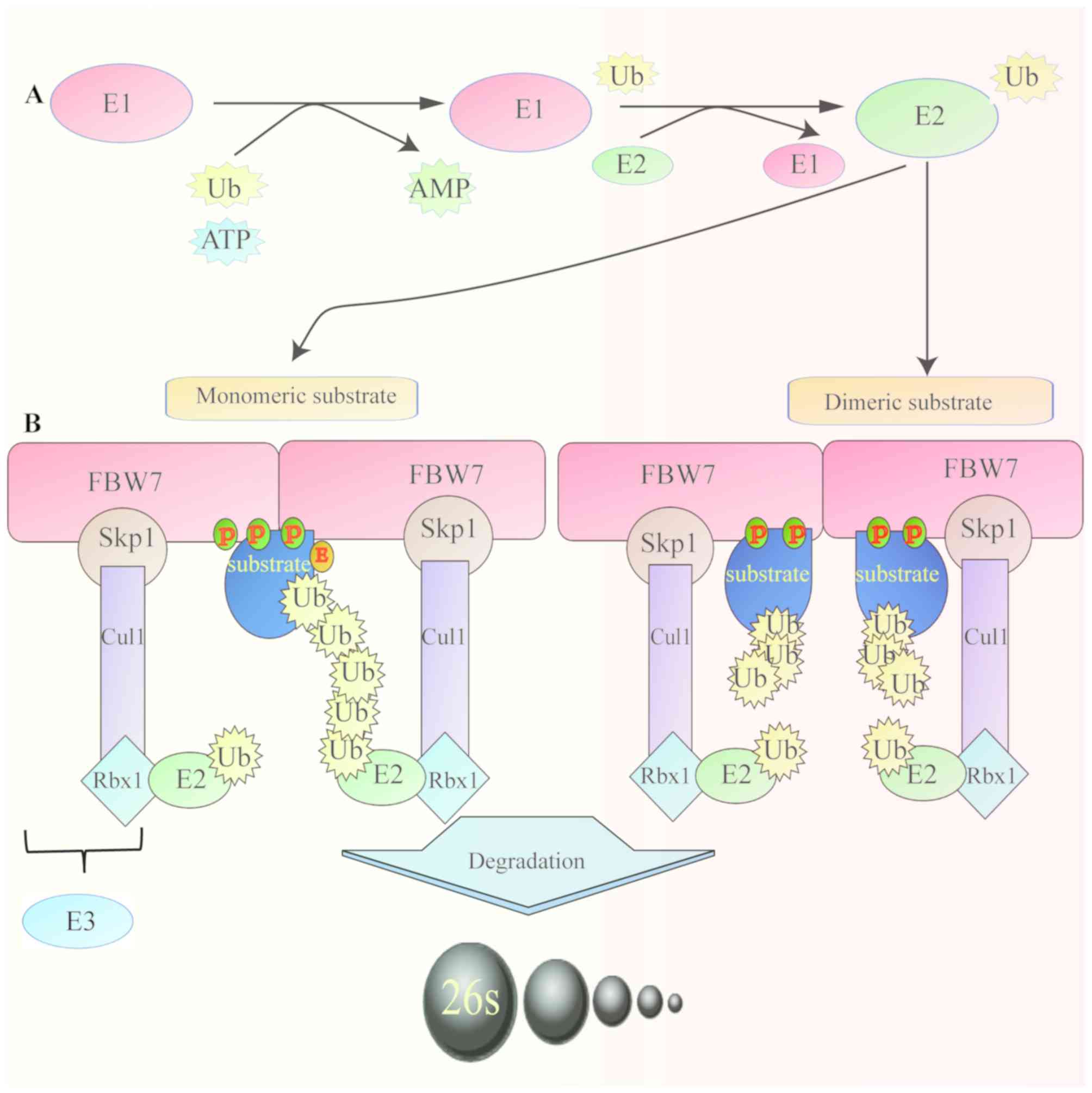
The ubiquitin-proteasome system (UPS) is the major
regulatory pathway of protein degradation in eukaryotic cells
(1). Defects in UPS function can
result in several diseases, including cancer (2). F-box and WD repeat domain-containing
protein 7 (FBW7), also known as FBW7, AGO or hCDC4, is an essential
component of the Skp1-Cul1-F-box (SCF)-type ubiquitin ligase (E3)
complex, which comprises four subunits: Skp1, cullin1, Rbx1 and an
F-box protein that determines substrate specificity (3). FBW7 is a member of the F-box protein
family (4). Previous studies
demonstrated that dysregulation of FBW7 serves a crucial role in
the development of hematological tumors (5–7)
(Fig. 1).
The target proteins degraded by FBW7 contain a
conserved sequence of phosphorylated amino acids named CDC4
phosphodegron (CDP). CDPs bind to the WD40 repeats, allowing the
recognition of the substrate by ubiquitin ligase for its subsequent
degradation (10). Furthermore,
dimerization of FBW7 increases the binding of FBW7 to substrates,
particularly to those with weak phosphodegrons (13). In addition, glycogen synthase 3β
(GSK3β) serves a crucial role in the degradation of substrate by
FBW7. GSK3β catalyzes the phosphorylation of threonine in the
substrate CDPs, promoting FBW7 binding and accelerating the
degradation of the substrate (14,15)
(Fig. 2).
FBW7 is an important substrate adaptor responsible
for recognition and binding of the substrate proteins in the
ubiquitin-proteasome degradation pathway. Many of its substrates
are oncogenes. For example, c-Myc is an important substrate for
ubiquitination by FBW7 (16,17).
In general, decreased expression of FBW7 results in
a significant increase in the intracellular content of c-Myc
protein, whereas overexpression of FBW7 promotes c-Myc
ubiquitination and degradation, decreasing its cellular level
(14). In the Burkitt's lymphoma
cell line, c-Myc is most commonly mutated at T58, causing the
failure of FBW7 to regulate c-Myc protein degradation, ultimately
leading to c-Myc protein accumulation and tumorigenesis (14). Cyclin E is another classical
substrate of FBW7. By binding to cyclin E-dependent kinase 2
(CDK2), cyclin E forms a complex that promotes the G/S phase
transition, leading to uncontrolled cell proliferation (18). FBW7 can also ubiquitinate various
other target proteins, including Notch1 (19), NF-κB (20), c-Jun (21), granulocyte colony stimulating factor
receptor (22), SHOC2 leucine rich
repeat scaffold protein (23),
brahma-related gene-1 (24), Aurora
A (25), Kruppel like factor 5
(26), heat shock transcription
factor 1 (27) and
CCAAT/enhancer-binding protein α (28) (Table
I).
The main mechanisms of FBW7 disruption in cancer are
deletion, mutation and promoter methylation (29–31) and
the gene mutation is the most common. A previous study demonstrated
that FBW7 is mutated in ~6% of human tumors, and in 31% of acute
T-cell lymphocytic leukemia cases (32). It has been demonstrated that
conditional FBW7 knockout in the T-cell lineage of mice leads
initially to thymic hyperplasia and subsequently to the development
of thymic lymphoma, indicating that the loss of FBW7 function is an
important factor responsible for tumorigenesis in the blood system
(33). In addition, FBW7 expression
and activity can be regulated by numerous genes, including p53
(34). FBW7 is a p53-dependent tumor
suppressor, and p53 activation can enhance the
ubiquitination-mediated degradation of oncoproteins (34). Furthermore, previous studies have
identified numerous micro (mi)RNAs that can regulate FBW7
expression, including miR-223 (35),
miR-25 (36), miR-182 (37), miR-503 (37) and miR-92a (38). In addition, in T-cell acute
lymphoblastic leukemia (T-ALL), miR-223 acts as an oncogene by
inhibiting the FBW7 expression. The expression of FBW7 is also
controlled by the RBP-J-interaction and tubulin-associated (RITA)
protein. Overexpression of RITA results in increased expression of
FBW7 and decreased expression of cyclin E, cyclin D1, CDK2, Hes-1
and NF-kBp65 (39). In addition,
NF-κB1 (40), Pin1 (41), family with sequence similarity 83
member D (FAM83D) (42) and Numb4
(43) are also regulators of FBW7
expression (Table II).
In order to determine whether FBW7 is differently
expressed in tumor tissues compared with normal tissues, the
Oncomine database (https://www.oncomine.org) was used to analyze FBW7
mRNA levels in different tumor and normal tissues. The results
indicated that FBW7 expression is higher in leukemia (Fig. 3A), suggesting that FBW7 may be
associated with the initiation and progression of certain
hematological tumors. The Timer database (https://cistrome.shinyapps.io/timer/) was then used to
analyze FBW7 mRNA levels in diffuse large B cell lymphoma (DLBCL).
The result demonstrated no significant difference in FBW7
expression level in DLBCL. This may be due to different expression
levels of FBW7 in various hematological tumors (Fig. 3B).
T-ALL is a highly proliferative hematologic
malignancy caused by the malignant transformation of T-cell
progenitors (44). Patients with
T-ALL typically present with aggressive clinical features
correlated with poor prognosis, including inhibition of normal
hematopoietic function, high white blood cell counts, pleural
effusions and central nervous system involvement (45). T-ALL accounts for 15 and 25% of the
total number of childhood and adult cases of acute lymphoblastic
leukemia, respectively (46). The
rate of T-ALL complete remission can reach 94%, and the long-term
survival rate can be as high as 85% (47); however, 20% pediatric patients and
40% adult patients are prone to recurrence and ultimately develop
refractory leukemia (48).
Understanding the underlying mechanisms of T-ALL development is
therefore critical. T-ALL is the only known malignant cancer that
can be induced by FBW7 deletion without the requirement of other
tumor-promoting factors (49,50).
Furthermore, T-ALL development is accelerated by the simultaneous
loss of p53 or Phosphatase and TENsin homolog or by concurrent
activation of Notch (49,51,52).
However, it was demonstrated that FBW7 hotspot mutation knock-in
mice (FBW7mut/+) do not develop spontaneous leukemia,
suggesting that the FBW7 missense mutation and FBW7 homologous
deletion present subtle differences (53). FBW7 deletion/mutation increases the
protein level of Notch and c-Myc (49,50,53,54).
However, Notch1 alone is not sufficient to induce or maintain T-ALL
in the absence of c-Myc function; however, c-Myc deletion in
established T-ALL specifically ablates leukemia-initiating cells
(LIC), and the inhibition of c-Myc induction by small molecule
inhibitors of bromodomain and extra-terminal motif/bromodomains can
suppress the proliferation of mouse and human T-ALL cells,
suggesting that c-Myc could be considered as a key oncogene driving
T-ALL (53). It was reported that
>60% of T-ALL cases present with abnormal activation of the
Notch1 signaling pathway, indicating that Notch1 might be the most
common oncogene in T-ALL (55).
Mutations of Notch1 occur mainly in the heterodimeric domain region
and the proline, glutamine, serine, and threonine domain (56,57).
Activating mutations of Notch1 gene result in sustained expression
of genes regulating T cell differentiation, including Hes family
bHLH transcription factor 1 and CD25, changing the expression
balance of c-Myc and p27 (58–60),
upregulating the NF-κB signaling pathway (5), enhancing the expression of the
anti-apoptotic X-linked inhibitor of apoptosis protein associated
with ubiquitination and degradation, and activating the
PKB/Akt/mTOR signaling pathway-mediated inhibition of p53 (61). All the aforementioned signaling
pathways inhibit apoptosis and cause abnormal proliferation of
non-functional T cells, thereby directly leading to the development
of T-ALL. In addition, Notch and NF-κB bind to the promoter of
miR-223, activating its expression (62). Notch-mediated upregulation of miR-223
subsequently inhibits FBW7 gene expression in T-ALL, and miR-223
and FBW7 expression are negatively correlated in T-ALL
patient-derived xenografts (62). In
addition, it has been demonstrated that FBW7 loss of function leads
to an upregulation of the glucocorticoid receptor in primary T-ALL
cells, thereby enhancing their sensitivity to glucocorticoids and
improving the prognosis of patients with T-ALL (63). MCL-1 is an anti-apoptotic protein of
the BCL-2 family that promotes cancer by inhibiting apoptosis
(64). Deletion of FBW7 in T-ALL
cell line results in an increased expression level of MCL-1. MCL-1
upregulation is sensitive to various kinase inhibitors, including
sorafenib, but resistant to the BCL2 inhibitor ABT-737. However,
when FBW7 function is restored or MCL-1 is deleted, cell
sensitivity to ABT-737 is restored (65). MCL-1 also serves a role in the
chemotherapy efficacy of anti-tubulin drugs. Treatment with
paclitaxel and similarly acting compounds induces MCL-1
phosphorylation, which is then recognized by FBW7 and degraded by
ubiquitination. Subsequently, the intracellular MCL-1 protein
content is significantly decreased and promotes therefore
apoptosis. In addition, when FBW7 is inactivated or downregulated
in the tumor, the stability of MCL-1 protein is enhanced, thereby
increasing resistance of the tumor to microtubule-targeted drugs
and decreasing the effectiveness of chemotherapy (66). The cellular content of MCL-1 protein
is also affected by the deubiquitinating enzyme named ubiquitin
specific peptidase 9 X-Linked (USP9X) (67). Since USP9X decreases the
ubiquitination of MCL-1 and increases its stability, USP9X
downregulation and decreased MCL-1 protein expression could
increase tumor cell sensitivity to ABT-737 treatment.
ATL is a malignant T cell monoclonal proliferative
disease caused by the human T-cell leukemia virus type 1 (HTLV-1).
ATL represents a rare type of lymphocytic leukemia/lymphoid tumor
affecting T cells (68). FBW7 acts
as a tumor suppressor in ATL cells; however mutations can transform
FBW7 into an oncogenic protein (69). Mutations in the WD40 domain of FBW7
were identified in 25% (8/32) of acute ATL cases (69). Furthermore, the FBW7 D510E and D527G
mutants are capable of ubiquitinating proteolysis of endogenous
cyclin E, MCL-1, and c-Myc. However, these mutants are ineffective
in degrading the Notch intracellular domain (NICD) in ATL cells,
resulting in the activation of Notch1 signaling. The increased
Notch1 signaling can subsequently promote ATL cell proliferation
and tumorigenesis (69). The same
mutants present significant carcinogenic activity when co-expressed
with HTLV-1 Tax, mutated p53 R276H or c-Myc38C (69). In addition, previous studies reported
the downregulation of FBW7 expression in ATL, which leads to c-Myc
accumulation and initiation of ATL cell proliferation (70). The c-Myc-FBW7 axis pathway could
therefore represent a potential target for the treatment of
ATL.
MM, which is an incurable malignant plasma cell
disease, is one of the most common hematological malignancies in
adults, accounting for 15% of malignant hematological tumors
(78). MM is primarily characterized
by clonal proliferation and abnormal accumulation of plasma cells
in the bone marrow, combined with the secretion of a large number
of monoclonal immunoglobulins (79).
The etiology of MM is complex. NF-κB may serve a crucial role in
supporting MM cell survival. The NF-κB signaling pathway is
activated in the majority of patients with MM, whereas NF-κB
signaling pathway blockage using some inhibitors of NF-κB kinase
subunit β (IKK2) can inhibit MM cell proliferation (80,81). In
non-canonical NF-κB pathway, IKKα is activated by NIK and directly
phosphorylates p100. Phosphorylated p100 is therefore degraded in a
26S proteasome-dependent manner (82). It has been demonstrated that p100
degradation in the nucleus is crucial to activate the non-canonical
NF-κB pathway (83). During this
reaction, FBW7 recognizes the CPD sequence of p100 and promotes
p100 degradation in a GSK-3 phosphorylation-dependent manner
(15). Furthermore, FBW7
overexpression enhances the activity of NF-κB, whereas FBW7
downregulation can upregulate p100 expression (15). In MM, FBW7 silencing leads to an
increased expression of p100 and partly activates MM cell apoptosis
(15,84). FBW7 may therefore be an oncogene in
MM. In addition, fibroblasts from patients with MM are crucial in
the progression of the disease and in drug resistance (85,86). For
example, it was demonstrated that miR-27b-3p is significantly
upregulated in the fibroblasts of bone marrow from patients with MM
where it controls FBW7 activity, which in turn regulates MCL-1 that
can promote fibroblast proliferation (87).
FBW7 is an important tumor suppressor that regulates
multiple oncogenes, including cyclin E, c-Myc, Notch, c-Jun and
mTOR. In addition, FBW7 is regulated by p53, certain miRNAs,
including miR-223, miR-25, miR-182, miR-503 and miR-92a, RITA,
FAM83D and Numb4. Mutation, deletion and hypermethylation are the
main mechanisms of FBW7 disruption in cancer, leading to tumor
progression. Mutation of FBW7 may promote tumorigenesis and
increase the tumor resistance to chemotherapy. Furthermore, the
carcinogenic effects of FBW7 have also been identified in certain
types of hematological tumor, including ATL and MM. These findings
have important implications for the understanding of hematopoietic
mechanisms, development of diagnostic reagents, and design and
optimization of therapeutic drugs. The detection of FBW7 mutations
is clinically relevant, and FBW7 could serve as a potential target
for the treatment of hematological tumors. Further investigation
into FBW7 mutations will have a positive impact on the prevention
of hematological tumors and the development of personalized
treatments.
Not applicable.
The present study was supported by the National
Science and Technology Major Project for New Drug (grant no.
2017ZX301033), the National Natural Science Foundation of China
(grant nos. 81874049, 81602179 and 81570198), the Co-construction
of Provincial and Department Project (grant no. WKJ-ZJ-1919) and
the Zhejiang Provincial Natural Science Foundation of China (grant
no. LY19H160036).
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
QZ, LH, XT and QX conceived and designed the review.
YG and ZX were involved in designing the review. QZ, LH, YG, ZX, XT
and QX were involved in the collection of references. QZ and LH
collected and assembled the data presented in Tables I and II. QZ and LH designed the figures. QZ and
LH wrote the manuscript. YG collected and assembled the data
present in Tables I and II. ZX was involved in designing the
figures and analysing the data of figure
3. All authors approved the final version of the
manuscript.
Not applicable.
Not applicable.
The authors declared that they have no competing
interests.
|
1
|
Hershko A, Ciechanover A and Varshavsky A:
The ubiquitin system. Nat Med. 6:1073–1081. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crusio KM, King B, Reavie LB and Aifantis
I: The ubiquitous nature of cancer: The role of the SCF(Fbw7)
complex in development and transformation. Oncogene. 29:4865–4873.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shimizu K, Nihira NT, Inuzuka H and Wei W:
Physiological functions of FBW7 in cancer and metabolism. Cell
Signal. 46:15–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Uddin S, Bhat AA, Krishnankutty R, Mir F,
Kulinski M and Mohammad RM: Involvement of F-BOX proteins in
progression and development of human malignancies. Semin Cancer
Biol. 36:18–32. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thompson BJ, Buonamici S, Sulis ML,
Palomero T, Vilimas T, Basso G, Ferrando A and Aifantis I: The
SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell
leukemia. J Exp Med. 204:1825–1835. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Welcker M and Clurman BE: FBW7 ubiquitin
ligase: A tumour suppressor at the crossroads of cell division,
growth and differentiation. Nat Rev Cancer. 8:83–93. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tan Y, Sangfelt O and Spruck C: The
Fbxw7/hCdc4 tumor suppressor in human cancer. Cancer Lett.
271:1–12. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Spruck CH, Strohmaier H, Sangfelt O,
Müller HM, Hubalek M, Müller-Holzner E, Marth C, Widschwendter M
and Reed SI: hCDC4 gene mutations in endometrial cancer. Cancer
Res. 62:4535–4539. 2002.PubMed/NCBI
|
|
9
|
Davis RJ, Welcker M and Clurman BE: Tumor
suppression by the Fbw7 ubiquitin ligase: Mechanisms and
opportunities. Cancer Cell. 26:455–464. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hao B, Oehlmann S, Sowa ME, Harper JW and
Pavletich NP: Structure of a Fbw7-Skp1-cyclin E complex:
Multisite-phosphorylated substrate recognition by SCF ubiquitin
ligases. Mol Cell. 26:131–143. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Welcker M, Larimore EA, Swanger J,
Bengoechea-Alonso MT, Grim JE, Ericsson J, Zheng N and Clurman BE:
Fbw7 dimerization determines the specificity and robustness of
substrate degradation. Genes Dev. 27:2531–2536. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Crusio K M, King B, Reavie L B and
Aifantis I: The ubiquitous nature of cancer: the role of the SCF
Fbw7 complex in development and transformation. Oncogene.
29:4865–4873. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang X, Orlicky S, Lin Z, Willems A,
Neculai D, Ceccarelli D, Mercurio F, Shilton BH, Sicheri F and
Tyers M: Suprafacial orientation of the SCFCdc4 dimer accommodates
multiple geometries for substrate ubiquitination. Cell.
129:1165–1176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Welcker M, Orian A, Jin J, Grim JE, Harper
JW, Eisenman RN and Clurman BE: The Fbw7 tumor suppressor regulates
glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein
degradation. Proc Natl Acad Sci USA. 101:9085–9090. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Busino L, Millman SE, Scotto L, Kyratsous
CA, Basrur V, O'Connor O, Hoffmann A, Elenitoba-Johnson KS and
Pagano M: Fbxw7α- and GSK3-mediated degradation of p100 is a
pro-survival mechanism in multiple myeloma. Nat Cell Biol.
14:375–385. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yada M, Hatakeyama S, Kamura T, Nishiyama
M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K and
Nakayama KI: Phosphorylation-dependent degradation of c-Myc is
mediated by the F-box protein Fbw7. EMBO J. 23:2116–2125. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Welcker M, Orian A, Grim JE, Eisenman RN
and Clurman BE: A nucleolar isoform of the Fbw7 ubiquitin ligase
regulates c-Myc and cell size. Curr Biol. 14:1852–1857. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Koepp DM, Schaefer LK, Ye X, Keyomarsi K,
Chu C, Harper JW and Elledge SJ: Phosphorylation-dependent
ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase.
Science. 294:173–177. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weng AP, Ferrando AA, Lee W, Morris JP IV,
Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT and Aster
JC: Activating mutations of NOTCH1 in human T cell acute
lymphoblastic leukemia. Science. 306:269–271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fukushima H, Matsumoto A, Inuzuka H, Zhai
B, Lau AW, Wan L, Gao D, Shaik S, Yuan M, Gygi SP, et al: SCF(Fbw7)
modulates the NFkB signaling pathway by targeting NFkB2 for
ubiquitination and destruction. Cell Rep. 1:434–443. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei W, Jin J, Schlisio S, Harper JW and
Kaelin WG Jr: The v-Jun point mutation allows c-Jun to escape
GSK3-dependent recognition and destruction by the Fbw7 ubiquitin
ligase. Cancer Cell. 8:25–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lochab S, Pal P, Kapoor I, Kanaujiya JK,
Sanyal S, Behre G and Trivedi AK: E3 ubiquitin ligase Fbw7
negatively regulates granulocytic differentiation by targeting
G-CSFR for degradation. Biochim Biophys Acta. 1833:2639–2652. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xie CM and Sun Y: The MTORC1-mediated
autophagy is regulated by the FBXW7-SHOC2-RPTOR axis. Autophagy.
15:1470–1472. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang L-Y, Zhao J, Chen H, Wan L, Inuzuka
H, Guo J, Fu X, Zhai Y, Lu Z, Wang X, et al: SCFFBW7-mediated
degradation of Brg1 suppresses gastric cancer metastasis. Nat
Commun. 9:35692018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Finkin S, Aylon Y, Anzi S, Oren M and
Shaulian E: Fbw7 regulates the activity of endoreduplication
mediators and the p53 pathway to prevent drug-induced polyploidy.
Oncogene. 27:4411–4421. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu N, Li H, Li S, Shen M, Xiao N, Chen Y,
Wang Y, Wang W, Wang R, Wang Q, et al: The Fbw7/human CDC4 tumor
suppressor targets proproliferative factor KLF5 for ubiquitination
and degradation through multiple phosphodegron motifs. J Biol Chem.
285:18858–18867. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kourtis N, Moubarak RS, Aranda-Orgilles B,
Lui K, Aydin IT, Trimarchi T, Darvishian F, Salvaggio C, Zhong J,
Bhatt K, et al: FBXW7 modulates cellular stress response and
metastatic potential through HSF1 post-translational modification.
Nat Cell Biol. 17:322–332. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bengoechea-Alonso MT and Ericsson J: The
ubiquitin ligase Fbxw7 controls adipocyte differentiation by
targeting C/EBPalpha for degradation. Proc Natl Acad Sci USA.
107:11817–11822. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Akhoondi S, Lindström L, Widschwendter M,
Corcoran M, Bergh J, Spruck C, Grandér D and Sangfelt O:
Inactivation of FBW7/hCDC4-beta expression by promoter
hypermethylation is associated with favorable prognosis in primary
breast cancer. Breast Cancer Res. 12:R1052010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mao JH, Kim IJ, Wu D, Climent J, Kang HC,
DelRosario R and Balmain A: FBXW7 targets mTOR for degradation and
cooperates with PTEN in tumor suppression. Science. 321:1499–1502.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Akhoondi S, Sun D, von der Lehr N,
Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D,
Marth C, et al: FBXW7/hCDC4 is a general tumor suppressor in human
cancer. Cancer Res. 67:9006–9012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Perry JM and Li L: Self-renewal versus
transformation: Fbxw7 deletion leads to stem cell activation and
leukemogenesis. Genes Dev. 22:1107–1109. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kimura T, Gotoh M, Nakamura Y and Arakawa
H: hCDC4b, a regulator of cyclin E, as a direct transcriptional
target of p53. Cancer Sci. 94:431–436. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mansour MR, Sanda T, Lawton LN, Li X,
Kreslavsky T, Novina CD, Brand M, Gutierrez A, Kelliher MA,
Jamieson CH, et al: The TAL1 complex targets the FBXW7 tumor
suppressor by activating miR-223 in human T cell acute
lymphoblastic leukemia. J Exp Med. 210:1545–1557. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xiang J, Hang JB, Che JM and Li HC: MiR-25
is up-regulated in non-small cell lung cancer and promotes cell
proliferation and motility by targeting FBXW7. Int J Clin Exp
Pathol. 8:9147–9153. 2015.PubMed/NCBI
|
|
37
|
Li L, Sarver AL, Khatri R, Hajeri PB,
Kamenev I, French AJ, Thibodeau SN, Steer CJ and Subramanian S:
Sequential expression of miR-182 and miR-503 cooperatively targets
FBXW7, contributing to the malignant transformation of colon
adenoma to adenocarcinoma. J Pathol. 234:488–501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou C, Shen L, Mao L, Wang B, Li Y and Yu
H: miR-92a is upregulated in cervical cancer and promotes cell
proliferation and invasion by targeting FBXW7. Biochem Biophys Res
Commun. 458:63–69. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang H, Yang Z, Liu C, Huang S, Wang H,
Chen Y and Chen G: RBP-J-interacting and tubulin-associated protein
induces apoptosis and cell cycle arrest in human hepatocellular
carcinoma by activating the p53-FBW7 pathway. Biochem Biophys Res
Commun. 454:71–77. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang H, Ma L, Li J, Yu Y, Zhang D, Wei J,
Jin H, Xu D, Gao J and Huang C: NF-κB1 inhibits c-Myc protein
degradation through suppression of FBW7 expression. Oncotarget.
5:493–505. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Min SH, Lau AW, Lee TH, Inuzuka H, Wei S,
Huang P, Shaik S, Lee DY, Finn G, Balastik M, et al: Negative
regulation of the stability and tumor suppressor function of Fbw7
by the Pin1 prolyl isomerase. Mol Cell. 46:771–783. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang Z, Liu Y, Zhang P, Zhang W, Wang W,
Curr K, Wei G and Mao JH: FAM83D promotes cell proliferation and
motility by downregulating tumor suppressor gene FBXW7. Oncotarget.
4:2476–2486. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang X, Xing H, Kim TM, Jung Y, Huang W,
Yang HW, Song S, Park PJ, Carroll RS and Johnson MD: Numb regulates
glioma stem cell fate and growth by altering epidermal growth
factor receptor and Skp1-Cullin-F-box ubiquitin ligase activity.
Stem Cells. 30:1313–1326. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Peirs S, Van der Meulen J, Van de Walle I,
Taghon T, Speleman F, Poppe B and Van Vlierberghe P: Epigenetics in
T-cell acute lymphoblastic leukemia. Immunol Rev. 263:50–67. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pui CH and Evans WE: Treatment of acute
lymphoblastic leukemia. N Engl J Med. 354:166–178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vadillo E, Dorantes-Acosta E, Pelayo R and
Schnoor M: T cell acute lymphoblastic leukemia (T-ALL): New
insights into the cellular origins and infiltration mechanisms
common and unique among hematologic malignancies. Blood Rev.
32:36–51. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Allen A, Sireci A, Colovai A, Pinkney K,
Sulis M, Bhagat G and Alobeid B: Early T-cell precursor
leukemia/lymphoma in adults and children. Leuk Res. 37:1027–1034.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pui CH, Robison LL and Look AT: Acute
lymphoblastic leukaemia. Lancet. 371:1030–1043. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Matsuoka S, Oike Y, Onoyama I, Iwama A,
Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, et al: Fbxw7
acts as a critical fail-safe against premature loss of
hematopoietic stem cells and development of T-ALL. Genes Dev.
22:986–991. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Onoyama I, Tsunematsu R, Matsumoto A,
Kimura T, de Alborán IM, Nakayama K and Nakayama KI: Conditional
inactivation of Fbxw7 impairs cell-cycle exit during T cell
differentiation and results in lymphomatogenesis. J Exp Med.
204:2875–2888. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kwon YW, Kim IJ, Wu D, Lu J, Stock WA Jr,
Liu Y, Huang Y, Kang HC, DelRosario R, Jen KY, et al: Pten
regulates Aurora-A and cooperates with Fbxw7 in modulating
radiation-induced tumor development. Mol Cancer Res. 10:834–844.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
King B, Trimarchi T, Reavie L, Xu L,
Mullenders J, Ntziachristos P, Aranda-Orgilles B, Perez-Garcia A,
Shi J, Vakoc C, et al: The ubiquitin ligase FBXW7 modulates
leukemia-initiating cell activity by regulating MYC stability.
Cell. 153:1552–1566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
King B, Trimarchi T, Reavie L, Xu L,
Mullenders J, Ntziachristos P, Aranda-Orgilles B, Perez-Garcia A,
Shi J and Vakoc C: Regulation of leukemia-initiating cell activity
by the ubiquitin ligase FBXW7. Cell. 153:1552–1566. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Thompson BJ, Jankovic V, Gao J, Buonamici
S, Vest A, Lee JM, Zavadil J, Nimer SD and Aifantis I: Control of
hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7.
J Exp Med. 205:1395–1408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tosello V and Ferrando AA: The NOTCH
signaling pathway: Role in the pathogenesis of T-cell acute
lymphoblastic leukemia and implication for therapy. Ther Adv
Hematol. 4:199–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Arruga F, Gizdic B, Bologna C, et al:
Mutations in NOTCH1 PEST domain orchestrate CCL19-driven homing of
chronic lymphocytic leukemia cells by modulating the tumor
suppressor gene DUSP22. 31:18822017.PubMed/NCBI
|
|
57
|
Chiang MY, Radojcic V and Maillard I:
Oncogenic Notch signaling in T-cell and B-cell lymphoproliferative
disorders. Curr Opin Hematol. 23:362–370. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Grabher C, von Boehmer H and Look AT:
Notch 1 activation in the molecular pathogenesis of T-cell acute
lymphoblastic leukaemia. Nat Rev Cancer. 6:347–359. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Suresh S and Irvine AE: The NOTCH
signaling pathway in normal and malignant blood cell production. J
Cell Commun Signal. 9:5–13. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Malecki MJ, Sanchez-Irizarry C, Mitchell
JL, Histen G, Xu ML, Aster JC and Blacklow SC: Leukemia-associated
mutations within the NOTCH1 heterodimerization domain fall into at
least two distinct mechanistic classes. Mol Cell Biol.
26:4642–4651. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
O'Neil J, Grim J, Strack P, Rao S,
Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters
R, et al: FBW7 mutations in leukemic cells mediate NOTCH pathway
activation and resistance to γ-secretase inhibitors. J Exp Med.
204:1813–1824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kumar V, Palermo R, Talora C, Campese AF,
Checquolo S, Bellavia D, Tottone L, Testa G, Miele E, Indraccolo S,
et al: Notch and NF-kB signaling pathways regulate miR-223/FBXW7
axis in T-cell acute lymphoblastic leukemia. Leukemia.
28:2324–2335. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Malyukova A, Brown S, Papa R, O'Brien R,
Giles J, Trahair TN, Dalla Pozza L, Sutton R, Liu T, Haber M, et
al: FBXW7 regulates glucocorticoid response in T-cell acute
lymphoblastic leukaemia by targeting the glucocorticoid receptor
for degradation. Leukemia. 27:1053–1062. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yang-Yen HF: Mcl-1: A highly regulated
cell death and survival controller. J Biomed Sci. 13:201–204. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Inuzuka H, Shaik S, Onoyama I, Gao D,
Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al:
SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for
ubiquitylation and destruction. Nature. 471:104–109. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wertz IE, Kusam S, Lam C, Okamoto T,
Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al:
Sensitivity to antitubulin chemotherapeutics is regulated by MCL1
and FBW7. Nature. 471:110–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Schwickart M, Huang X, Lill JR, Liu J,
Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F,
Eastham-Anderson J, et al: Deubiquitinase USP9X stabilizes MCL1 and
promotes tumour cell survival. Nature. 463:103–107. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Katsuya H, Ishitsuka K, Utsunomiya A,
Hanada S, Eto T, Moriuchi Y, Saburi Y, Miyahara M, Sueoka E, Uike
N, et al ATL-Prognostic Index Project, : Treatment and survival
among 1594 patients with ATL. Blood. 126:2570–2577. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Yeh C-H, Bellon M, Pancewicz-Wojtkiewicz J
and Nicot C: Oncogenic mutations in the FBXW7 gene of adult T-cell
leukemia patients. Proc Natl Acad Sci USA. 113:6731–6736. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mihashi Y, Mizoguchi M, Takamatsu Y,
Ishitsuka K, Iwasaki H, Koga M, Urabe K, Momosaki S, Sakata T,
Kiyomi F, et al: C-MYC and its main ubiquitin ligase, FBXW7,
influence cell proliferation and prognosis in adult T-cell
leukemia/lymphoma. Am J Surg Pathol. 41:1139–1149. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chiorazzi N, Rai KR and Ferrarini M:
Chronic lymphocytic leukemia. N Engl J Med. 352:804–815. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kipps TJ, Stevenson FK, Wu CJ, Croce CM,
Packham G, Wierda WG, O'Brien S, Gribben J and Rai K: Chronic
lymphocytic leukaemia. Nat Rev Dis Primers. 3:160962017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Bosch F and Dalla-Favera R: Chronic
lymphocytic leukaemia: From genetics to treatment. Nat Rev Clin
Oncol. 16:684–701. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Landau DA, Tausch E, Taylor-Weiner AN,
Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M,
Böttcher S, et al: Mutations driving CLL and their evolution in
progression and relapse. Nature. 526:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Jeromin S, Weissmann S, Haferlach C,
Dicker F, Bayer K, Grossmann V, Alpermann T, Roller A, Kohlmann A,
Haferlach T, et al: SF3B1 mutations correlated to cytogenetics and
mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated
CLL patients. Leukemia. 28:108–117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Quijada-Álamo M, Hernández-Sánchez M,
Robledo C, Hernández-Sánchez JM, Benito R, Montaño A,
Rodríguez-Vicente AE, Quwaider D, Martín AÁ, García-Álvarez M, et
al: Next-generation sequencing and FISH studies reveal the
appearance of gene mutations and chromosomal abnormalities in
hematopoietic progenitors in chronic lymphocytic leukemia. J
Hematol Oncol. 10:832017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Close V, Close W, Kugler SJ, Reichenzeller
M, Yosifov DY, Bloehdorn J, Pan L, Tausch E, Westhoff MA, Döhner H,
et al: FBXW7 mutations reduce binding of NOTCH1, leading to cleaved
NOTCH1 accumulation and target gene activation in CLL. Blood.
133:830–839. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Palumbo A and Anderson K: Multiple
myeloma. N Engl J Med. 364:1046–1060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Sonneveld P, Avet-Loiseau H, Lonial S,
Usmani S, Siegel D, Anderson KC, Chng WJ, Moreau P, Attal M, Kyle
RA, et al: Treatment of multiple myeloma with high-risk
cytogenetics: A consensus of the International Myeloma Working
Group. Blood. 127:2955–2962. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Annunziata CM, Davis RE, Demchenko Y,
Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W,
et al: Frequent engagement of the classical and alternative
NF-kappaB pathways by diverse genetic abnormalities in multiple
myeloma. Cancer Cell. 12:115–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jourdan M, Moreaux J, De Vos J, Hose D,
Mahtouk K, Abouladze M, Robert N, Baudard M, Rème T, Romanelli A,
et al: Targeting NF-kappaB pathway with an IKK2 inhibitor induces
inhibition of multiple myeloma Cell Proliferation. Br J Haematol.
138:160–168. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Sun SC: The non-canonical NF-κB pathway in
immunity and inflammation. Nat Rev Immunol. 17:545–558. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Senftleben U, Cao Y, Xiao G, Greten FR,
Krähn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, et al: Activation
by IKKalpha of a second, evolutionary conserved, NF-kappa B
signaling pathway. Science. 293:1495–1499. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Busino L, Millman SE and Pagano M:
SCF-mediated degradation of p100 (NF-κB2): Mechanisms and relevance
in multiple myeloma. Sci Signal. 5:pt142012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Frassanito MA, Rao L, Moschetta M, Ria R,
Di Marzo L, De Luisi A, Racanelli V, Catacchio I, Berardi S, Basile
A, et al: Bone marrow fibroblasts parallel multiple myeloma
progression in patients and mice: In vitro and in vivo studies.
Leukemia. 28:904–916. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Frassanito MA, De Veirman K, Desantis V,
Di Marzo L, Vergara D, Ruggieri S, Annese T, Nico B, Menu E,
Catacchio I, et al: Halting pro-survival autophagy by TGFβ
inhibition in bone marrow fibroblasts overcomes bortezomib
resistance in multiple myeloma patients. Leukemia. 30:640–648.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Frassanito MA, Desantis V, Di Marzo L,
Craparotta I, Beltrame L, Marchini S, Annese T, Visino F, Arciuli
M, Saltarella I, et al: Bone marrow fibroblasts overexpress miR-27b
and miR-214 in step with multiple myeloma progression, dependent on
tumour cell-derived exosomes. J Pathol. 247:241–253. 2019.
View Article : Google Scholar : PubMed/NCBI
|