II (Ang II)-treated cardiomyocytes to reduce
cardiomyocyte apoptosis and improve TAC-induced cardiac remodeling
in mice (72). Therefore, it may
be suggested that SGLT2is improves damaged cardiac function by
continual myocardial cell apoptosis.
Many of the mechanisms are effectuated through the
mitochondrial pathway. However, only a small number of studies have
assessed the role of SGLT2is in myocardial cell apoptosis induced
by the death receptor pathway. Hence, this mechanism should be
evaluated to understand the anti-apoptotic mechanism of
SGLT2is.
Autophagy is a process that degrades and
recirculates damaged organelles, misfolded proteins and other
macromolecules through lysosomal-dependent pathways to maintain
cell homeostasis and function (73). Previous studies have shown a
crucial role of basal autophagy in cardiac development and in the
maintenance of normal cardiac function (74–77).
However, insufficient or excessive autophagy can affect the
development of pathological cardiac remodeling (78–80).
Furthermore, the activation of autophagy leads to the death of
cardiomyocytes in MI and I/R injury and has been shown to have a
dual effect in numerous research models, which may be related to
the Beclin 1 (BECN1) or AMPK-mammalian target of rapamycin (mTOR)
pathways (81–83). Several studies have demonstrated
that SGLT2is exerts cardioprotective effects through the activation
or inhibition of autophagy.
In mouse models of coronary artery ligation-induced
diabetic and non-diabetic MI, EMPA-treated mice demonstrated a
significant decrease in cardiomyocyte death due to excessive
autophagy, which reduced autophagic flux by targeting the
Na+/H+ exchanger 1 (NHE1) on cardiomyocytes
(63). EMPA exerts myocardial
protective effects through mitochondrial autophagy and the novel
BECN1-Toll-like receptor (TLR)9-SIRT3 axis (84). Furthermore, EMPA exerts
cardioprotective effects in non-diabetic mice with MI with acute
hyperglycemia by suppressing beclin 1 (BCN1)-dependent autophagy
rather than targeting NHE1 in cardiomyocytes (85). Previous studies have demonstrated
that BCN1 promotes the crosstalk between apoptosis and autophagy
(86). In another study, EMPA was
reported to inhibit ER stress-induced autophagy by inhibiting the
PERK/activating transcription factor 4/BCN1 signaling pathway,
thereby alleviating myocardial I/R injury and cardiomyocyte
apoptosis (87). Furthermore,
overexpression of p62 and light chain 3II/I activates autophagy
when EMPA is administered. It reduces cardiac lipid toxicity in
Zucker diabetic fatty (ZDF) rats (88).
Likewise, DAPA represses cardiac remodeling and
hypoxia-induced apoptosis in heart failure through the activation
of autophagy via the AMPK/mTOR pathway (89). It also protects against myocardial
I/R injury by limiting NLR family pyrin domain containing 3 (NLRP3)
inflammatory vesicle activation and regulating autophagy (90). The dose of DAPA administered in
this study was 40 mg/kg/day, which is 20X higher compared with the
allometric-adapted dose used in human clinical trials. It cannot be
ruled out that the final result is related to high doses. SGLT2is
exert cardioprotective effects through the activation and
inhibition of autophagy via the interference of varied pathological
conditions and detection time. SGLT2is regulate autophagy through
the AMPK pathway, ER stress and inflammasomes. These pathways also
regulate the processes of cell apoptosis, inflammation and
angiogenesis. However, the association between these pathological
processes and autophagy or the precise mechanisms of SGLT2is are
yet to be elucidated.
Ferroptosis is a form of programmed cell death
different from cell apoptosis, cell necrosis and cell autophagy. It
is mediated by iron-dependent lipid peroxides and characterized by
reduced intracellular glutathione (GSH) expression, reduced GSH
peroxidase 4 (GPX4) activity and the accumulation of ROS and lipid
peroxides (91–93). Several studies have shown the role
of ferroptosis in numerous cardiovascular diseases, such as
cardiomyopathy, MI, myocardial I/R injury, atherosclerosis and
heart failure (94–98). Furthermore, SGLT2is exert
cardioprotective effects through the ferroptosis pathway. In model
rats, CANA can treat heart failure with preserved ejection fraction
(HFpEF) by reducing iron intake and iron overload, reducing lipid
peroxidation, increasing GSH production and inhibiting oxidative
stress to regulate ferroptosis (99).
Furthermore, advanced glycation end-products inhibit
the expression of solute carrier family 7 member 11 and ferritin in
diabetic cardiomyopathy and reduce GSH levels. This elevates lipid
peroxidation levels and ferroptosis, which in turn triggers cardiac
inflammation and cardiac remodeling, including cardiomyocyte
hypertrophy, pro-fibrotic response, fibrosis and ultimately cardiac
dysfunction (100). Another study
reported that CANA may reduce ferroptosis and improve myocardial
oxidative stress in diabetic cardiomyopathy mice by regulating iron
metabolism and the systemic cystine-glutamate antiporter
(Xc−)/GSH/GPX4 axis (101). However, the relationship and
specific mechanism of CANA in regulating iron metabolism and the
Xc−/GSH/GPX4 axis require further evaluation. In
addition, CANA has been reported to inhibit inflammation and
ferroptosis through the activation of the AMPK pathway, thereby
reducing lipotoxicity in cardiomyocytes (102).
Furthermore, EMPA prevents DNA oxidation and
ferroptosis in trastuzumab-induced C57BL/6J mice, which attenuates
cardiotoxicity (103). Likewise,
DAPA suppresses the MAPK signaling pathway in a model of myocardial
I/R injury, reducing ferroptosis and exerting protective benefits
on the heart (104).
Inflammation is a leading factor affecting cardiac
remodeling and the progression of heart failure (105,106). Toll-like receptors (TLRs), a
family of transmembrane receptors, are recognized by
danger-associated molecular patterns (DAMPs) in MI and activate
nuclear factor-B (NF-κB), which in turn activates a cascade of
inflammatory mediators, including cell adhesion molecules,
chemokines, and inflammatory cytokines (107,108). Furthermore, the inflammasome,
which are polymeric protein structures, form molecular platforms
that are activated when cells are infected or stressed, stimulates
the inflammatory response by activating several inflammatory
cytokines, such as IL-1 and IL-18 (109). Thus, targeting specific
cytokines, growth factors or inflammatory pathways could alleviate
adverse cardiac remodeling.
The activation of multiple pro-inflammatory
cytokines, such as TNF-α, IL-1 and IL-6, mediates cardiac
remodeling through their effects on cardiomyocytes, fibroblasts and
immune cells (110). These
cytokines can induce cardiomyocyte hypertrophy and apoptosis
(111–113). Pro-inflammatory cytokines enhance
the activity of matrix metalloproteinases and decrease the
production of extracellular matrix (ECM) components in fibroblasts,
which causes the ECM to degrade (114–116). Thus, pro-inflammatory cytokines
serve a role in pathological cardiac remodeling. Furthermore, the
pleiotropic anti-inflammatory factor IL-10 decreases the expression
of TNF-α, IL-1 and IL-6 to reduce cardiac inflammation (117,118).
Furthermore, SGLT2is downregulates pro-inflammatory
cytokines and improves cardiac function in cardiovascular diseases.
DAPA decreased the levels of inflammatory cytokines IL-6 and TNF-α
in HFpEF pigs administered deoxycorticosterone acetate and Ang II
to construct an ejection fraction-preserving heart failure model
(41). Likewise, EMPA decreased
TNF-α and IL-6 levels in patients with HfpEF and ZDF obese rats,
reduced inflammation and enhanced myocardial function (119). Furthermore, EMPA markedly
decreased the level of TNF-α and reduced myocardial fibrosis in
hypertensive heart failure rats (42). DAPA decreased the levels of the
pro-inflammatory cytokines IL-1, IL-6 and TNF-α in viral
myocarditis mice infected with Coxsackievirus B3. DAPA facilitated
macrophage polarization through STAT3-related pathways to reduce
myocarditis (120). Furthermore,
DAPA improves cardiac hypertrophy in streptozocin-induced type 2
diabetic rats by inhibiting the nuclear translocation of NF-κB and
reducing the expression of calpain-1 in cardiomyocytes, decreasing
IL-6 and TNF-α levels and upregulating IL-10 levels (58). In addition, DAPA regulates
malondialdehyde, TNF-α and ROS levels by blocking the C-X3-C motif
chemokine ligand 1/receptor 1 axis and NF-κB activity, thereby
reducing lipopolysaccharide-induced inflammation and oxidative
stress (121). However, this
study was performed in vitro using H9C2 cells and requires
validation in patients.
The NLRP3 inflammasome accelerates the process of
fibrosis by stimulating the production of proinflammatory cytokines
IL-1β and IL-18 (122). Several
factors, including MI, stress, obesity, diabetes and metabolic
syndrome, activate the NLRP3 inflammasome and promote inflammation
(123–125). DAPA exerts anti-inflammatory
effects on the development of diabetic cardiomyopathy in type 2
diabetic mice by decreasing the expression of NLRP3 inflammasome,
IL-1β, IL-6 and TNF-α (126,127). Likewise, EMPA inhibits cardiac
fibrosis and inflammation in non-diabetic mice treated with
Doxorubicin via the NLRP3 and MyD88 signaling pathways and
inhibition of NLRP3 and NF-κB inhibits the pro-inflammatory
cytokine storms in Doxorubicin-treated cardiomyocytes (128). Furthermore, DAPA decreases
p38-dependent TLR4 expression to prevent NLRP3 activation, which
then enhances cardiac function in Doxorubicin-induced dilated
cardiomyopathy (129). Finally,
CANA reduces type 17 T-helper cell infiltration and protects
cardiomyocytes from apoptosis by inhibiting the NLRP3 inflammasome
pathway, which reduces myocarditis-induced cardiac inflammation
(68).
Macrophages also serve a role in the inflammatory
response during cardiac remodeling (130). SGLT2is reduce cardiac fibrosis by
regulating macrophage M2 polarization in infarcted rat hearts via
the STAT3 signaling pathway (131). Inflammatory and NF-κB signaling
pathways are triggered in patients with arrhythmogenic
cardiomyopathy (ACM) (132). DAPA
reduces cardiac fibrosis and inflammation in ACM mice by reversing
hypoxia-inducible factor (HIF)-2α signaling, inhibiting the NF-κB
signaling pathway (133).
Accumulating evidence indicates that the role of SGLT2is in
controlling inflammation is associated with fat reduction, which is
efficacious in epicardial adipose tissue (134). Although the aforementioned
studies have reported that SGLT2is serve a myocardial-protective
role through anti-inflammatory mechanisms, another study on EMPA
reported conflicting results; EMPA did not show any effect on the
NLRP3 inflammasome pathway or interleukin-1β levels (135). The present review concluded that
the effectiveness of SGLT2is in inhibiting inflammation is
indeterminate, and more comprehensive information is essential to
draw further conclusions.
Oxidative stress is a redox imbalance caused by the
excessive production of ROS and/or an impaired antioxidant response
(136). The primary ROS sources
in the heart are mitochondria, NADPH oxidase (NOX), xanthine
oxidase (XO) and uncoupled nitric oxide synthase (NOS) (137). A large number of heart cells can
be affected by NOX via redox signal transduction. NOX regulates
redox-sensitive target proteins to limit the production of ROS
(138). Under physiological
circumstances, normal ROS signaling controls the growth and
maturation of cardiomyocytes, the processing of cardiac calcium,
excitatory systolic coupling and vascular tone (139). However, oxidative stress
effectuated by a sharp rise in ROS causes cardiac hypertrophy,
fibrosis, apoptosis and contractile failure under pathological
circumstances (140).
Furthermore, oxidative stress is considered a key factor in the
development of pathological cardiac remodeling and heart failure,
as this disrupts mitochondrial activity by inducing oxidative
damage to mitochondrial DNA, RNA, lipids and proteins. Oxidative
stress also impairs myocardial cell systolic function by inducing
mitochondria-associated oxidative modifications of
excitation-contraction-coupled core proteins (141). Several studies have shown that
the cardiac benefits of SGLT2is are reduced oxidative stress in
vivo and ameliorated mitochondrial dysfunction through multiple
signaling pathways.
EMPA improves mitochondrial function by inhibiting
mitochondrial fission in type 2 diabetic hearts, as demonstrated by
an increase in the expression of mitochondrial fusion-related
proteins mitofusin-1 and optic atrophy 1 and the inhibition of DRP1
expression in type 2 diabetic db/db mice and H9C2 cardiomyocytes.
In the present study, oxidative stress was reduced by increasing
the expression of nuclear factor erythroid 2-related factor 2
(Nrf2) and its downstream genetic targets (59).
DAPA protects cardiomyocytes from
hyperglycemia-induced damage by inhibiting NOX-mediated oxidative
stress (142), whereas treatment
with EMPA reduces LV hypertrophy and fibrosis after TAC and MI in
non-diabetic mouse models and Sprague Dawley (SD) rats with
coronary artery ligation-induced oxidative stress. Hypertrophy and
fibrosis were improved by upregulating mitochondrial biogenesis,
enhancing mitochondrial oxidative phosphorylation, reducing ROS
production, attenuating apoptosis and increasing autophagy
(39,143). EMPA treatment in diet-induced
obese mice reduced cardiac fat accumulation and mitochondrial
injury, improved myocardial hypertrophy and cardiac fibrosis and
reduced cardiac dysfunction. This effect may be reduced by
Sestrin2-mediated AMPK-mTOR signaling and Nrf2/heme oxygenase
1-mediated oxidative stress responses (144). In addition, EMPA inhibited
high-fructose diet-induced cardiac dysfunction in type 2 diabetic
SD rats by attenuating mitochondria-driven oxidative stress
(145). DAPA reduces oxidative
stress, mitochondrial dysfunction, fibrosis, hypertrophy and
inflammation in Doxorubicin-stimulated rats via inhibition of
PI3K/AKT/Nrf2 signaling (61).
This study used only male and no female animals, while females may
be more sensitive to Doxorubicin and mimic the clinical state.
Modifications in myocardial energy metabolism
contribute to the development of pathological cardiac remodeling
(146). It is often manifested by
a switch of the heart back to the fetal genetic program and a shift
in preference of metabolic substrate from fatty acids to glucose
(147,148). Cardiac remodeling is associated
with reduced lipid oxidation capacity and increased glucose
dependence (149). Although the
conversion of myocardial metabolic substrates from fatty acids to
glucose lowers oxygen consumption, it can be detrimental to cardiac
performance and aggravate heart failure because of insufficient
energy production (150,151). Furthermore, preserving fatty acid
oxidation during stress overload prevents the effects of glucose on
cardiac remodeling (152,153). Thus, promoting the use of fatty
acids and other metabolic substrates to regulate energy metabolism
may be a promising therapeutic strategy for heart failure and to
improve cardiac remodeling (154).
EMPA reduces excessive glycolysis in TAC-induced
cardiac overload mice by binding to glucose transporter (GLUT)
proteins, such as GLUT1 and GLUT4, which increases the expression
of CD36, restores fatty acid uptake and improves mitochondrial
oxidative phosphorylation. The reduced glucose uptake may also lead
to an impaired pentose phosphate pathway, which in turn activates
AMP-activated protein kinases and blocks mTOR complex 1 (mTORC1) to
reduce cardiac hypertrophy (57).
Contrastingly, EMPA has been shown to improve
diabetic cardiac remodeling in diabetic cardiomyopathic rats by
reducing fatty acid and increasing glucose metabolism (155), although this may be related to
increased fatty acids in diabetic heart disease, which leads to
lipid toxicity and insulin resistance (146,156), hence the contrasting results
reported. EMPA significantly elevated cardiac metabolism and
cardiac ATP production in coronary artery-ligated non-diabetic male
SD rats by increasing ketone body bioavailability and myocardial
oxidation of glucose and fatty acids (39). However, this study did not provide
direct evidence that EMPA-treated hearts were associated with
increased ketone body oxidation, nor did it quantify the
relationship between ketone body oxidation and increased myocardial
ATP levels.
Similarly, EMPA altered the myocardial fuel
metabolic substrates from glucose to ketone bodies, free fatty
acids and branched-chain amino acids in non-diabetic pigs induced
by 2 h of proximal balloon occlusion of the left anterior
descending branch. This improved myocardial energy, enhanced LV
systolic function and improved unfavorable LV remodeling (38).
Angiogenesis is the physiological and pathological
process of forming new microvessels from pre-existing capillaries
in response to hypoxia. Angiogenesis involves endothelial cell
proliferation, migration, differentiation, tube formation and
regulation of angiogenic factors (157–159). The development of cardiac
remodeling is significantly influenced by microvascular density
(157,160). Several studies have demonstrated
that promoting angiogenesis increases the density of microvessels
and arteriolar, thus reducing cardiac remodeling (161–165).
Previous studies have shown that EMPA promotes
myocardial microcirculatory perfusion and cardiac function by
reducing AMPK-mediated mitochondrial fission and oxidative stress
and stabilizing F-actin (166).
Studies in a mouse model of diabetes-related hindlimb ischemia
found that DAPA promotes vascular endothelial cell proliferation
and migration through the prolyl hydroxylase domain protein
2/HIF-1α axis, the secretion of multiple angiogenic factors, the
formation of neovascularization and increases in blood perfusion
(167). EMPA improves systolic
dysfunction during LV pressure overload in mice by activating the
AKT/endothelial NOS (eNOS)/NO pathway to prevent endothelial
apoptosis and maintain capillarization (168). In the event of myocardial I/R
injury in non-diabetic mice, EMPA inhibits the DNA-dependent
protein kinase catalytic subunit/fission 1 protein/mitochondrial
fission pathway, protecting the microvascular system (169). However, the microvascular
function in vivo is difficult to evaluate. In this study,
only electron microscopy was used to observe the structural changes
of microvessels in mice treated with EMPA, which is insufficient.
However, coronary blood flow reserve can also be used. Another
study demonstrated that DAPA reduces cardiac endothelial
dysfunction and microvascular injury by inhibition of the
XO/sarco(endo) plasmic reticulum calcium ATPase
2/calmodulin-dependent kinase II/coffilin pathway in I/R injury
mice (170). EMPA also improves
endothelial cell dysfunction induced by a mutant aldehyde
dehydrogenase 2 unable to metabolize acetaldehyde by inhibiting
NHE1 and activating the AKT kinase and eNOS pathways (171). EMPA attenuates cardiac
microvascular I/R injury through the activation of the
AMP-activated protein kinase α1 (AMPKα1)/UNC-52-like kinase 1/FUN14
domain containing 1/mitophagy pathway (172).
The development of cardiac fibrosis is regulated by
members of the TGF-β family, particularly TGF-β1, which activates
Smad-dependent or non-Smad-mediated signaling pathways (173). TGF-β1 is a key cytokine mediating
the conversion of cardiac fibroblasts into myofibroblasts that is
regulated by numerous substances (174). Previous studies have shown that
EMPA significantly decreases TGF-β1/Smad2 levels and upregulates
the expression of the negative feedback regulator Smad7 to
alleviate cardiac oxidative stress and fibrosis in diabetic mice
(175). Furthermore, the
antifibrotic activity of Smad7 on TGF-β and epidermal growth factor
receptor 2 reduces myofibroblast activation and the production of
structural and matrix proteins (176). Early administration of EMPA
during MI reduces myocardial fibrosis and inhibits the TGF-1/Smad3
fibrotic pathway (177). This
study explored the effects of EMPA on early cardiac physiology and
fibrosis after myocardial infarction. Only samples taken after 4
weeks of administration were examined, and earlier samples were not
evaluated, so the results may differ. DAPA reduces TGF-β1 levels
and increases the expression of the negative feedback regulator
Smad7 in Ang II-induced cardiac remodeling (178).
MAPK is a class of highly conserved serine/threonine
protein kinases regulated by a cascade of tertiary phosphorylation
activation (183,184). MAPK is divided into four
subgroups: Extracellular signal-regulated kinase 1/2 (ERK1/2),
c-Jun N-terminal kinase (JNK), p38 MAPK and ERK5 (185,186). Under pathological conditions, the
MAPK signaling pathway is activated by numerous extracellular
stimulation signals and is essential for cell proliferation,
differentiation, apoptosis and stress response (184,187). These findings suggested that the
MAPK signaling pathway regulates cardiac remodeling due to multiple
pathologies (188–191).
Furthermore, TAC activated ERK1/2, p38 and JNK in
mice and treatment with DAPA inhibited the expression of JNK and
p38 to reduce cardiac remodeling (56). Likewise, DAPA attenuated palmitic
acid-induced cell hypertrophy and apoptosis and improved cardiac
dysfunction and remodeling in high-fat diet-induced obese mice.
This protective effect both in vivo and in vitro is
mediated by the NHE1/MAPK signaling pathway (192), and whether DAPA exerts
cardio-protective effects through NHE1 requires further evaluation.
Furthermore, this suggests that the protective effect of EMPA on
the heart may be mediated through stimulation of the ERK1/2
signaling pathway in I/R injury (69).
mTOR, a class of atypical serine/threonine protein
kinases, is a member of the phosphatidylinositol 3-kinase
(PI3K)-related protein kinase family. The interaction of mTOR with
different proteins forms two macromolecular complexes with
different structures and functions, mTORC1 and mTORC2 (193). mTOR also integrates multiple
extracellular signals, such as nutrient levels, energy and growth
factors, and serves a role in cell growth, proliferation, survival,
protein synthesis, autophagy and metabolism (194,195). Several studies have shown its
crucial role in the physiological and pathological processes of the
heart (196–201). Furthermore, the Akt and AMPK
pathways are regulators of mTORC1, with AMPK negatively regulating
the mTOR signaling pathway (202).
Another study reported that Ertugliflozin reduces LV
fibrosis in mice with cardiac hypertrophy by activating the
AMPK/mTOR pathway and inhibiting its downstream targets p70S6K and
4E-BP1 (203). This target
mediates translation to promote mTORC1 synthesis and causes
mTORC1-induced myocardial hypertrophy (204). Likewise, EMPA modulates autophagy
in cardiomyocytes to ameliorate sunitinib-induced cardiac
dysfunction, an effect mediated by the activation of
sunitinib-inhibited AMPK and reducing Sunitinib-activated mTOR
levels (37). AMPK/mTOR is one of
the main pathways regulating autophagy, which can be regulated by
direct phosphorylation of UNC-51-like kinases 1 (205). Furthermore, EMPA improves
obesity-related cardiac dysfunction by increasing the AMPK level
and endothelial nitric oxide synthase phosphorylation, and
inhibiting Akt and mTOR phosphorylation (144). Previous research has demonstrated
that the heart can be protected by inhibiting the PI3K/AKT/mTOR
pathway (206). CANA alleviates
cardiomyocyte lipotoxicity in diabetic cardiomyopathy mouse models
by blocking the mTOR/HIF-1 pathway (207). Likewise, CANA is an
SGLT1i/SGLT2is and its impact on the mTOR signaling pathway should
be excluded from SGLT1 interference.
Serum and glucocorticoid-induced protein kinase 1
(SGK1) are the main mediators of cardiac remodeling through the
activation of epithelial sodium channel (ENaC) proteins responsible
for promoting fibrosis and upregulating NHE1 activity (208,209). DAPA attenuates LVDD and
myocardial fibrosis by modulating SGK1 signaling and ENaC protein
(210). Due to the use of pigs in
this study, the sample size was small and the results had
statistical limitations. The JAK/STAT signaling pathway is a
promoter of fibroblast activation and ischemic-induced cardiac
dysfunction (211,212). CANA attenuates fibrosis by
reducing JAK/STAT signaling, activating AMPK and through
antioxidant signaling (213).
Characteristics of diabetic cardiomyopathy include decreased cyclic
guanosine monophosphate (cGMP) levels and altered soluble guanylate
cyclase enzyme (sGC)-cGMP- dependent protein kinase (PKG)
signaling, which regulate systolic and diastolic dysfunction under
diabetic conditions (214,215).
Furthermore, EMPA improves cardiac function by preventing oxidative
stress-induced injury via the sGC/cGMP/PKG pathway (216).
The present review provided a comprehensive summary
of the molecular mechanisms through which SGLT2is attenuate
pathological cardiac remodeling in animal and in vitro
cellular models. The molecular pathways of SGLT2is in cardiac
remodeling in terms of cardiac hypertrophy, cardiac fibrosis,
inflammation, apoptosis, autophagy, ferroptosis, oxidative stress
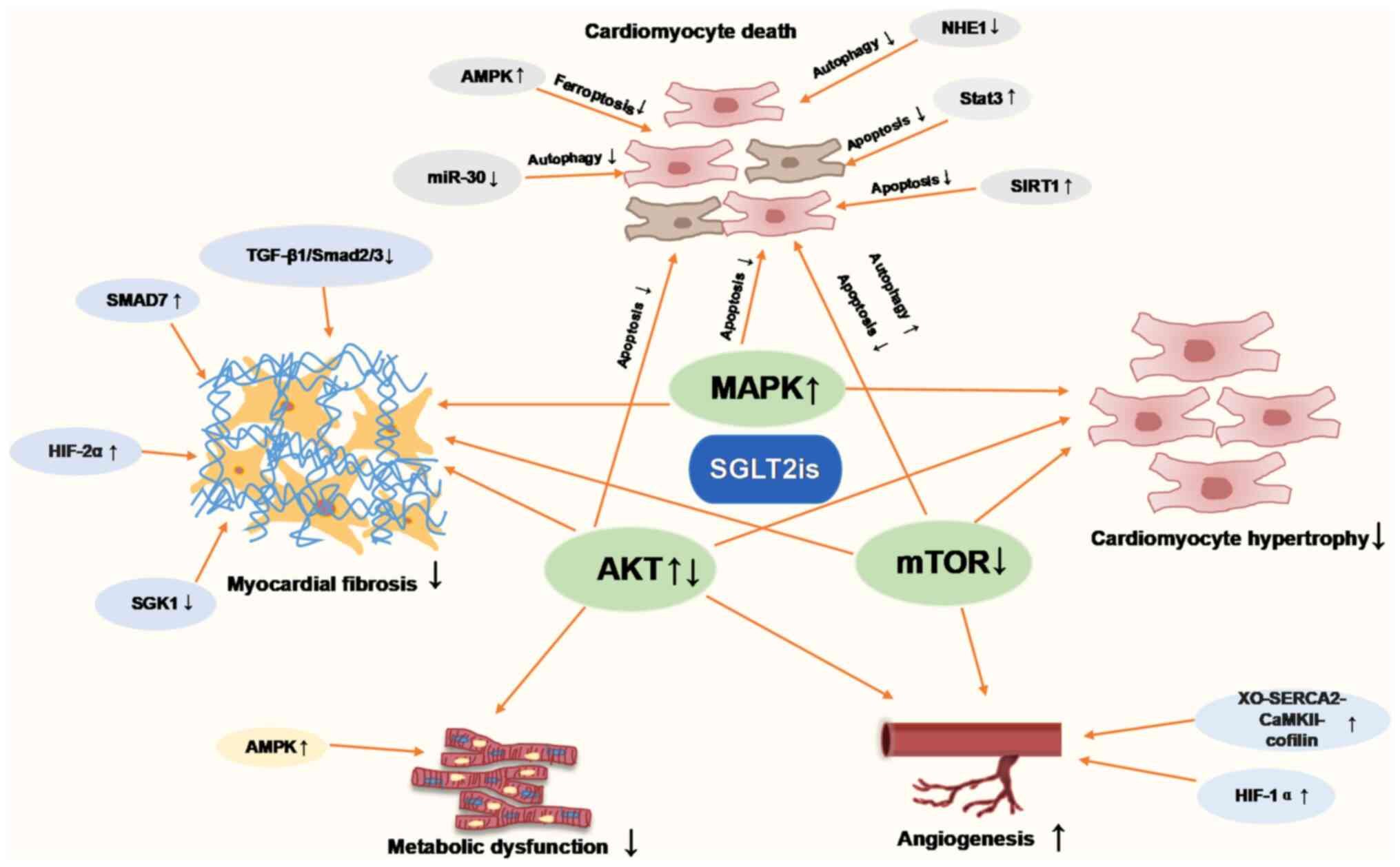
and energy metabolism, were summarized in Fig. 2. Thus, which supports the potential
use of SGLT2i as a therapeutic which can inhibit numerous
mechanisms of cardiac remodeling, such as MI, I/R and diabetic
cardiomyopathy. SGLT2is are directly or indirectly involved in
regulating molecular pathways of cardiac remodeling. Of note, the
interaction between inflammation and oxidative stress increases the
production of ROS and pro-inflammatory mediators, and SGLT2is
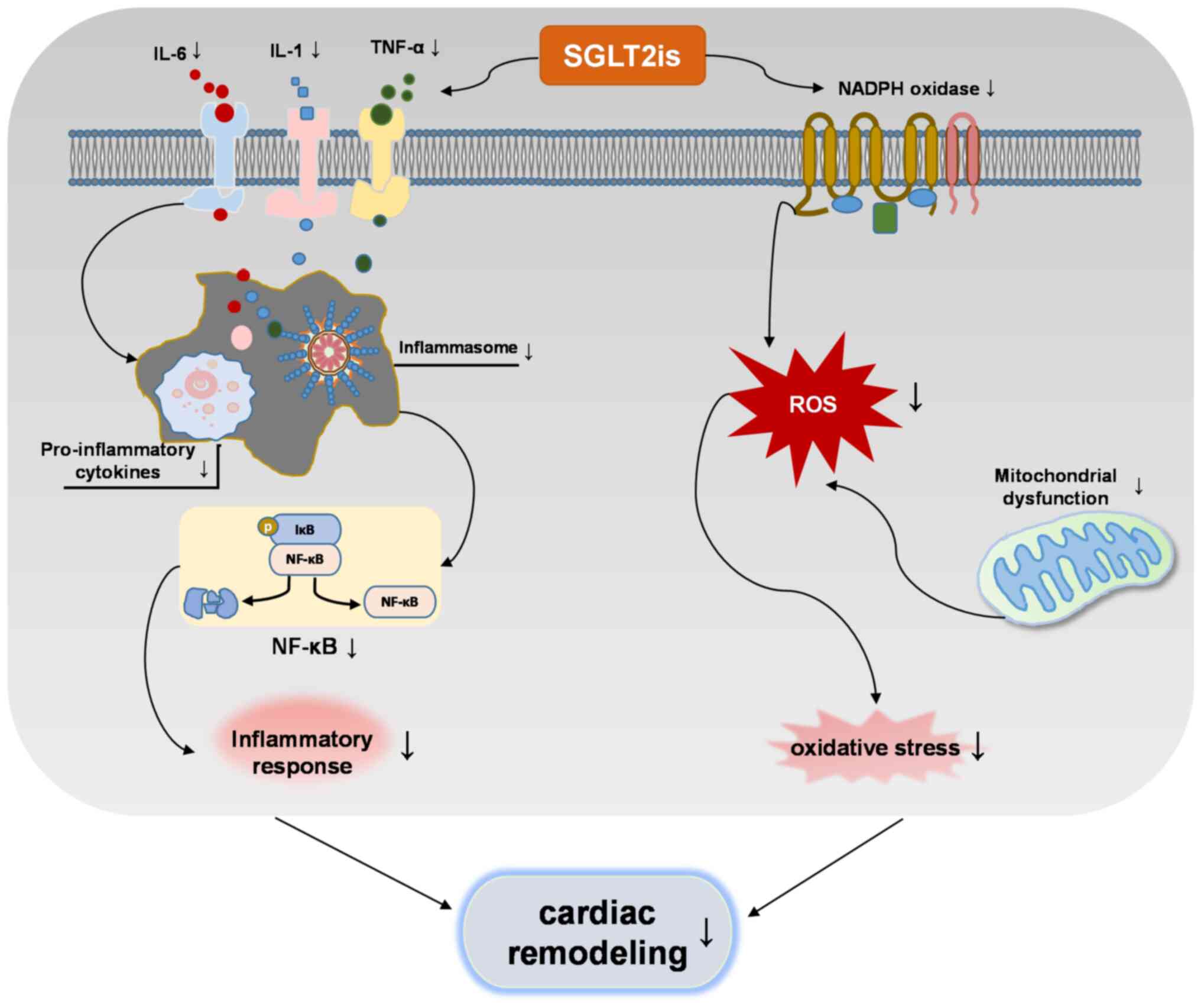
inhibit this interaction to regulate cardiac remodeling (Fig. 3). Based on this summary, it is
speculated that SGLT2is exert inhibitory effects on cardiac
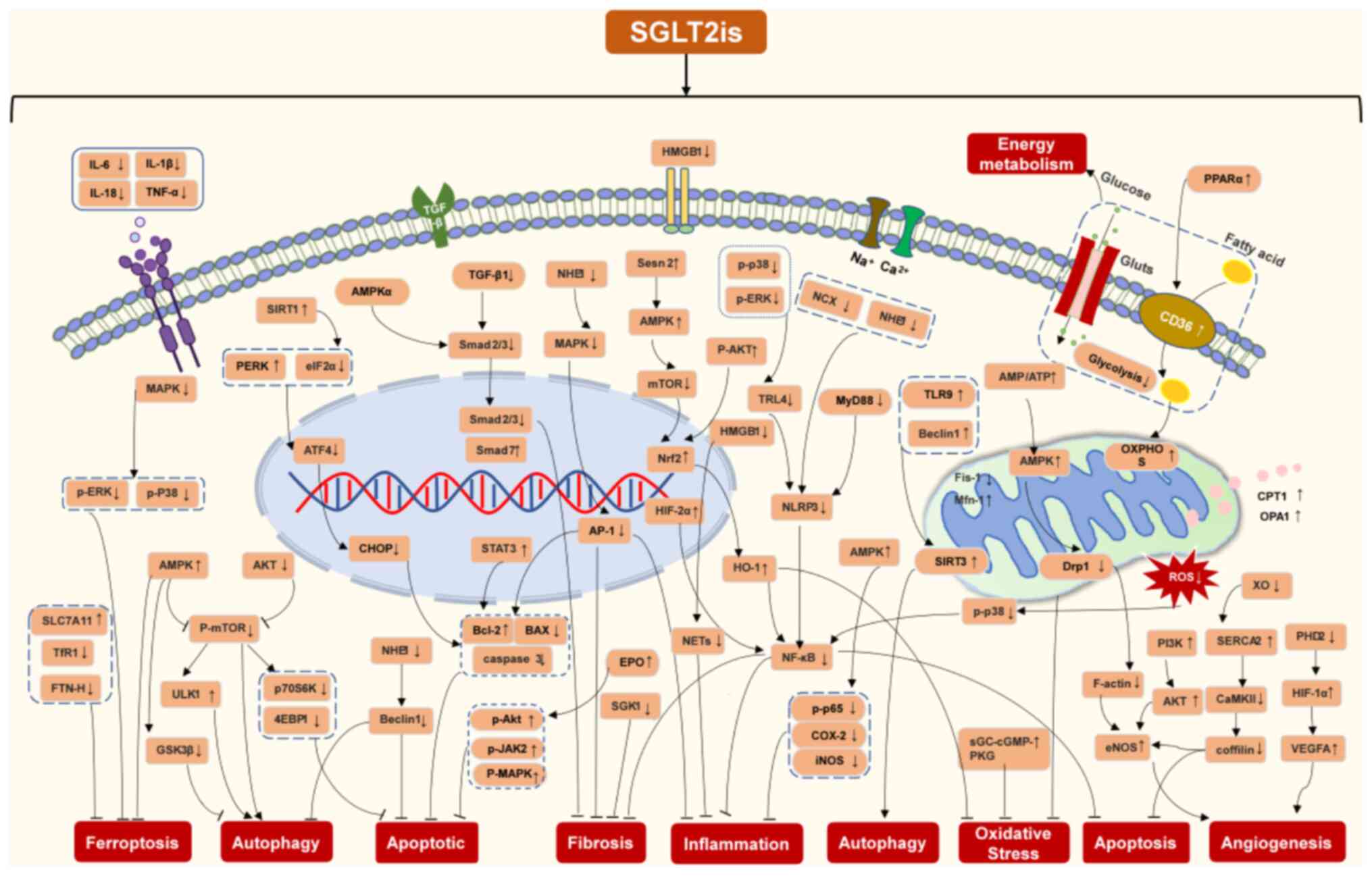
remodeling (Fig. 4).
To date, the effect of SGLT2is on cardiac remodeling
has been evaluated by several approaches, but studies on how it
functions in the heart require further evaluation. In addition, the
epigenetic mechanisms of SGLT2is in cardiac remodeling have not
been reported. In recent years, the impact of epigenetics on
disease development has received significant attention and studies
on cardiac diseases suggest that the epigenetic mechanisms of
SGLT2is require further assessment in future studies.
Regarding diabetic and non-diabetic pathological
cardiac remodeling, few studies have simultaneously compared
whether both occur through the same mechanism. SGLT2is have been
clinically approved for use in non-diabetic heart failure, while in
diabetic heart disease, their role may be influenced by SGLT2
targets. Thus, exploring the mechanism of action of SGLT2is in
non-diabetic cardiac remodeling may provide a basis for clinical
application in the heart.
The present review emphasizes that SGLT2is are not
only effective in controlling blood sugar in diabetes but can also
mitigate heart damage, suggesting their dual use in managing both
conditions.
Not applicable.
This work was supported by the National Natural Science
Foundation of China (grant no. 82104156).
Not applicable.
BC, YF and JG conceived, designed and planned the
study. All authors collected and read the literature. BC and JG
were responsible for the literature review and preparing the first
draft of the manuscript. HY, XW and JG revised the manuscript. All
authors have read and approved the final version of the manuscript.
Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Cohn JN, Ferrari R and Sharpe N: Cardiac
remodeling-concepts and clinical implications: A consensus paper
from an international forum on cardiac remodeling. Behalf of an
international forum on cardiac remodeling. J Am Coll Cardiol.
35:569–582. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang D, Liu HQ, Liu FY, Tang N, Guo Z, Ma
SQ, An P, Wang MY, Wu HM, Yang Z, et al: The roles of
noncardiomyocytes in cardiac remodeling. Int J Biol Sci.
16:2414–2429. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu
C, Zhu JX, Yang Z, Deng W and Tang QZ: Mechanisms contributing to
cardiac remodelling. Clin Sci (Lond). 131:2319–2345. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gao J, Xu W, Wang J, Wang K and Li P: The
role and molecular mechanism of non-coding RNAs in pathological
cardiac remodeling. Int J Mol Sci. 18:6082017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang LS, Liu Y, Chen Y, Ren JL, Zhang YR,
Yu YR, Jia MZ, Ning ZP, Du J, Tang CS and Qi YF: Intermedin
alleviates pathological cardiac remodeling by upregulating klotho.
Pharmacol Res. 159:1049262020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takefuji M, Wirth A, Lukasova M, Takefuji
S, Boettger T, Braun T, Althoff T, Offermanns S and Wettschureck N:
G(13)-mediated signaling pathway is required for pressure
overload-induced cardiac remodeling and heart failure. Circulation.
126:1972–1982. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McCarroll CS, He W, Foote K, Bradley A,
McGlynn K, Vidler F, Nixon C, Nather K, Fattah C, Riddell A, et al:
Runx1 deficiency protects against adverse cardiac remodeling after
myocardial infarction. Circulation. 137:57–70. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bujak M, Ren G, Kweon HJ, Dobaczewski M,
Reddy A, Taffet G, Wang XF and Frangogiannis NG: Essential role of
Smad3 in infarct healing and in the pathogenesis of cardiac
remodeling. Circulation. 116:2127–2138. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ni L, Yuan C, Chen G, Zhang C and Wu X:
SGLT2i: Beyond the glucose-lowering effect. Cardiovasc Diabetol.
19:982020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Home PD, Pocock SJ, Beck-Nielsen H, Curtis
PS, Gomis R, Hanefeld M, Jones NP, Komajda M and McMurray JJV;
RECORD Study Team, : Rosiglitazone evaluated for cardiovascular
outcomes in oral agent combination therapy for type 2 diabetes
(RECORD): A multicentre, randomised, open-label trial. Lancet.
373:2125–2135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wilcox T, De Block C, Schwartzbard AZ and
Newman JD: Diabetic agents, from metformin to SGLT2 inhibitors and
GLP1 receptor agonists: JACC focus seminar. J Am Coll Cardiol.
75:1956–1974. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zinman B, Wanner C, Lachin JM, Fitchett D,
Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ,
et al: Empagliflozin, cardiovascular outcomes, and mortality in
type 2 diabetes. N Engl J Med. 373:2117–2128. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Neal B, Perkovic V, Mahaffey KW, de Zeeuw
D, Fulcher G, Erondu N, Shaw W, Law G, Desai M and Matthews DR;
CANVAS Program Collaborative Group, : Canagliflozin and
cardiovascular and renal events in type 2 diabetes. N Engl J Med.
377:644–657. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wiviott SD, Raz I, Bonaca MP, Mosenzon O,
Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, et
al: Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N
Engl J Med. 380:347–357. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McDonagh TA, Metra M, Adamo M, Gardner RS,
Baumbach A, Böhm M, Burri H, Butler J, Čelutkienė J, Chioncel O, et
al: 2021 ESC guidelines for the diagnosis and treatment of acute
and chronic heart failure. Eur Heart J. 42:3599–3726. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Heidenreich PA, Bozkurt B, Aguilar D,
Allen LA, Byun JJ, Colvin MM, Deswal A, Drazner MH, Dunlay SM,
Evers LR, et al: 2022 AHA/ACC/HFSA guideline for the management of
heart failure: A report of the American college of
cardiology/American heart association joint committee on clinical
practice guidelines. Circulation. 145:e895–e1032. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Braunwald E: Gliflozins in the management
of cardiovascular disease. N Engl J Med. 386:2024–2034. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Van Steenbergen A, Balteau M, Ginion A,
Ferté L, Battault S, Ravenstein CM, Balligand JL, Daskalopoulos EP,
Gilon P, Despa F, et al: Sodium-myoinositol cotransporter-1, SMIT1,
mediates the production of reactive oxygen species induced by
hyperglycemia in the heart. Sci Rep. 7:411662017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cowie MR and Fisher M: SGLT2 inhibitors:
Mechanisms of cardiovascular benefit beyond glycaemic control. Nat
Rev Cardiol. 17:761–772. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tripolt NJ, Kolesnik E, Pferschy PN,
Verheyen N, Ablasser K, Sailer S, Alber H, Berger R, Kaulfersch C,
Leitner K, et al: Impact of EMpagliflozin on cardiac function and
biomarkers of heart failure in patients with acute MYocardial
infarction-the EMMY trial. Am Heart J. 221:39–47. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
von Lewinski D, Tripolt NJ, Sourij H,
Pferschy PN, Oulhaj A, Alber H, Gwechenberger M, Martinek M, Seidl
S, Moertl D, et al: Ertugliflozin to reduce arrhythmic burden in
ICD/CRT patients (ERASe-trial)-a phase III study. Am Heart J.
246:152–160. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee MMY, Brooksbank KJM, Wetherall K,
Mangion K, Roditi G, Campbell RT, Berry C, Chong V, Coyle L,
Docherty KF, et al: Effect of empagliflozin on left ventricular
volumes in patients with type 2 diabetes, or prediabetes, and heart
failure with reduced ejection fraction (SUGAR-DM-HF). Circulation.
143:516–525. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aimo A, Vergaro G, González A, Barison A,
Lupón J, Delgado V, Richards AM, de Boer RA, Thum T, Arfsten H, et
al: Cardiac remodelling-part 2: Clinical, imaging and laboratory
findings. A review from the study group on biomarkers of the heart
failure association of the European society of cardiology. Eur J
Heart Fail. 24:944–958. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Singh JSS, Fathi A, Vickneson K, Mordi I,
Mohan M, Houston JG, Pearson ER, Struthers AD and Lang CC: Research
into the effect of SGLT2 inhibition on left ventricular remodelling
in patients with heart failure and diabetes mellitus (REFORM) trial
rationale and design. Cardiovasc Diabetol. 15:972016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Santos-Gallego CG, Vargas-Delgado AP,
Requena-Ibanez JA, Garcia-Ropero A, Mancini D, Pinney S, Macaluso
F, Sartori S, Roque M, Sabatel-Perez F, et al: Randomized trial of
empagliflozin in nondiabetic patients with heart failure and
reduced ejection fraction. J Am Coll Cardiol. 77:243–255. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Verma S, Mazer CD, Yan AT, Mason T, Garg
V, Teoh H, Zuo F, Quan A, Farkouh ME, Fitchett DH, et al: Effect of
empagliflozin on left ventricular mass in patients with type 2
diabetes mellitus and coronary artery disease: The EMPA-HEART
cardiolink-6 randomized clinical trial. Circulation. 140:1693–1702.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Omar M, Jensen J, Ali M, Frederiksen PH,
Kistorp C, Videbæk L, Poulsen MK, Tuxen CD, Möller S, Gustafsson F,
et al: Associations of empagliflozin with left ventricular volumes,
mass, and function in patients with heart failure and reduced
ejection fraction: A substudy of the empire HF randomized clinical
trial. JAMA Cardiol. 6:836–840. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hwang IC, Cho GY, Yoon YE, Park JJ, Park
JB, Lee SP, Kim HK, Kim YJ and Sohn DW: Different effects of SGLT2
inhibitors according to the presence and types of heart failure in
type 2 diabetic patients. Cardiovasc Diabetol. 19:692020.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Soga F, Tanaka H, Tatsumi K, Mochizuki Y,
Sano H, Toki H, Matsumoto K, Shite J, Takaoka H, Doi T and Hirata
KI: Impact of dapagliflozin on left ventricular diastolic function
of patients with type 2 diabetic mellitus with chronic heart
failure. Cardiovasc Diabetol. 17:1322018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brown AJM, Gandy S, McCrimmon R, Houston
JG, Struthers AD and Lang CC: A randomized controlled trial of
dapagliflozin on left ventricular hypertrophy in people with type
two diabetes: The DAPA-LVH trial. Eur Heart J. 41:3421–3432. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lan NSR, Yeap BB, Fegan PG, Green G,
Rankin JM and Dwivedi G: Empagliflozin and left ventricular
diastolic function following an acute coronary syndrome in patients
with type 2 diabetes. Int J Cardiovasc Imaging. 37:517–527. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
von Lewinski D, Kolesnik E, Tripolt NJ,
Pferschy PN, Benedikt M, Wallner M, Alber H, Berger R, Lichtenauer
M, Saely CH, et al: Empagliflozin in acute myocardial infarction:
The EMMY trial. Eur Heart J. 43:4421–4432. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ersbøll M, Jürgens M, Hasbak P, Kjær A,
Wolsk E, Zerahn B, Brandt-Jacobsen NH, Gæde P, Rossing P, Faber J,
et al: Effect of empagliflozin on myocardial structure and function
in patients with type 2 diabetes at high cardiovascular risk: The
SIMPLE randomized clinical trial. Int J Cardiovasc Imaging.
38:579–587. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Palmiero G, Cesaro A, Galiero R, Loffredo
G, Caturano A, Vetrano E, Rinaldi L, Salvatore T, Ruggiero R,
Rosaria Di Palo M, et al: Impact of gliflozins on cardiac
remodeling in patients with type 2 diabetes mellitus & reduced
ejection fraction heart failure: A pilot prospective study. GLISCAR
study. Diabetes Res Clin Pract. 200:1106862023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Russo V, Malvezzi Caracciolo D'Aquino M,
Caturano A, Scognamiglio G, Pezzullo E, Fabiani D, Del Giudice C,
Carbone A, Bottino R, Caso V, et al: Improvement of global
longitudinal strain and myocardial work in type 2 diabetes patients
on sodium-glucose cotransporter 2 inhibitors therapy. J Cardiovasc
Pharmacol. 82:196–200. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nagueh SF: Left ventricular diastolic
function: Understanding pathophysiology, diagnosis, and prognosis
with echocardiography. JACC Cardiovasc Imaging. 13:228–244. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ren C, Sun K, Zhang Y, Hu Y, Hu B, Zhao J,
He Z, Ding R, Wang W and Liang C: Sodium-glucose CoTransporter-2
inhibitor empagliflozin ameliorates sunitinib-induced cardiac
dysfunction via regulation of AMPK-mTOR signaling pathway-mediated
autophagy. Front Pharmacol. 12:6641812021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Santos-Gallego CG, Requena-Ibanez JA, San
Antonio R, Ishikawa K, Watanabe S, Picatoste B, Flores E,
Garcia-Ropero A, Sanz J, Hajjar RJ, et al: Empagliflozin
ameliorates adverse left ventricular remodeling in nondiabetic
heart failure by enhancing myocardial energetics. J Am Coll
Cardiol. 73:1931–1944. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yurista SR, Silljé HHW, Oberdorf-Maass SU,
Schouten EM, Pavez Giani MG, Hillebrands JL, van Goor H, van
Veldhuisen DJ, de Boer RA and Westenbrink BD: Sodium-glucose
co-transporter 2 inhibition with empagliflozin improves cardiac
function in non-diabetic rats with left ventricular dysfunction
after myocardial infarction. Eur J Heart Fail. 21:862–873. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Habibi J, Aroor AR, Sowers JR, Jia G,
Hayden MR, Garro M, Barron B, Mayoux E, Rector RS, Whaley-Connell A
and DeMarco VG: Sodium glucose transporter 2 (SGLT2) inhibition
with empagliflozin improves cardiac diastolic function in a female
rodent model of diabetes. Cardiovasc Diabetol. 16:92017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang N, Feng B, Ma X, Sun K, Xu G and
Zhou Y: Dapagliflozin improves left ventricular remodeling and
aorta sympathetic tone in a pig model of heart failure with
preserved ejection fraction. Cardiovasc Diabetol. 18:1072019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee HC, Shiou YL, Jhuo SJ, Chang CY, Liu
PL, Jhuang WJ, Dai ZK, Chen WY, Chen YF and Lee AS: The
sodium-glucose co-transporter 2 inhibitor empagliflozin attenuates
cardiac fibrosis and improves ventricular hemodynamics in
hypertensive heart failure rats. Cardiovasc Diabetol. 18:452019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lahnwong S, Palee S, Apaijai N,
Sriwichaiin S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC and
Chattipakorn N: Acute dapagliflozin administration exerts
cardioprotective effects in rats with cardiac ischemia/reperfusion
injury. Cardiovasc Diabetol. 19:912020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kräker K, Herse F, Golic M, Reichhart N,
Crespo-Garcia S, Strauß O, Grune J, Kintscher U, Ebrahim M, Bader
M, et al: Effects of empagliflozin and target-organ damage in a
novel rodent model of heart failure induced by combined
hypertension and diabetes. Sci Rep. 10:140612020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Santos-Gallego CG, Requena-Ibanez JA, San
Antonio R, Garcia-Ropero A, Ishikawa K, Watanabe S, Picatoste B,
Vargas-Delgado AP, Flores-Umanzor EJ, Sanz J, et al: Empagliflozin
ameliorates diastolic dysfunction and left ventricular
fibrosis/stiffness in nondiabetic heart failure: A multimodality
study. JACC Cardiovasc Imaging. 14:393–407. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lin YW, Chen CY, Shih JY, Cheng BC, Chang
CP, Lin MT, Ho CH, Chen ZC, Fisch S and Chang WT: Dapagliflozin
improves cardiac hemodynamics and mitigates arrhythmogenesis in
mitral regurgitation-induced myocardial dysfunction. J Am Heart
Assoc. 10:e0192742021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dini FL, Galeotti GG, Terlizzese G,
Fabiani I, Pugliese NR and Rovai I: Left ventricular mass and
thickness: Why does it matter? Heart Fail Clin. 15:159–166. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Matsutani D, Sakamoto M, Kayama Y, Takeda
N, Horiuchi R and Utsunomiya K: Effect of canagliflozin on left
ventricular diastolic function in patients with type 2 diabetes.
Cardiovasc Diabetol. 17:732018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Querejeta R, López B, González A, Sánchez
E, Larman M, Martínez Ubago JL and Díez J: Increased collagen type
I synthesis in patients with heart failure of hypertensive origin:
Relation to myocardial fibrosis. Circulation. 110:1263–1268. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Frangogiannis NG: Cardiac fibrosis: Cell
biological mechanisms, molecular pathways and therapeutic
opportunities. Mol Aspects Med. 65:70–99. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Frangogiannis NG: Cardiac fibrosis.
Cardiovasc Res. 117:1450–1488. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Travers JG, Tharp CA, Rubino M and
McKinsey TA: Therapeutic targets for cardiac fibrosis: From old
school to next-gen. J Clin Invest. 132:e1485542022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Aluja D, Delgado-Tomás S, Ruiz-Meana M,
Barrabés JA and Inserte J: Calpains as potential therapeutic
targets for myocardial hypertrophy. Int J Mol Sci. 23:41032022.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yerra VG, Batchu SN, Kabir G, Advani SL,
Liu Y, Siddiqi FS, Connelly KA and Advani A: Empagliflozin disrupts
a Tnfrsf12a-mediated feed forward loop that promotes left
ventricular hypertrophy. Cardiovasc Drugs Ther. 36:619–632. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shi L, Zhu D, Wang S, Jiang A and Li F:
Dapagliflozin attenuates cardiac remodeling in mice model of
cardiac pressure overload. Am J Hypertens. 32:452–459. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li X, Lu Q, Qiu Y, do Carmo JM, Wang Z, da
Silva AA, Mouton A, Omoto ACM, Hall ME, Li J and Hall JE: Direct
Cardiac actions of the sodium glucose co-transporter 2 inhibitor
empagliflozin improve myocardial oxidative phosphorylation and
attenuate pressure-overload heart failure. J Am Heart Assoc.
10:e0182982021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu L, Luo H, Liang Y, Tang J and Shu Y:
Dapagliflozin ameliorates STZ-induced cardiac hypertrophy in type 2
diabetic rats by inhibiting the calpain-1 expression and nuclear
transfer of NF-kappaB. Comput Math Methods Med.
2022:32930542022.PubMed/NCBI
|
|
59
|
Wang J, Huang X, Liu H, Chen Y, Li P, Liu
L, Li J, Ren Y, Huang J, Xiong E, et al: Empagliflozin ameliorates
diabetic cardiomyopathy via attenuating oxidative stress and
improving mitochondrial function. Oxid Med Cell Longev.
2022:11224942022.PubMed/NCBI
|
|
60
|
Kimura T, Nakamura K, Miyoshi T, Yoshida
M, Akazawa K, Saito Y, Akagi S, Ohno Y, Kondo M, Miura D, et al:
Inhibitory effects of tofogliflozin on cardiac hypertrophy in dahl
salt-sensitive and salt-resistant rats fed a high-fat diet. Int
Heart J. 60:728–735. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hsieh PL, Chu PM, Cheng HC, Huang YT, Chou
WC, Tsai KL and Chan SH: Dapagliflozin mitigates doxorubicin-caused
myocardium damage by regulating AKT-mediated oxidative stress,
cardiac remodeling, and inflammation. Int J Mol Sci. 23:101462022.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Asensio Lopez MDC, Lax A, Hernandez
Vicente A, Saura Guillen E, Hernandez-Martinez A, Fernandez Del
Palacio MJ, Bayes-Genis A and Pascual Figal DA: Empagliflozin
improves post-infarction cardiac remodeling through GTP enzyme
cyclohydrolase 1 and irrespective of diabetes status. Sci Rep.
10:135532020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jiang K, Xu Y, Wang D, Chen F, Tu Z, Qian
J, Xu S, Xu Y, Hwa J, Li J, et al: Cardioprotective mechanism of
SGLT2 inhibitor against myocardial infarction is through reduction
of autosis. Protein Cell. 13:336–359. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Crow MT, Mani K, Nam YJ and Kitsis RN: The
mitochondrial death pathway and cardiac myocyte apoptosis. Circ
Res. 95:957–970. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental mechanisms of regulated cell death and
implications for heart disease. Physiol Rev. 99:1765–1817. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Abbate A, Bussani R, Amin MS, Vetrovec GW
and Baldi A: Acute myocardial infarction and heart failure: Role of
apoptosis. Int J Biochem Cell Biol. 38:1834–1840. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu Y, Wu M, Xu J, Xu B and Kang L:
Empagliflozin prevents from early cardiac injury post myocardial
infarction in non-diabetic mice. Eur J Pharm Sci. 161:1057882021.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Long Q, Li L, Yang H, Lu Y, Yang H, Zhu Y,
Tang Y, Liu C and Yuan J: SGLT2 inhibitor, canagliflozin,
ameliorates cardiac inflammation in experimental autoimmune
myocarditis. Int Immunopharmacol. 110:1090242022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
El-Sayed N, Mostafa YM, AboGresha NM,
Ahmed AAM, Mahmoud IZ and El-Sayed NM: Dapagliflozin attenuates
diabetic cardiomyopathy through erythropoietin up-regulation of
AKT/JAK/MAPK pathways in streptozotocin-induced diabetic rats. Chem
Biol Interact. 347:1096172021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Fan ZG, Xu Y, Chen X, Ji MY and Ma GS:
Appropriate dose of dapagliflozin improves cardiac outcomes by
normalizing mitochondrial fission and reducing cardiomyocyte
apoptosis after acute myocardial infarction. Drug Des Devel Ther.
16:2017–2030. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chang WT, Shih JY, Lin YW, Chen ZC, Kan
WC, Lin TH and Hong CS: Dapagliflozin protects against
doxorubicin-induced cardiotoxicity by restoring STAT3. Arch
Toxicol. 96:2021–2032. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ren FF, Xie ZY, Jiang YN, Guan X, Chen QY,
Lai TF and Li L: Dapagliflozin attenuates pressure overload-induced
myocardial remodeling in mice via activating SIRT1 and inhibiting
endoplasmic reticulum stress. Acta Pharmacol Sin. 43:1721–1732.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zein L, Fulda S, Kögel D and van Wijk SJL:
Organelle-specific mechanisms of drug-induced autophagy-dependent
cell death. Matrix Biol. 100-101:54–64. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhang J, Liu J, Huang Y, Chang JYF, Liu L,
McKeehan WL, Martin JF and Wang F: FRS2α-mediated FGF signals
suppress premature differentiation of cardiac stem cells through
regulating autophagy activity. Circ Res. 110:e29–e39. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Morales PE, Arias-Durán C, Ávalos-Guajardo
Y, Aedo G, Verdejo HE, Parra V and Lavandero S: Emerging role of
mitophagy in cardiovascular physiology and pathology. Mol Aspects
Med. 71:1008222020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Gatica D, Chiong M, Lavandero S and
Klionsky DJ: Molecular mechanisms of autophagy in the
cardiovascular system. Circ Res. 116:456–467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Munasinghe PE, Riu F, Dixit P, Edamatsu M,
Saxena P, Hamer NS, Galvin IF, Bunton RW, Lequeux S, Jones G, et
al: Type-2 diabetes increases autophagy in the human heart through
promotion of Beclin-1 mediated pathway. Int J Cardiol. 202:13–20.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Shirakabe A, Zhai P, Ikeda Y, Saito T,
Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B and Sadoshima J:
Drp1-dependent mitochondrial autophagy plays a protective role
against pressure overload-induced mitochondrial dysfunction and
heart failure. Circulation. 133:1249–1263. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Nishida K and Otsu K: Autophagy during
cardiac remodeling. J Mol Cell Cardiol. 95:11–18. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Nah J, Zhai P, Huang CY, Fernández ÁF,
Mareedu S, Levine B and Sadoshima J: Upregulation of Rubicon
promotes autosis during myocardial ischemia/reperfusion injury. J
Clin Invest. 130:2978–2991. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ikeda S, Zablocki D and Sadoshima J: The
role of autophagy in death of cardiomyocytes. J Mol Cell Cardiol.
165:1–8. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Gatica D, Chiong M, Lavandero S and
Klionsky DJ: The role of autophagy in cardiovascular pathology.
Cardiovasc Res. 118:934–950. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wang CY, Chen CC, Lin MH, Su HT, Ho MY,
Yeh JK, Tsai ML, Hsieh IC and Wen MS: TLR9 binding to Beclin 1 and
mitochondrial SIRT3 by a sodium-glucose co-transporter 2 inhibitor
protects the heart from doxorubicin toxicity. Biology (Basel).
9:3692020.PubMed/NCBI
|
|
85
|
Deng R, Jiang K, Chen F, Miao Y, Lu Y, Su
F, Liang J, Qian J, Wang D, Xiang Y and Shen L: Novel
cardioprotective mechanism for empagliflozin in nondiabetic
myocardial infarction with acute hyperglycemia. Biomed
Pharmacother. 154:1136062022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wang CC, Li Y, Qian XQ, Zhao H, Wang D,
Zuo GX and Wang K: Empagliflozin alleviates myocardial I/R injury
and cardiomyocyte apoptosis via inhibiting ER stress-induced
autophagy and the PERK/ATF4/Beclin1 pathway. J Drug Target.
30:858–872. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Aragón-Herrera A, Feijóo-Bandín S, Otero
Santiago M, Barral L, Campos-Toimil M, Gil-Longo J, Costa Pereira
TM, Garcia-Caballero T, Rodriguez-Segade S, Rodriguez J, et al:
Empagliflozin reduces the levels of CD36 and cardiotoxic lipids
while improving autophagy in the hearts of Zucker diabetic fatty
rats. Biochem Pharmacol. 170:1136772019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ma H and Ma Y: Dapagliflozin inhibits
ventricular remodeling in heart failure rats by activating
autophagy through AMPK/mTOR pathway. Comput Math Methods Med.
2022:62602022022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yu YW, Que JQ, Liu S, Huang KY, Qian L,
Weng YB, Rong FN, Wang L, Zhou YY, Xue YJ and Ji KT: Sodium-glucose
co-transporter-2 inhibitor of dapagliflozin attenuates myocardial
ischemia/reperfusion injury by limiting NLRP3 inflammasome
activation and modulating autophagy. Front Cardiovasc Med.
8:7682142022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Qin Y, Qiao Y, Wang D, Tang C and Yan G:
Ferritinophagy and ferroptosis in cardiovascular disease:
Mechanisms and potential applications. Biomed Pharmacother.
141:1118722021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Huang X, Song Y, Wei L, Guo J, Xu W and Li
M: The emerging roles of ferroptosis in organ fibrosis and its
potential therapeutic effect. Int Immunopharmacol. 116:1098122023.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Feng Y, Madungwe NB, Imam Aliagan AD,
Tombo N and Bopassa JC: Liproxstatin-1 protects the mouse
myocardium against ischemia/reperfusion injury by decreasing VDAC1
levels and restoring GPX4 levels. Biochem Biophys Res Commun.
520:606–611. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Baba Y, Higa JK, Shimada BK, Horiuchi KM,
Suhara T, Kobayashi M, Woo JD, Aoyagi H, Marh KS, Kitaoka H and
Matsui T: Protective effects of the mechanistic target of rapamycin
against excess iron and ferroptosis in cardiomyocytes. Am J Physiol
Heart Circ Physiol. 314:H659–H668. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Bai T, Li M, Liu Y, Qiao Z and Wang Z:
Inhibition of ferroptosis alleviates atherosclerosis through
attenuating lipid peroxidation and endothelial dysfunction in mouse
aortic endothelial cell. Free Radic Biol Med. 160:92–102. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Chen X, Xu S, Zhao C and Liu B: Role of
TLR4/NADPH oxidase 4 pathway in promoting cell death through
autophagy and ferroptosis during heart failure. Biochem Biophys Res
Commun. 516:37–43. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ma S, He LL, Zhang GR, Zuo QJ, Wang ZL,
Zhai JL, Zhang TT, Wang Y, Ma HJ and Guo YF: Canagliflozin
mitigates ferroptosis and ameliorates heart failure in rats with
preserved ejection fraction. Naunyn Schmiedebergs Arch Pharmacol.
395:945–962. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Wang X, Chen X, Zhou W, Men H, Bao T, Sun
Y, Wang Q, Tan Y, Keller BB, Tong Q, et al: Ferroptosis is
essential for diabetic cardiomyopathy and is prevented by
sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 12:708–722.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Du S, Shi H, Xiong L, Wang P and Shi Y:
Canagliflozin mitigates ferroptosis and improves myocardial
oxidative stress in mice with diabetic cardiomyopathy. Front
Endocrinol (Lausanne). 13:10116692022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zhang W, Lu J, Wang Y, Sun P, Gao T, Xu N,
Zhang Y and Xie W: Canagliflozin attenuates lipotoxicity in
cardiomyocytes by inhibiting inflammation and ferroptosis through
activating AMPK pathway. Int J Mol Sci. 24:8582023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Min J, Wu L, Liu Y, Song G, Deng Q, Jin W,
Yu W, Abudureyimu M, Pei Z and Ren J: Empagliflozin attenuates
trastuzumab-induced cardiotoxicity through suppression of DNA
damage and ferroptosis. Life Sci. 312:1212072023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chen W, Zhang Y, Wang Z, Tan M, Lin J,
Qian X, Li H and Jiang T: Dapagliflozin alleviates myocardial
ischemia/reperfusion injury by reducing ferroptosis via MAPK
signaling inhibition. Front Pharmacol. 14:10782052023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Aimo A, Castiglione V, Borrelli C, Saccaro
LF, Franzini M, Masi S, Emdin M and Giannoni A: Oxidative stress
and inflammation in the evolution of heart failure: From
pathophysiology to therapeutic strategies. Eur J Prev Cardiol.
27:494–510. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Halade GV and Lee DH: Inflammation and
resolution signaling in cardiac repair and heart failure.
EBioMedicine. 79:1039922022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Prabhu SD and Frangogiannis NG: The
biological basis for cardiac repair after myocardial infarction:
From inflammation to fibrosis. Circ Res. 119:91–112. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Timmers L, Pasterkamp G, de Hoog VC,
Arslan F, Appelman Y and de Kleijn DPV: The innate immune response
in reperfused myocardium. Cardiovasc Res. 94:276–283. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Frieler RA and Mortensen RM: Immune cell
and other noncardiomyocyte regulation of cardiac hypertrophy and
remodeling. Circulation. 131:1019–1030. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Haudek SB, Taffet GE, Schneider MD and
Mann DL: TNF provokes cardiomyocyte apoptosis and cardiac
remodeling through activation of multiple cell death pathways. J
Clin Invest. 117:2692–2701. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ing DJ, Zang J, Dzau VJ, Webster KA and
Bishopric NH: Modulation of cytokine-induced cardiac myocyte
apoptosis by nitric oxide, Bak, and Bcl-x. Circ Res. 84:21–33.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Meléndez GC, McLarty JL, Levick SP, Du Y,
Janicki JS and Brower GL: Interleukin 6 mediates myocardial
fibrosis, concentric hypertrophy, and diastolic dysfunction in
rats. Hypertension. 56:225–231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sivasubramanian N, Coker ML, Kurrelmeyer
KM, MacLellan WR, DeMayo FJ, Spinale FG and Mann DL: Left
ventricular remodeling in transgenic mice with cardiac restricted
overexpression of tumor necrosis factor. Circulation. 104:826–831.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Saxena A, Chen W, Su Y, Rai V, Uche OU, Li
N and Frangogiannis NG: IL-1 induces proinflammatory leukocyte
infiltration and regulates fibroblast phenotype in the infarcted
myocardium. J Immunol. 191:4838–4848. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Chou CH, Hung CS, Liao CW, Wei LH, Chen
CW, Shun CT, Wen WF, Wan CH, Wu XM, Chang YY, et al: IL-6
trans-signalling contributes to aldosterone-induced cardiac
fibrosis. Cardiovasc Res. 114:690–702. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Didion SP, Kinzenbaw DA, Schrader LI, Chu
Y and Faraci FM: Endogenous interleukin-10 inhibits angiotensin
II-induced vascular dysfunction. Hypertension. 54:619–624. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Szalay G, Sauter M, Hald J, Weinzierl A,
Kandolf R and Klingel K: Sustained nitric oxide synthesis
contributes to immunopathology in ongoing myocarditis attributable
to interleukin-10 disorders. Am J Pathol. 169:2085–2093. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Kolijn D, Pabel S, Tian Y, Lódi M, Herwig
M, Carrizzo A, Zhazykbayeva S, Kovács A, Fülöp GÁ, Falcão-Pires I,
et al: Empagliflozin improves endothelial and cardiomyocyte
function in human heart failure with preserved ejection fraction
via reduced pro-inflammatory-oxidative pathways and protein kinase
Gα oxidation. Cardiovasc Res. 117:495–507. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yan P, Song X, Tran J, Zhou R, Cao X, Zhao
G and Yuan H: Dapagliflozin alleviates coxsackievirus B3-induced
acute viral myocarditis by regulating the macrophage polarization
through Stat3-related pathways. Inflammation. 45:2078–2090. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Faridvand Y, Nemati M,
Zamani-Gharehchamani E, Nejabati HR, Zamani ARN, Nozari S, Safaie
N, Nouri M and Jodati A: Dapagliflozin protects H9c2 cells against
injury induced by lipopolysaccharide via suppression of
CX3CL1/CX3CR1 axis and NF-κB activity. Curr Mol Pharmacol.
15:862–869. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Pinar AA, Scott TE, Huuskes BM, Tapia
Cáceres FE, Kemp-Harper BK and Samuel CS: Targeting the NLRP3
inflammasome to treat cardiovascular fibrosis. Pharmacol Ther.
209:1075112020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Toldo S and Abbate A: The NLRP3
inflammasome in acute myocardial infarction. Nat Rev Cardiol.
15:203–214. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Suetomi T, Willeford A, Brand CS, Cho Y,
Ross RS, Miyamoto S and Brown JH: Inflammation and NLRP3
inflammasome activation initiated in response to pressure overload
by Ca2+/calmodulin-dependent protein kinase II δ
signaling in cardiomyocytes are essential for adverse cardiac
remodeling. Circulation. 138:2530–2544. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Sokolova M, Ranheim T, Louwe MC, Halvorsen
B, Yndestad A and Aukrust P: NLRP3 inflammasome: A novel player in
metabolically induced inflammation-potential influence on the
myocardium. J Cardiovasc Pharmacol. 74:276–284. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Ye Y, Bajaj M, Yang HC, Perez-Polo JR and
Birnbaum Y: SGLT-2 inhibition with dapagliflozin reduces the
activation of the Nlrp3/ASC inflammasome and attenuates the
development of diabetic cardiomyopathy in mice with type 2
diabetes. Further augmentation of the effects with saxagliptin, a
DPP4 inhibitor. Cardiovasc Drugs Ther. 31:119–132. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Chen H, Tran D, Yang HC, Nylander S,
Birnbaum Y and Ye Y: Dapagliflozin and ticagrelor have additive
effects on the attenuation of the activation of the NLRP3
inflammasome and the progression of diabetic cardiomyopathy: An
AMPK-mTOR interplay. Cardiovasc Drugs Ther. 34:443–461. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Quagliariello V, De Laurentiis M, Rea D,
Barbieri A, Monti MG, Carbone A, Paccone A, Altucci L, Conte M,
Canale ML, et al: The SGLT-2 inhibitor empagliflozin improves
myocardial strain, reduces cardiac fibrosis and pro-inflammatory
cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc
Diabetol. 20:1502021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Hu J, Xu J, Tan X, Li D, Yao D, Xu B and
Lei Y: Dapagliflozin protects against dilated cardiomyopathy
progression by targeting NLRP3 inflammasome activation. Naunyn
Schmiedebergs Arch Pharmacol. 396:1461–1470. 2023.PubMed/NCBI
|
|
130
|
Pullen AB, Jadapalli JK, Rhourri-Frih B
and Halade GV: Re-evaluating the causes and consequences of
non-resolving inflammation in chronic cardiovascular disease. Heart
Fail Rev. 25:381–391. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Lee TM, Chang NC and Lin SZ:
Dapagliflozin, a selective SGLT2 inhibitor, attenuated cardiac
fibrosis by regulating the macrophage polarization via STAT3
signaling in infarcted rat hearts. Free Radic Biol Med.
104:298–310. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Chelko SP, Asimaki A, Lowenthal J,
Bueno-Beti C, Bedja D, Scalco A, Amat-Alarcon N, Andersen P, Judge
DP, Tung L and Saffitz JE: Therapeutic modulation of the immune
response in arrhythmogenic cardiomyopathy. Circulation.
140:1491–1505. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Yang Z, Li T, Xian J, Chen J, Huang Y,
Zhang Q, Lin X, Lu H and Lin Y: SGLT2 inhibitor dapagliflozin
attenuates cardiac fibrosis and inflammation by reverting the
HIF-2alpha signaling pathway in arrhythmogenic cardiomyopathy.
FASEB J. 36:e224102022. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Salvatore T, Galiero R, Caturano A,
Vetrano E, Rinaldi L, Coviello F, Di Martino A, Albanese G,
Colantuoni S, Medicamento G, et al: Dysregulated epicardial adipose
tissue as a risk factor and potential therapeutic target of heart
failure with preserved ejection fraction in diabetes. Biomolecules.
12:1762022. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Gordon M, Meagher P and Connelly KA:
Effect of empagliflozin and liraglutide on the nucleotide-binding
and oligomerization domain-like receptor family pyrin
domain-containing 3 inflammasome in a rodent model of type 2
diabetes mellitus. Can J Diabetes. 45:553–556. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Michaeloudes C, Abubakar-Waziri H, Lakhdar
R, Raby K, Dixey P, Adcock IM, Mumby S, Bhavsar PK and Chung KF:
Molecular mechanisms of oxidative stress in asthma. Mol Aspects
Med. 85:1010262022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Weissman D and Maack C: Redox signaling in
heart failure and therapeutic implications. Free Radic Biol Med.
171:345–364. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zhang M, Perino A, Ghigo A, Hirsch E and
Shah AM: NADPH oxidases in heart failure: Poachers or gamekeepers?
Antioxid Redox Signal. 18:1024–1041. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Burgoyne JR, Mongue-Din H, Eaton P and
Shah AM: Redox signaling in cardiac physiology and pathology. Circ
Res. 111:1091–1106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
van der Pol A, van Gilst WH, Voors AA and
van der Meer P: Treating oxidative stress in heart failure: Past,
present and future. Eur J Heart Fail. 21:425–435. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Li A, Zheng N and Ding X: Mitochondrial
abnormalities: A hub in metabolic syndrome-related cardiac
dysfunction caused by oxidative stress. Heart Fail Rev.
27:1387–1394. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Xing YJ, Liu BH, Wan SJ, Cheng Y, Zhou SM,
Sun Y, Yao XM, Hua Q, Meng XJ, Cheng JH, et al: A SGLT2 inhibitor
dapagliflozin alleviates diabetic cardiomyopathy by suppressing
high glucose-induced oxidative stress in vivo and in vitro. Front
Pharmacol. 12:7081772021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Li X, Flynn ER, do Carmo JM, Wang Z, da
Silva AA, Mouton AJ, Omoto ACM, Hall ME and Hall JE: Direct cardiac
actions of sodium-glucose cotransporter 2 inhibition improve
mitochondrial function and attenuate oxidative stress in pressure
overload-induced heart failure. Front Cardiovasc Med. 9:8592532022.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Sun X, Han F, Lu Q, Li X, Ren D, Zhang J,
Han Y, Xiang YK and Li J: Empagliflozin ameliorates obesity-related
cardiac dysfunction by regulating sestrin2-mediated AMPK-mTOR
signaling and redox homeostasis in high-fat diet-induced obese
mice. Diabetes. 69:1292–1305. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Bugga P, Mohammed SA, Alam MJ, Katare P,
Meghwani H, Maulik SK, Arava S and Banerjee SK: Empagliflozin
prohibits high-fructose diet-induced cardiac dysfunction in rats
via attenuation of mitochondria-driven oxidative stress. Life Sci.
307:1208622022. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal
JS and Stanley WC: Myocardial fatty acid metabolism in health and
disease. Physiol Rev. 90:207–258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Taegtmeyer H, Sen S and Vela D: Return to
the fetal gene program: A suggested metabolic link to gene
expression in the heart. Ann N Y Acad Sci. 1188:191–198. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Ingwall JS: Energy metabolism in heart
failure and remodelling. Cardiovasc Res. 81:412–419. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Rosca MG, Tandler B and Hoppel CL:
Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell
Cardiol. 55:31–41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Zhang L, Jaswal JS, Ussher JR,
Sankaralingam S, Wagg C, Zaugg M and Lopaschuk GD: Cardiac
insulin-resistance and decreased mitochondrial energy production
precede the development of systolic heart failure after
pressure-overload hypertrophy. Circ Heart Fail. 6:1039–1048. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
De Jong KA and Lopaschuk GD: Complex
energy metabolic changes in heart failure with preserved ejection
fraction and heart failure with reduced ejection fraction. Can J
Cardiol. 33:860–871. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Kolwicz SC Jr, Olson DP, Marney LC,
Garcia-Menendez L, Synovec RE and Tian R: Cardiac-specific deletion
of acetyl CoA carboxylase 2 prevents metabolic remodeling during
pressure-overload hypertrophy. Circ Res. 111:728–738. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Choi YS, de Mattos ABM, Shao D, Li T,
Nabben M, Kim M, Wang W, Tian R and Kolwicz SC Jr: Preservation of
myocardial fatty acid oxidation prevents diastolic dysfunction in
mice subjected to angiotensin II infusion. J Mol Cell Cardiol.
100:64–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Chess DJ, Lei B, Hoit BD, Azimzadeh AM and
Stanley WC: Effects of a high saturated fat diet on cardiac
hypertrophy and dysfunction in response to pressure overload. J
Card Fail. 14:82–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Trang NN, Chung CC, Lee TW, Cheng WL, Kao
YH, Huang SY, Lee TI and Chen YJ: Empagliflozin and liraglutide
differentially modulate cardiac metabolism in diabetic
cardiomyopathy in rats. Int J Mol Sci. 22:11772021. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Jia G, DeMarco VG and Sowers JR: Insulin
resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat
Rev Endocrinol. 12:144–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Lai J and Chen C: The role of
epoxyeicosatrienoic acids in cardiac remodeling. Front Physiol.
12:6424702021. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Shi W, Xin Q, Yuan R, Yuan Y, Cong W and
Chen K: Neovascularization: The main mechanism of MSCs in ischemic
heart disease therapy. Front Cardiovasc Med. 8:6333002021.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Shu HY, Peng YZ, Hang WJ, Zhang M, Shen L,
Wang DW and Zhou N: Trimetazidine enhances myocardial angiogenesis
in pressure overload-induced cardiac hypertrophy mice through
directly activating Akt and promoting the binding of HSF1 to VEGF-A
promoter. Acta Pharmacol Sin. 43:2550–2561. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Ni Y, Deng J, Bai H, Liu C, Liu X and Wang
X: CaMKII inhibitor KN-93 impaired angiogenesis and aggravated
cardiac remodelling and heart failure via inhibiting
NOX2/mtROS/p-VEGFR2 and STAT3 pathways. J Cell Mol Med. 26:312–325.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Blom JN, Wang X, Lu X, Kim MY, Wang G and
Feng Q: Inhibition of intraflagellar transport protein-88 promotes
epithelial-to-mesenchymal transition and reduces cardiac remodeling
post-myocardial infarction. Eur J Pharmacol. 933:1752872022.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Wei T, Huang G, Gao J, Huang C, Sun M, Wu
J, Bu J and Shen W: Sirtuin 3 deficiency accelerates hypertensive
cardiac remodeling by impairing angiogenesis. J Am Heart Assoc.
6:e0061142017. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Gogiraju R, Hubert A, Fahrer J, Straub BK,
Brandt M, Wenzel P, Münzel T, Konstantinides S, Hasenfuss G and
Schäfer K: Endothelial leptin receptor deletion promotes cardiac
autophagy and angiogenesis following pressure overload by
suppressing Akt/mTOR signaling. Circ Heart Fail. 12:e0056222019.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Gu J, Wang S, Guo H, Tan Y, Liang Y, Feng
A, Liu Q, Damodaran C, Zhang Z, Keller BB, et al: Inhibition of p53
prevents diabetic cardiomyopathy by preventing early-stage
apoptosis and cell senescence, reduced glycolysis, and impaired
angiogenesis. Cell Death Dis. 9:822018. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Zhou H, Wang S, Zhu P, Hu S, Chen Y and
Ren J: Empagliflozin rescues diabetic myocardial microvascular
injury via AMPK-mediated inhibition of mitochondrial fission. Redox
Biol. 15:335–346. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Nugrahaningrum DA, Marcelina O, Liu C, Wu
S and Kasim V: Dapagliflozin promotes neovascularization by
improving paracrine function of skeletal muscle cells in diabetic
hindlimb ischemia mice through PHD2/HIF-1α axis. Front Pharmacol.
11:11042020. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Nakao M, Shimizu I, Katsuumi G, Yoshida Y,
Suda M, Hayashi Y, Ikegami R, Hsiao YT, Okuda S, Soga T and
Minamino T: Empagliflozin maintains capillarization and improves
cardiac function in a murine model of left ventricular pressure
overload. Sci Rep. 11:183842021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Zou R, Shi W, Qiu J, Zhou N, Du N, Zhou H,
Chen X and Ma L: Empagliflozin attenuates cardiac microvascular
ischemia/reperfusion injury through improving mitochondrial
homeostasis. Cardiovasc Diabetol. 21:1062022. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Ma L, Zou R, Shi W, Zhou N, Chen S, Zhou
H, Chen X and Wu Y: SGLT2 inhibitor dapagliflozin reduces
endothelial dysfunction and microvascular damage during cardiac
ischemia/reperfusion injury through normalizing the
XO-SERCA2-CaMKII-coffilin pathways. Theranostics. 12:5034–5050.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Guo H, Yu X, Liu Y, Paik DT, Justesen JM,
Chandy M, Jahng JWS, Zhang T, Wu W, Rwere F, et al: SGLT2 inhibitor
ameliorates endothelial dysfunction associated with the common
ALDH2 alcohol flushing variant. Sci Transl Med. 15:eabp99522023.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Cai C, Guo Z, Chang X, Li Z, Wu F, He J,
Cao T, Wang K, Shi N, Zhou H, et al: Empagliflozin attenuates
cardiac microvascular ischemia/reperfusion through activating the
AMPKalpha1/ULK1/FUNDC1/mitophagy pathway. Redox Biol.
52:1022882022. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Hanna A and Frangogiannis NG: The role of
the TGF-beta superfamily in myocardial infarction. Front Cardiovasc
Med. 6:1402019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Tarbit E, Singh I, Peart JN and Rose'Meyer
RB: Biomarkers for the identification of cardiac fibroblast and
myofibroblast cells. Heart Fail Rev. 24:1–15. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Li C, Zhang J, Xue M, Li X, Han F, Liu X,
Xu L, Lu Y, Cheng Y, Li T, et al: SGLT2 inhibition with
empagliflozin attenuates myocardial oxidative stress and fibrosis
in diabetic mice heart. Cardiovasc Diabetol. 18:152019. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Humeres C, Shinde AV, Hanna A, Alex L,
Hernández SC, Li R, Chen B, Conway SJ and Frangogiannis NG: Smad7
effects on TGF-β and ErbB2 restrain myofibroblast activation and
protect from postinfarction heart failure. J Clin Invest.
132:e1469262022. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Daud E, Ertracht O, Bandel N, Moady G,
Shehadeh M, Reuveni T and Atar S: The impact of empagliflozin on
cardiac physiology and fibrosis early after myocardial infarction
in non-diabetic rats. Cardiovasc Diabetol. 20:1322021. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Zhang Y, Lin X, Chu Y, Chen X, Du H, Zhang
H, Xu C, Xie H, Ruan Q, Lin J, et al: Dapagliflozin: a
sodium-glucose cotransporter 2 inhibitor, attenuates angiotensin
II-induced cardiac fibrotic remodeling by regulating TGFβ1/Smad
signaling. Cardiovasc Diabetol. 20:1212021. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Qi H, Liu Y, Li S, Chen Y, Li L, Cao Y, E
M, Shi P, Song C, Li B and Sun H: Activation of AMPK attenuated
cardiac fibrosis by inhibiting CDK2 via p21/p27 and miR-29 family
pathways in rats. Mol Ther Nucleic Acids. 8:277–290. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Hinson JT, Chopra A, Lowe A, Sheng CC,
Gupta RM, Kuppusamy R, O'Sullivan J, Rowe G, Wakimoto H, Gorham J,
et al: Integrative analysis of PRKAG2 cardiomyopathy iPS and
microtissue models identifies AMPK as a regulator of metabolism,
survival, and fibrosis. Cell Rep. 17:3292–3304. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Tian J, Zhang M, Suo M, Liu D, Wang X, Liu
M, Pan J, Jin T and An F: Dapagliflozin alleviates cardiac fibrosis
through suppressing EndMT and fibroblast activation via
AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J Cell Mol
Med. 25:7642–7659. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Chen X, Yang Q, Bai W, Yao W, Liu L, Xing
Y, Meng C, Qi P, Dang Y and Qi X: Dapagliflozin attenuates
myocardial fibrosis by inhibiting the TGF-β1/Smad signaling pathway
in a normoglycemic rabbit model of chronic heart failure. Front
Pharmacol. 13:8731082022. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Hepworth EMW and Hinton SD:
Pseudophosphatases as regulators of MAPK signaling. Int J Mol Sci.
22:125952021. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Rose BA, Force T and Wang Y:
Mitogen-activated protein kinase signaling in the heart: Angels
versus demons in a heart-breaking tale. Physiol Rev. 90:1507–1546.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Xu Z, Sun J, Tong Q, Lin Q, Qian L, Park Y
and Zheng Y: The role of ERK1/2 in the development of diabetic
cardiomyopathy. Int J Mol Sci. 17:20012016. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Zhang Q, Wang L, Wang S, Cheng H, Xu L,
Pei G, Wang Y, Fu C, Jiang Y, He C and Wei Q: Signaling pathways
and targeted therapy for myocardial infarction. Signal Transduct
Target Ther. 7:782022. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Pearson G, Robinson F, Beers Gibson T, Xu
BE, Karandikar M, Berman K and Cobb MH: Mitogen-activated protein
(MAP) kinase pathways: Regulation and physiological functions.
Endocr Rev. 22:153–183. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Streicher JM, Ren S, Herschman H and Wang
Y: MAPK-activated protein kinase-2 in cardiac hypertrophy and
cyclooxygenase-2 regulation in heart. Circ Res. 106:1434–1443.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Liu X, Chen K, Zhuang Y, Huang Y, Sui Y,
Zhang Y, Lv L and Zhang G: Paeoniflorin improves pressure
overload-induced cardiac remodeling by modulating the MAPK
signaling pathway in spontaneously hypertensive rats. Biomed
Pharmacother. 111:695–704. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Ren J, Zhang S, Kovacs A, Wang Y and
Muslin AJ: Role of p38alpha MAPK in cardiac apoptosis and
remodeling after myocardial infarction. J Mol Cell Cardiol.
38:617–623. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Yang N, Zou C, Luo W, Xu D, Wang M, Wang
Y, Wu G, Shan P and Liang G: Sclareol attenuates angiotensin
II-induced cardiac remodeling and inflammation via inhibiting MAPK
signaling. Phytother Res. 37:578–591. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Lin K, Yang N, Luo W, Qian JF, Zhu WW, Ye
SJ, Yuan CX, Xu DY, Liang G, Huang WJ and Shan PR: Direct
cardio-protection of dapagliflozin against obesity-related
cardiomyopathy via NHE1/MAPK signaling. Acta Pharmacol Sin.
43:2624–2635. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Yang H, Rudge DG, Koos JD, Vaidialingam B,
Yang HJ and Pavletich NP: mTOR kinase structure, mechanism and
regulation. Nature. 497:217–223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 169:361–371. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Gangloff YG, Mueller M, Dann SG, Svoboda
P, Sticker M, Spetz JF, Um SH, Brown EJ, Cereghini S, Thomas G and
Kozma SC: Disruption of the mouse mTOR gene leads to early
postimplantation lethality and prohibits embryonic stem cell
development. Mol Cell Biol. 24:9508–9516. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Murakami M, Ichisaka T, Maeda M, Oshiro N,
Hara K, Edenhofer F, Kiyama H, Yonezawa K and Yamanaka S: mTOR is
essential for growth and proliferation in early mouse embryos and
embryonic stem cells. Mol Cell Biol. 24:6710–6718. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Shioi T, McMullen JR, Tarnavski O,
Converso K, Sherwood MC, Manning WJ and Izumo S: Rapamycin
attenuates load-induced cardiac hypertrophy in mice. Circulation.
107:1664–1670. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Das A, Salloum FN, Filippone SM, Durrant
DE, Rokosh G, Bolli R and Kukreja RC: Inhibition of mammalian
target of rapamycin protects against reperfusion injury in diabetic
heart through STAT3 signaling. Basic Res Cardiol. 110:312015.
View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Zhao X, Lu S, Nie J, Hu X, Luo W, Wu X,
Liu H, Feng Q, Chang Z, Liu Y, et al: Phosphoinositide-dependent
kinase 1 and mTORC2 synergistically maintain postnatal heart growth
and heart function in mice. Mol Cell Biol. 34:1966–1975. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Xia Y, Wen HY, Young ME, Guthrie PH,
Taegtmeyer H and Kellems RE: Mammalian target of rapamycin and
protein kinase A signaling mediate the cardiac transcriptional
response to glutamine. J Biol Chem. 278:13143–13150. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Sciarretta S, Volpe M and Sadoshima J:
Mammalian target of rapamycin signaling in cardiac physiology and
disease. Circ Res. 114:549–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Moellmann J, Mann PA, Kappel BA, Kahles F,
Klinkhammer BM, Boor P, Kramann R, Ghesquiere B, Lebherz C, Marx N
and Lehrke M: The sodium-glucose co-transporter-2 inhibitor
ertugliflozin modifies the signature of cardiac substrate
metabolism and reduces cardiac mTOR signalling, endoplasmic
reticulum stress and apoptosis. Diabetes Obes Metab. 24:2263–2272.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Sciarretta S, Forte M, Frati G and
Sadoshima J: New insights into the role of mTOR signaling in the
cardiovascular system. Circ Res. 122:489–505. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Chen L, Liu P, Feng X and Ma C:
Salidroside suppressing LPS-induced myocardial injury by inhibiting
ROS-mediated PI3K/Akt/mTOR pathway in vitro and in vivo. J Cell Mol
Med. 21:3178–3189. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Sun P, Wang Y, Ding Y, Luo J, Zhong J, Xu
N, Zhang Y and Xie W: Canagliflozin attenuates lipotoxicity in
cardiomyocytes and protects diabetic mouse hearts by inhibiting the
mTOR/HIF-1α pathway. iScience. 24:1025212021. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Das S, Aiba T, Rosenberg M, Hessler K,
Xiao C, Quintero PA, Ottaviano FG, Knight AC, Graham EL, Bostrom P,
et al: Pathological role of serum- and glucocorticoid-regulated
kinase 1 in adverse ventricular remodeling. Circulation.
126:2208–2219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Lang F and Shumilina E: Regulation of ion
channels by the serum- and glucocorticoid-inducible kinase SGK1.
FASEB J. 27:3–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Lee SG, Kim D, Lee JJ, Lee HJ, Moon RK,
Lee YJ, Lee SJ, Lee OH, Kim C, Oh J, et al: Dapagliflozin
attenuates diabetes-induced diastolic dysfunction and cardiac
fibrosis by regulating SGK1 signaling. BMC Med. 20:3092022.
View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Mascareno E, El-Shafei M, Maulik N, Sato
M, Guo Y, Das DK and Siddiqui MA: JAK/STAT signaling is associated
with cardiac dysfunction during ischemia and reperfusion.
Circulation. 104:325–329. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Chakraborty D, Šumová B, Mallano T, Chen
CW, Distler A, Bergmann C, Ludolph I, Horch RE, Gelse K, Ramming A,
et al: Activation of STAT3 integrates common profibrotic pathways
to promote fibroblast activation and tissue fibrosis. Nat Commun.
8:11302017. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Sabe SA, Xu CM, Sabra M, Harris DD,
Malhotra A, Aboulgheit A, Stanley M, Abid MR and Sellke FW:
Canagliflozin improves myocardial perfusion, fibrosis, and function
in a swine model of chronic myocardial ischemia. J Am Heart Assoc.
12:e0286232023. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Yu LM, Di WC, Dong X, Li Z, Zhang Y, Xue
XD, Xu YL, Zhang J, Xiao X, Han JS, et al: Melatonin protects
diabetic heart against ischemia-reperfusion injury, role of
membrane receptor-dependent cGMP-PKG activation. Biochim Biophys
Acta Mol Basis Dis. 1864:563–578. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Mátyás C, Németh BT, Oláh A, Hidi L,
Birtalan E, Kellermayer D, Ruppert M, Korkmaz-Icöz S, Kökény G,
Horváth EM, et al: The soluble guanylate cyclase activator
cinaciguat prevents cardiac dysfunction in a rat model of type-1
diabetes mellitus. Cardiovasc Diabetol. 14:1452015. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Xue M, Li T, Wang Y, Chang Y, Cheng Y, Lu
Y, Liu X, Xu L, Li X, Yu X, et al: Empagliflozin prevents
cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice.
Clin Sci (Lond). 133:1705–1720. 2019. View Article : Google Scholar : PubMed/NCBI
|