Introduction
Breast cancer is the predominant malignant tumor
amongst women (1). Although China
has a low incidence of breast cancer compared with Europe and North
America, incidence has increased in recent years whilst the age of
onset has reduced (2). The World
Health Organization notes breast cancer as a serious threat to
women's health (3). Breast cancer
metastasis is a key factor that affects the prognosis and clinical
outcomes for patients, therefore effective control of tumor
invasion and metastasis is critical to improve the prognosis for
patients with breast cancer. Multiple studies (4–6) have
demonstrated that the occurrence and development of breast cancer
involves the inactivation of tumor suppressor genes and mutations
in oncogenes. Furthermore, aberrant regulation of cellular
apoptosis serves an important role in carcinogenesis (7).
For tumors to develop, an adequate supply of
nutrients and oxygen is required (8). However, as the tumors continue to
grow, nutrient and oxygen consumption requirements increase, while
the supply begins to decline. At this point, the tumor requires new
blood vessels to maintain its growth. Vascular endothelial growth
factor (VEGF) is the most important factor in vascular regulation
(9). In tumor development, hypoxia
activates hypoxia inducible factor 1α (HIF-1α) and VEGF to induce
neovascularization (10). VEGF is
required for the regeneration of blood vessels in embryos and in
adults following injury (11).
High expression levels of VEGF have been demonstrated to be
negatively correlated with the patient survival rate in a variety
of solid tumors (12,13).
p53 is the most frequent tumor suppressor gene
mutated in human cancers (14).
However, in approximately 50–60% of the tumors harboring p53
mutations, recovery of wild-type p53 expression suppresses tumor
growth and metastasis (15). A
previous study demonstrated that during hypoxia, p53 has a
dual-directional regulatory function for VEGF. In the early stages
of hypoxia, p53 and HIF-1α interactions promote the expression of
VEGF, however, during long-term hypoxia, p53 exerts an inhibitory
effect on VEGF (16). Agani et
al (17) observed that in
Hep3B human hepatoblastoma and RKO colon cancer cell cells, the
activation of endogenous p53 and exogenous p53 did not impact VEGF
mRNA levels. However, studies have proposed that wild-type p53
inhibits VEGF transcription in vitro (18). p53 acts via specific regulatory
protein 1 and v-src sarcoma viral oncogene homolog kinase to
achieve its inhibitory effect on VEGF, thus influencing the
formation of blood vessels (17–19).
Although a cross-talk between p53 and VEGF has been suggested, the
underlying mechanisms remain to be fully elucidated.
Gene therapy has received increased attention
however, due to the complexity of tumors, single gene treatment
strategies are not optional. Therefore, in the present study, a
co-expression plasmid for double gene therapy in tumor cells was
investigated. Wild-type p53 and VEGF short-interfering RNA
(si-VEGF) plasmids were used alone and in combination in MDA-MB-231
cells, with the effects and underlying mechanisms investigated.
Materials and methods
Cell culture and plasmids
MDA-MB-231 breast cancer cells (Chinese Academy of
Sciences Cell Bank, Shanghai, China) were cultured in Dulbecco's
modified Eagle's medium with 10% (v/v) fetal bovine serum (FBS; GE
Healthcare Life Sciences, Logan, UT, USA) in 95% air and 5%
CO2 at 37°C. The eukaryotic expression vectors (Jilin
University, Jilin, China) pcDNA3.1-p53 (p53), pGCsiRNA-VEGF
(si-VEGF), pcDNA3.1-p53/U6 siRNA-VEGF (Pvp53) and pGCsiRNA-scramble
(si-scramble; scramble siRNA sequence as a negative control) were
used in these experiments. The MDA-MB-231 cells were plated into
24-well culture plates (4×104 cells/well). Plasmids were
then transfected into cells using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
After 48 h, the cell morphology of all groups was observed using
light microscopy (Oympus BX41-PHD-P11; Olympus Corporation, Tokyo,
Japan).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assays
Transfected MDA-MB-231 cells were plated into
96-well flat bottom microplates (8×103 cells/well). At
24, 48 and 72 h, MTT (5 mg/ml; Sangon Biotech Co., Ltd., Shanghai,
China) was added to each well and the cells were incubated at 37°C
for 4 h. The medium was then removed and 200 µl dimethyl
sulfoxide (Sangon Biotech Co., Ltd.) was added to dissolve the
reduced formazan product. The plates were read in an enzyme-linked
immunosorbent assay reader (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) at 490 nm. The proliferation inhibition rate was
calculated according to the absorbance values.
Scratch wound assay
A scratch wound was made by scraping the monolayer
of transfected cells across the cover glass with a sterile cell
lifter (3008; Corning, Inc., Corning, NY, USA). Following wounding,
the culture medium supplemented with 10% FBS was replaced with
culture medium supplemented with 2% FBS, in order to maintain cell
survival but prevent proliferation. Cells were then allowed to
migrate for 48 h. Cell migration was evaluated by measuring the
size of the scratch-wound 48 h following wounding using an Oympus
BX41-PHD-P11 microscope.
Flow cytometric analysis (FCM)
Transfected MDA-MB-231 cells were washed three times
with phosphate-buffered saline (Sangon Biotech Co., Ltd.). The
cells were resuspended in 400 µl DNA binding buffer (Beckman
Coulter Inc., Brea, CA, USA). Subsequently, 5 µl annexin V
fluorescein isothiocyanate and 10 µl propidium iodide
(Beckman Coulter Inc.) were added and the samples were incubated
for 15 min and 10 min at room temperature in the dark,
respectively. The apoptotic rate was measured by FCM using an
Epics-XL-MCL flow cytometer (Beckman Coulter, Inc.).
Reverse transcription-polymerase chain
reaction (RT-PCR)
For RT-PCR analysis, total RNA from cells was
extracted using Invitrogen TRIzol reagent (Thermo Fisher
Scientific, Inc.). A total of 5 µg total RNA (purified
following DNase I treatment; Thermo Fisher Scientific, Inc.) from
each sample was reverse transcribed to complementary cDNA using
SMART® MMLV Reverse Transcriptase kit (cat. no. 639523;
Takara Bio, Inc., Otsu, Japan). The resultant cDNAs (100 ng) were
used in the PCR with the gene-specific primers of interest
(Table I). PCR was conducted using
2X EasyTaq PCR SuperMix (cat. no. AS111-02; Beijing Transgen
Biotech Co., Ltd., Beijing, China). The reaction conditions are
shown in Table II. Primer
concentrations for each gene were obtained by performing a series
of pre-experiments. PCR products were separated by agarose gel
(Invitrogen; Thermo Fisher Scientific, Inc.) electrophoresis and
visualized by ethidium bromide staining.
 | Table IPrimer sequences for reverse
transcription-polymerase chain reaction. |
Table I
Primer sequences for reverse
transcription-polymerase chain reaction.
| Gene | Forward primer
sequence | Reverse primer
sequence |
|---|
| GAPDH |
5′-GGGTGATGCTGGTGCTGAGTATGT-3′ |
5′-AAGAATGGGAGTTGCTGTTGAAGTC-3′ |
| p53 |
5′-CCTCCTCAGCATCTTATCCG-3′ |
5′-CACAAACACGCACCTCAAA-3′ |
| VEGF |
5′-GAGGGCAGAATCATCACGAA-3′ |
5′-GGCTCCAGGGCATTAGACA-3′ |
| Bcl-2 |
5′-GACTTCGCCGAGATGTCCAGC-3′ |
5′-TGTGGCCCAGATAGGCACCC-3′ |
| Bax |
5′-GGCCCACCAGCTCTGAGCAGA-3′ |
5′-GCCACGTGGGGGTCCCAAAGT-3′ |
| Table IIPolymerase chain reaction
thermocycling conditions. |
Table II
Polymerase chain reaction
thermocycling conditions.
| Gene | Denaturation | Annealing | Extension | Cycle no. |
|---|
| GAPDH | 94°C 30 sec | 56°C, 45 sec | 72°C, 45 sec | 25 |
| p53 | 94°C 30 sec | 56°C, 45 sec | 72°C, 45 sec | 28 |
| VEGF | 94°C 30 sec | 55°C, 30 sec | 72°C, 30 sec | 30 |
| Bcl-2 | 94°C 30 sec | 60°C, 45 sec | 72°C, 45 sec | 30 |
| Bax | 94°C 30 sec | 56°C, 45 sec | 72°C, 45 sec | 30 |
Western blot analysis
Cells were harvested and lysed with lysis buffer
(Takara Bio, Inc.). Following centrifugation at 12,000 × g for 20
min at 4°C, the protein content of the supernatants was determined
using Bradford reagent (Bio-Rad Laboratories, Inc.). A total of 30
µg protein from each sample was separated by 12%
polyacrylamide gel (Sangon Biotech Co., Ltd.) electrophoresis and
transferred to polyvinylidene fluoride membranes (Invitrogen) as
described previously (3,4). The following antibodies were used for
western blot analysis: Rabbit polyclonal anti-β-actin (1:3,000;
cat. no. AP0060), rabbit polyclonal anti-p21 (1:500; cat. no.
BS1269), rabbit polyclonal anti-Bax (1:500; cat. no. BS2538),
rabbit polyclonal anti-Bcl-2 (1:500; cat. no. BS1511) and rabbit
polyclonal anti-p53 (1:500; cat. no. BS1273); Bioworld Technology,
Inc., St. Louis Park, MN, USA), rabbit polyclonal cleaved caspase-3
(1:1,000; cat. no. 9661; Cell Signaling Technology, Inc., Danvers,
MA, USA), rabbit polyclonal anti-caspase 8 (1:200; cat. no.
sc-7890) and rabbit polyclonal anti-cleaved caspase-8 (1:200; cat.
no. sc-7890) Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
rabbit polyclonal anti-VEGF (1:1,000; cat. no. 19003-1-AP;
ProteinTech Group, Inc., Chicago, IL, USA), mouse monoclonal
anti-matrix metal-loproteinase-2 (MMP-2; 1:200; cat. no. sc-13594;
Santa Cruz Biotechnology, Inc.), mouse monoclonal anti-MMP-9
(1:200; cat. no. sc-21733) and anti-rabbit (1:1,000; cat. no.
sc-2054) and anti-mouse (1:1,000; cat. no. sc-2005) IgG (Santa Cruz
Biotechnology, Inc.). Proteins were detected using an enhanced
chemiluminescence kit (cat. no. 120702-74; Advansta, Inc., Menio
Park, CA, USA).
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical comparisons of data were performed using analysis of
variance to determine statistical significance. Statistical
analyses were conducted using SPSS 11.0 (SPSS, Inc., Chicago, IL,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
p53 and VEGF gene and protein expression
in MDA-MB-231 cells transfected with the Pvp53 co-expression
plasmid
The expression of the p53 and VEGF genes and
proteins in MDA-MB-231 cells transfected with Pvp53 plasmid for 48
h was examined by RT-PCR and western blot analysis. p53 mRNA
expression was significantly increased in cells transfected with
p53 alone and Pvp53, whilst VEGF mRNA expression was reduced in
cells transfected with si-VEGF and Pvp53 plasmids. In addition,
VEGF mRNA expression in Pvp53-transfected cells was reduced
compared with the si-VEGF plasmid alone (Fig. 1A and B). This result suggests that
p53 is able to inhibit the expression of VEGF and is consistent
with previous studies (18). The
western blot results of p53 and VEGF expression were consistent
with the mRNA results (Fig. 1C and
D). To examine p53 function, the protein expression of p21 was
measured. The protein expression of p21 was increased in cells
transfected with p53 and Pvp53 plasmids, however was not altered in
cells treated with the si-VEGF plasmid (Fig. 1C and D).
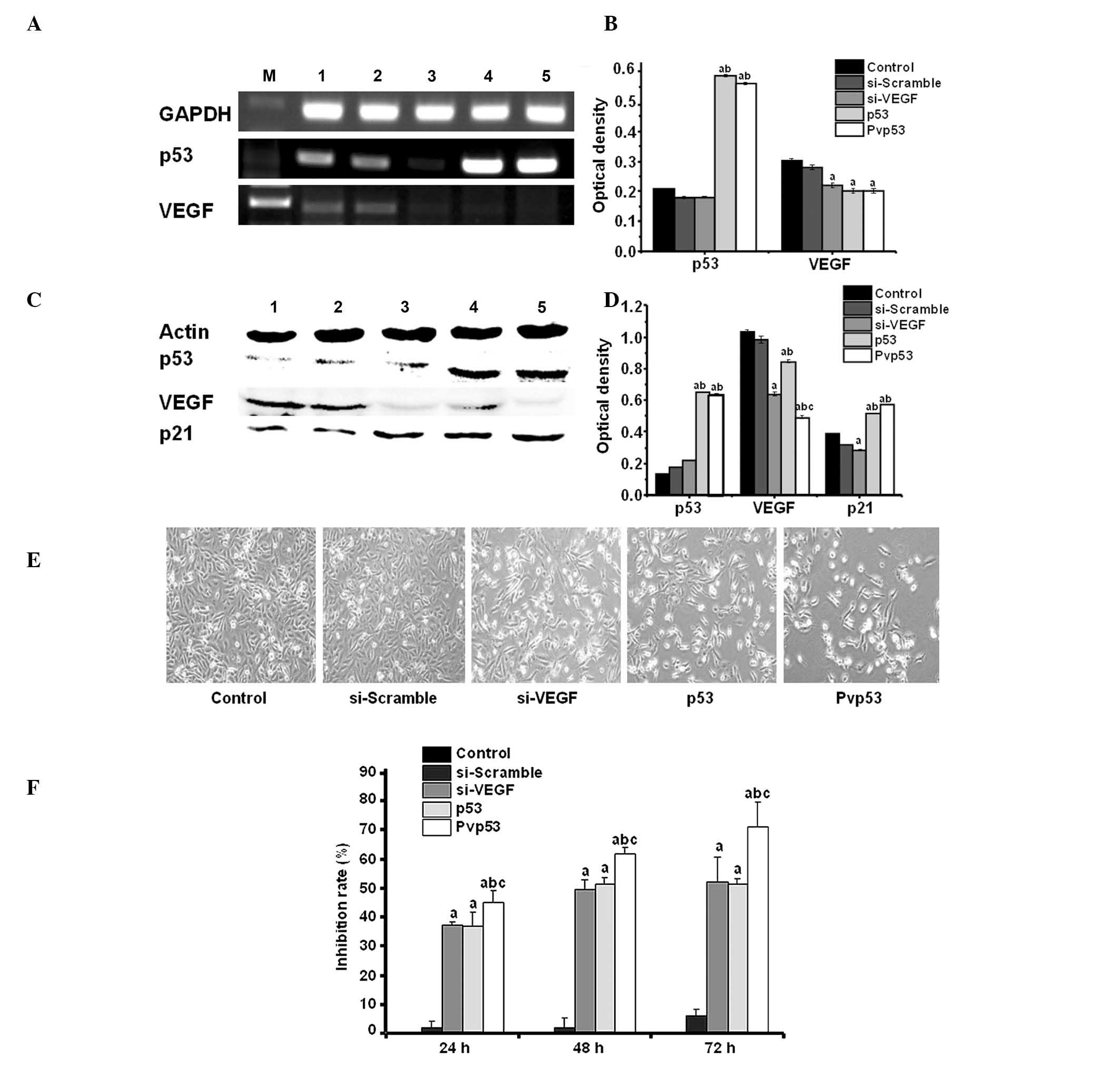 | Figure 1Effects on cell morphology and
survival by Pvp53 transfection. (A and B) Expression of p53 and
VEGF mRNA was measured using reverse transcription-polymerase chain
reaction. (C and D) Protein expression levels of p53 and VEGF were
measured by western blotting. M, marker; lane 1, control; lane 2,
si-scramble; lane 3, si-VEGF; lane 4, p53; lane 5, Pvp53. (E)
Images of cell morphology (magnification, ×200). (F) Inhibitory
rate on cell proliferation at different time points. Data are
presented as the mean ± standard deviation obtained from three
independent experiments. aP<0.05 vs. control or
si-scramble group; bP<0.05 vs. si-VEGF group;
cP<0.05 vs. p53 group. VEGF, vascular endothelial
growth factor; si, short interfering RNA; GAPDH, glyceraldehyde
3-phosphate dehydrogenase. |
Cell morphology and survival in
Pvp53-transfected cells
The cell morphology of transfected cells was
investigated using light microscopy. Compared with the control, 48
h following transfection, the cells of the three transfection
groups (p53, si-VEGF and Pvp53) were observed to be rounded, with
an irregular shape and exhibiting refractive index variation. The
alterations were greatest in cells transfected with the Pvp53
plasmid (Fig. 1E). The MTT assay
indicated that all three plasmids inhibited the growth of
MDA-MB-231 cells and the survival rate of the cells was reduced in
a time-dependent manner. The Pvp53 plasmid exerted the greatest
inhibitory effect on cell survival (Fig. 1F, Table III).
| Table IIIInhibitory rate of cell
proliferation. |
Table III
Inhibitory rate of cell
proliferation.
| Time (h)
|
|---|
| Group | 24 | 48 | 72 |
|---|
| Control | 0.0±0.0 | 0.0±1.5 | 0.0±3.0 |
| si-scramble | 1.6±2.3 | 1.7±4.7 | 5.8±2.4 |
| si-VEGF | 24.3±1.0a | 39.7±3.1a | 42.4±8.5a |
| p53 | 23.7±5.1a | 41.6±2.5a | 40.2±3.4a |
| Pvp53 | 31.8±4.3a,b,c | 52.0±2.4a,b,c | 63.9±4.1a,b,c |
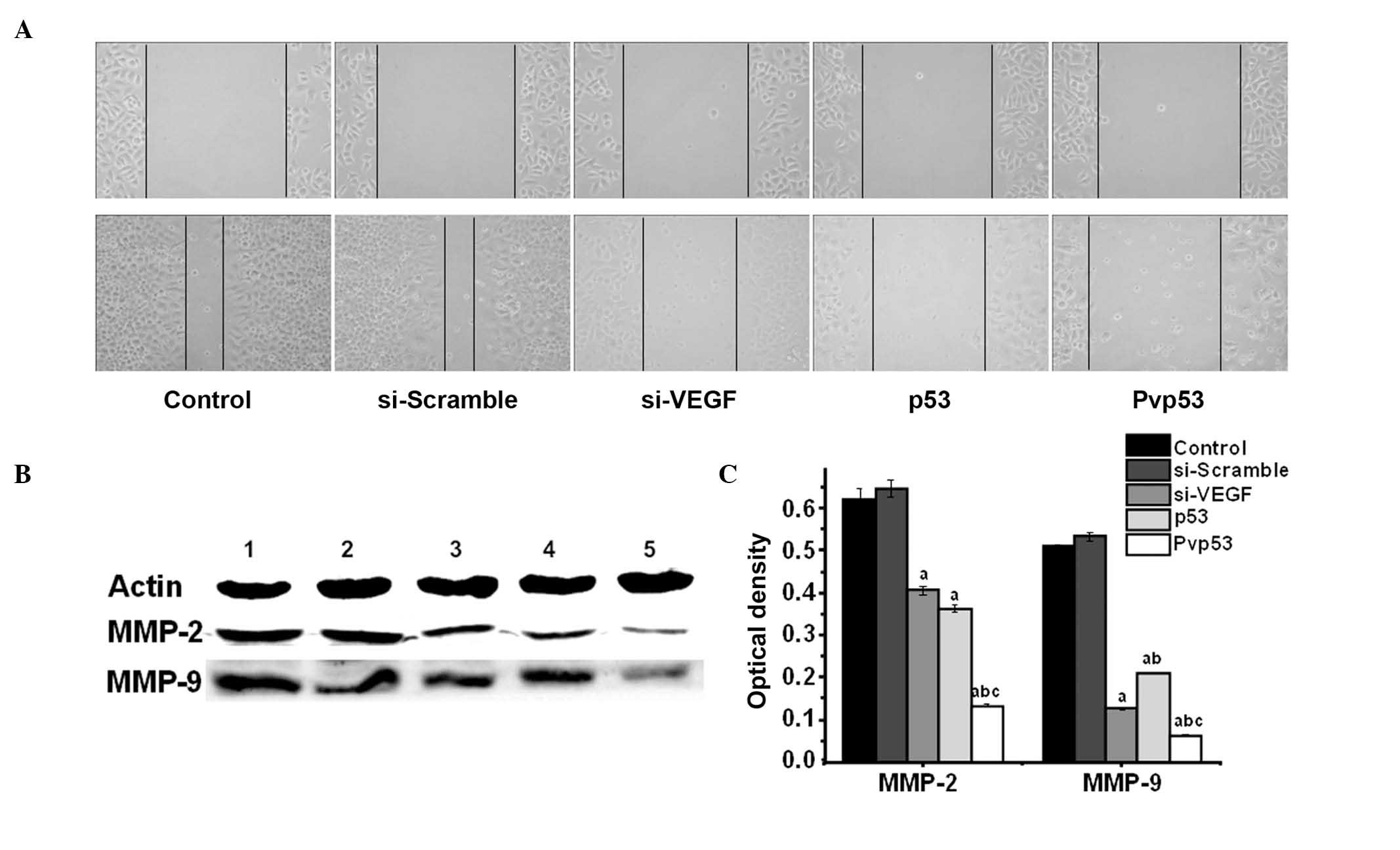
Effects of Pvp53 co-expression plasmid on
wound-induced migration of MDA-MB-231 cells
To examine cell motility, a scratch-wound assay was
used. A scratch was introduced into the confluent monolayer, and
the movement of cells into the injured area was monitored by
microscopy (Fig. 2A). A
significant reduction in the motility of the cells in the Pvp53
group was observed at 48 h compared with the p53 and si-VEGF
groups.
To investigate the mechanisms involved in the
inhibition of motility following transfection, the protein
expression levels of MMP-2 and MMP-9 were measured. The three
transfection groups exhibited reduced levels of MMP-2 and MMP-9,
however Pvp53 demonstrated the greatest reduction in MMP2 and MMP-9
expression levels (Fig. 2B and
C).
Effects of Pvp53 on cell apoptosis and
associated mechanisms
To evaluate apoptosis in MDA-MB-231 cells
transfected with Pvp53, cell apoptosis was measured by FCM. The
right upper quadrant indicates late apoptosis, and the right lower
quadrant indicates early apoptosis. Early apoptosis and late
apoptosis were used to define the apoptotic rate. Quantitative
analysis using FCM demonstrated the apoptotic rate of the control
and si-scramble groups were 1.6% and 1.8%, respectively. The
apoptotic rate of the si-VEGF group, p53 group and Pvp53 group were
14.9%, 19.3% and 24.0%, respectively. Compared with the control
group and si-Scramble group, the apoptotic rates of the si-VEGF,
p53 and Pvp53 groups significantly increased, and the highest
apoptotic rates were observed in the Pvp53 group (Fig. 3A).
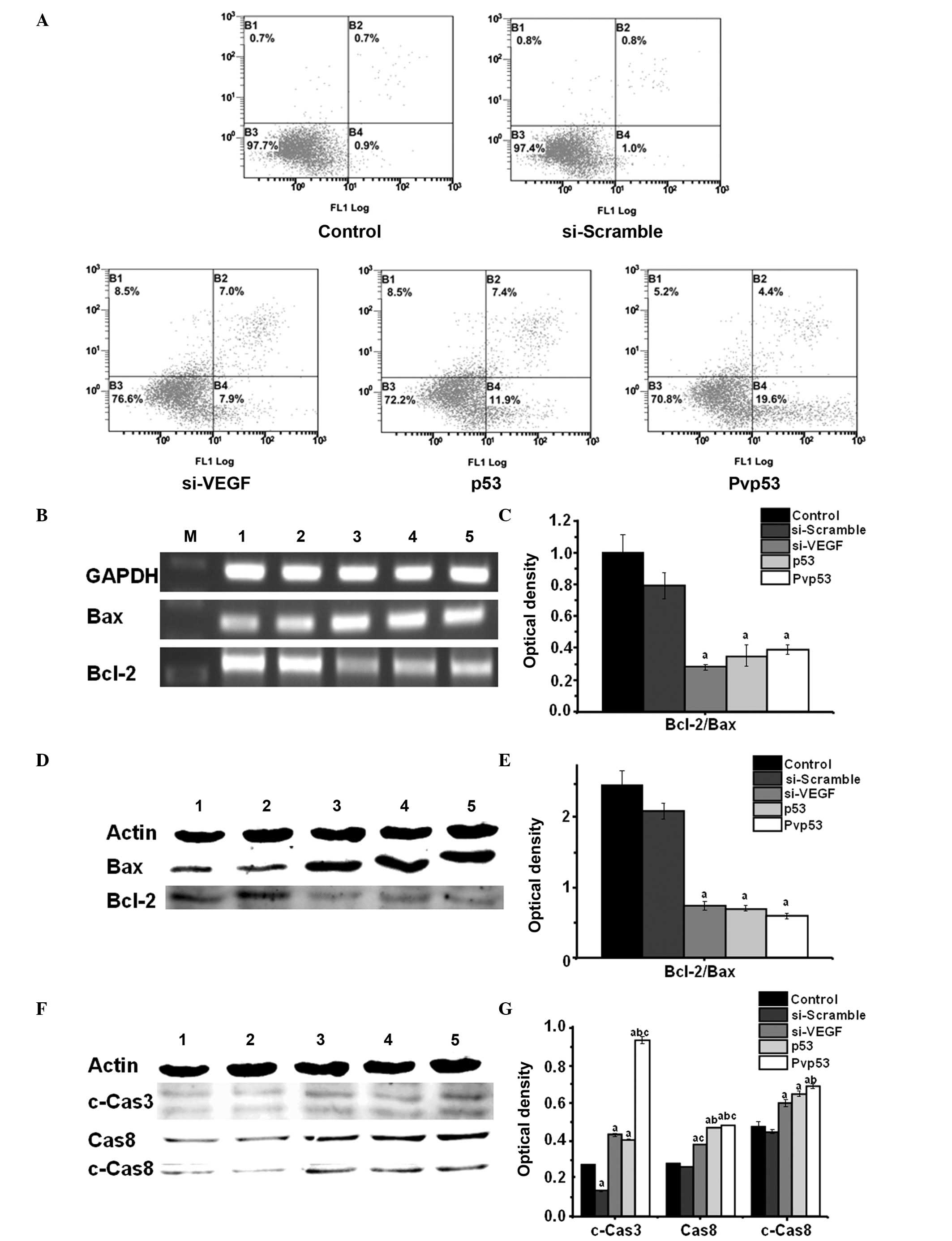 | Figure 3Effects of Pvp53 transfection on cell
apoptosis. (A) Flow cytometric analysis of apoptotic cells using
annexin V-fluorescein isothiocyanate and propidium iodide staining
at 48 h following transfection. (B and C) Expression levels of
Bcl-2/Bax mRNA were measured using reverse transcription-polymerase
chain reaction. Protein expression levels of (D and E) Bcl-2/Bax,
(F and G) Cas8, c-Cas8 and c-Cas3 were measured using western
blotting. M, marker; lane 1, control; lane 2, si-scramble; lane 3,
si-VEGF; lane 4, p53; lane 5, Pvp53. aP<0.05 vs.
control or si-scramble group; bP<0.05 vs. si-VEGF
group; cP<0.05 vs. p53 group. si, short interfering
RNA; VEGF, vascular endothelial growth factor; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; Cas, caspase; c-Cas,
cleaved-Cas. |
To determine the potential mechanisms underpinning
the inhibition of cell growth, the expression levels of
apoptosis-associated proteins were examined. Plasmid transfections
resulted in a significant increase in the expression levels of Bax,
cleaved caspase-3 and 8 and Bax, and in addition reduced expression
of Bcl-2 was observed (Fig.
3B–F).
Discussion
Maintenance of homeostasis is an essential process
for cell survival. Cells are required to respond to an
ever-changing environment to maintain homeostasis, and the tumor
suppressor p53 is a vital component in the response to stressors
such as DNA damage, oncogene activity and hypoxia (20).
p53 and VEGF serve important roles in tumorigenesis.
Previous studies have confirmed an association between p53 and
VEGF, with p53 serving a role in the regulation of VEGF (16,21,22).
In the current study, the Pvp53 plasmid was constructed to silence
the expression of VEGF, while inducing high expression levels of
wild-type p53. In the p53 plasmid group, expression of VEGF was
reduced, and compared with the si-VEGF plasmid, the VEGF silencing
phenomenon was more overt in the Pvp53 group (Fig. 1A–D). This indicates that p53 is
able to inhibit expression of VEGF, which is consistent with the
observations of Farhang Ghahremani et al (18).
The tumor suppressor gene p53, termed the “guardian
of the genome”, serves an essential role in preserving genomic
stability by preventing genome mutations (23). The major function of p53 involves
the blockage of cell cycle progression in response to DNA damage
(24), however the majority of
tumors possess p53 mutations (13). Breast tumors with p53 mutations are
predominantly classified into basal-like or the HER2-amplified
subgroup, while the luminal subgroup of breast cancers almost
exclusively expresses wild-type p53 (13). Typically, mutant p53 is stably
expressed and accumulates in cells, with mutations in p53 able to
alter the function of the p53 protein, and some mutations resulting
in oncogenic activity (20).
Previous studies have demonstrated that mutant p53 has the
potential ability to induce tumorigenesis, predominantly through
the inhibition of transcription, which inactivates additional
tumor-suppressor genes (25–28).
Mutations in p53 are diverse and alter the core molecular pathways
involved in drug responses (29).
Types of p53 mutations, widely termed gain of function mutations,
convert the protein from a tumor suppressor to an oncogene
(20).
The p53 protein is a multifunctional protein that is
able to induce DNA damage and apoptosis, and has an important role
in controlling cellular responses to numerous stress signals
(30). p53 is posttranslationally
modified and degraded by prote-ases, and is normally expressed at
low levels and is unable to bind specifically to DNA (30). Under conditions of stress, p53
accumulates via multiple mechanisms, including enhanced
translation, reduced proteolytic degradation and post-translational
modification (30). p53 activates
the expression of pro-apoptotic proteins by transcriptional
regulation, and indirectly acts on the mitochondrial pathway to
induce apoptosis (31).
Furthermore, p53 additionally promotes apoptosis by activating
pro-apoptotic proteins (Bax, Bak) in a transcription-dependent
manner, by binding to apoptosis-inhibiting proteins (Bcl-2, Bcl-XL)
or by acting directly on the outer mitochondrial membrane,
resulting in permeability alterations and promoting apoptosis
(31). The current study indicated
that compared with p53 and si-VEGF single plasmid transfections,
the Pvp53 co-expression plasmid significantly reduced the ratio of
Bcl-2/Bax, and increased the expression of cleaved caspase-3 and 8,
thereby promoting breast cancer cell apoptosis (Fig. 3). This result suggests that Pvp53
is able to activate the caspase pathway, indicating a pro-apoptotic
function.
One of the hallmark cancer cell phenotypic
alterations is angiogenesis. Tumor growth and metastasis is
dependent upon the formation of new blood vessels, and
tumor-derived VEGF serves an important role in the formation of new
blood vessels (32-34). Numerous studies (35-37)
have demonstrated that high expression of VEGF is observed in
almost all malignant tumors, however normal tissues do not express
VEGF or express only minimal levels. In addition, previous studies
have demonstrated an association between the expression of VEGF and
gastric carcinogenesis, development and prognosis (38-40).
VEGF is able to induce the proliferation of endothelial cells,
enhance vascular permeability and alter the state of the
extracellular matrix in addition to the expression of genes
(13). Studies have indicated that
wild-type p53 is able to inhibit VEGF expression, tumor growth and
metastasis (13,41,42).
Mutant p53 increases the expression of VEGF, thereby promoting
tumor angiogenesis, growth and metastasis (43). p53 is able to transactivate
pro-apoptotic and cell cycle arresting genes (44). The p53 downstream gene, p21, is a
universal inhibitor of cyclin-dependent kinases (CDK), and belongs
to the CIP/KIP family of CDK inhibitors (45). Furthermore, p21 possess additional
functions, such as inducing apoptosis or enhancing the apoptotic
response to chemotherapeutic agents (46-48).
In addition, p21 is a inhibitor of VEGF and angiogenesis (16). In the current study, compared with
the si-VEGF plasmid group, the p53 plasmid group exhibited
downregulation of VEGF expression, indicating that p53 has a
suppressive effect on VEGF, which may be associated with the high
expression of p21 (Fig. 1C and D).
These results demonstrate that p53 has a role in the regulation of
VEGF expression, and thus part of its antitumor effect may be
achieved by inhibiting the expression of VEGF.
To investigate the effect and mechanism of the Pvp53
plasmid on tumor cells, the alterations in cell migration were
investigated. In a variety of tumor cells that highly express MMPs,
it has been observed that the basement membrane is degraded and
that certain growth factors involved in tumor development are
activated (31). In the current
study, the scratch-wound assay indicated that Pvp53 significantly
inhibited the migration of MDA-MB-231 cells. Compared with the
si-VEGF and p53 single gene transfection group, the Pvp53
co-expression group markedly affected the expression of MMP-2 and
MMP-9 (Fig. 2). These results
indicate that p53 and VEGF are able to regulate the expression of
MMPs. In addition, Pvp53 is able to inhibit the migration of
MDA-MB-231 cells by inhibiting the expression of MMP-2 and MMP-9,
with the expression of MMPs being inversely proportional to the
expression of p53, which was directly proportional to VEGF.
In conclusion, compared with the single gene
approaches, the Pvp53 co-expression plasmid exhibited an enhanced
inhibitory effect on the proliferation and migration of MDA-MB-231
cells, and possessed greater pro-apoptotic ability.
Acknowledgments
The current study was supported by a Research Fund
for the Scientific and Technological Development Plan Project in
Jilin Province (grant no. 20150414025GH) and the Scientific and
Technological Research Planning Project of Education in Jilin
Province [grant no. (2014)B030].
References
|
1
|
DeNardo DG and Coussens LM: Inflammation
and breast cancer. Balancing immune response: Crosstalk between
adaptive and innate immune cells during breast cancer progression.
Breast Cancer Res. 9:2122007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Khan TM, Leong JP, Ming LC and Khan AH:
Association of knowledge and cultural perceptions of Malaysian
women with delay in diagnosis and treatment of breast cancer: A
systematic review. Asian Pac J Cancer Prev. 16:5349–5357. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ramdzan ZM and Nepveu A: CUX1, a
haploinsufficient tumour suppressor gene overexpressed in advanced
cancers. Nat Rev Cancer. 14:673–682. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang K, Zhang Q, Li D, Ching K, Zhang C,
Zheng X, Ozeck M, Shi S, Li X, Wang H, Rejto P, et al: PEST domain
mutations in Notch receptors comprise an oncogenic driver segment
in triple-negative breast cancer sensitive to a γ-secretase
inhibitor. Clin Cancer Res. 21:1487–1496. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gurpinar E and Vousden KH: Hitting
cancers' weak spots: Vulnerabilities imposed by p53 mutation.
Trends Cell Biol. 25:486–495. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Desmoulière A, Guyot C and Gabbiani G: The
stroma reaction myofibroblast: A key player in the control of tumor
cell behavior. Int J Dev Biol. 48:509–517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Siemann DW and Horsman MR: Modulation of
the tumor vasculature and oxygenation to improve therapy. Pharmacol
Ther. 153:107–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sia D, Alsinet C, Newell P and Villanueva
A: VEGF signaling in cancer treatment. Curr Pharm Des.
20:2834–2842. 2014. View Article : Google Scholar
|
|
10
|
Ji K, Wang B, Shao YT, Zhang L, Liu YN,
Shao C, Li XJ, Li X, Hu JD, Zhao XJ, et al: Synergistic suppression
of prostatic cancer cells by coexpression of both murine double
minute 2 small interfering RNA and wild-type p53 gene in vitro and
in vivo. J Pharmacol Exp Ther. 338:173–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Gao L, Li Y, Lin G, Shao Y, Ji K,
Yu H, Hu J, Kalvakolanu DV, Kopecko DJ, et al: Effects of
plasmid-based Stat3-specific short hairpin RNA and GRIM-19 on PC-3M
tumor cell growth. Clin Cancer Res. 14:559–568. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moreira IS, Fernandes PA and Ramos MJ:
Vascular endothelial growth factor (VEGF) inhibition--a critical
review. Anticancer Agents Med Chem. 7:223–245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Poon RT, Fan ST and Wong J: Clinical
implications of circulating angiogenic factors in cancer patients.
J Clin Oncol. 19:1207–1225. 2001.PubMed/NCBI
|
|
14
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mukhopadhyay D, Tsiokas L and Sukhatme VP:
Wild-type p53 and v-Src exert opposing influences on human vascular
endothelial growth factor gene expression. Cancer Res.
55:6161–6165. 1995.PubMed/NCBI
|
|
17
|
Agani F, Kirsch DG, Friedman SL, Kastan MB
and Semenza GL: p53 does not repress hypoxia-induced transcription
of the vascular endothelial growth factor gene. Cancer Res.
57:4474–4477. 1997.PubMed/NCBI
|
|
18
|
Farhang Ghahremani M, Goossens S and Haigh
JJ: The p53 family and VEGF regulation: “It's complicated”. Cell
Cycle. 12:1331–1332. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oren M and Rotter V: Mutant p53
gain-of-function in cancer. Cold Spring Harb Perspect Biol.
2:a0011072010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brachova P, Mueting SR, Devor EJ and
Leslie KK: Oncomorphic TP53 Mutations in Gynecologic Cancers Lose
the Normal Protein:Protein Interactions with the microRNA
Microprocessing Complex. J Cancer Ther. 5:506–516. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zietz C, Rössle M, Haas C, Sendelhofert A,
Hirschmann A, Stürzl M and Löhrs U: MDM-2 oncoprotein
overexpression, p53 gene mutation, and VEGF up-regulation in
angiosarcomas. Am J Pathol. 153:1425–1433. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pal S, Datta K and Mukhopadhyay D: Central
role of p53 on regulation of vascular permeability factor/vascular
endothelial growth factor (VPF/VEGF) expression in mammary
carcinoma. Cancer Res. 61:6952–6957. 2001.PubMed/NCBI
|
|
23
|
Abusail MS, Dirweesh AMA, Salih RAA and
Gadelkarim AH: Expression of EGFR and p53 in head and neck tumors
among Sudanese patients. Asian Pac J Cancer Prev. 14:6415–6418.
2013. View Article : Google Scholar
|
|
24
|
Xu CT, Zheng F, Dai X, Du JD, Liu HR, Zhao
L and Li WM: Association between TP53 Arg72Pro polymorphism and
hepatocellular carcinoma risk: A meta-analysis. Asian Pac J Cancer
Prev. 13:4305–4309. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goh AM, Coffill CR and Lane DP: The role
of mutant p53 in human cancer. J Pathol. 223:116–126. 2011.
View Article : Google Scholar
|
|
26
|
Muller PAJ, Vousden KH and Norman JC: p53
and its mutants in tumor cell migration and invasion. J Cell Biol.
192:209–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Brosh R and Rotter V: When mutants gain
new powers: News from the mutant p53 field. Nat Rev Cancer.
9:701–713. 2009.PubMed/NCBI
|
|
28
|
Yi YW, Kang HJ, Kim HJ, Kong Y, Brown ML
and Bae I: Targeting mutant p53 by a SIRT1 activator YK-3-237
inhibits the proliferation of triple-negative breast cancer cells.
Oncotarget. 4:984–994. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Khoo KH, Verma CS and Lane DP: Drugging
the p53 pathway: Understanding the route to clinical efficacy. Nat
Rev Drug Discov. 13:217–236. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song Y, Li X, Li Y, Li N, Shi X, Ding H,
Zhang Y, Li X, Liu G and Wang Z: Non-esterified fatty acids
activate the ROS-p38-p53/Nrf2 signaling pathway to induce bovine
hepa-tocyte apoptosis in vitro. Apoptosis. 19:984–997. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu J, Zhang L, Hwang PM, Kinzler KW and
Vogelstein B: PUMA induces the rapid apoptosis of colorectal cancer
cells. Mol Cell. 7:673–682. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moens S, Goveia J, Stapor PC, Cantelmo AR
and Carmeliet P: The multifaceted activity of VEGF in angiogenesis
- Implications for therapy responses. Cytokine Growth Factor Rev.
25:473–482. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stamati K, Priestley JV, Mudera V and
Cheema U: Laminin promotes vascular network formation in 3D in
vitro collagen scaffolds by regulating VEGF uptake. Exp Cell Res.
327:68–77. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cannon JE, Upton PD, Smith JC and Morrell
NW: Intersegmental vessel formation in zebrafish: Requirement for
VEGF but not BMP signalling revealed by selective and non-selective
BMP antagonists. Br J Pharmacol. 161:140–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen J, Tang D, Wang S, Li QG, Zhang JR,
Li P, Lu Q, Niu G, Gao J, Ye NY and Wang DR: High expressions of
galectin-1 and VEGF are associated with poor prognosis in gastric
cancer patients. Tumour Biol. 35:2513–2519. 2014. View Article : Google Scholar
|
|
36
|
Zhao J, Chen L, Shu B, Tang J, Zhang L,
Xie J, Qi S and Xu Y: Granulocyte/macrophage colony-stimulating
factor influences angiogenesis by regulating the coordinated
expression of VEGF and the Ang/Tie system. PLoS One. 9:e926912014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao D, Pan C, Sun J, Gilbert C,
Drews-Elger K, Azzam DJ, Picon-Ruiz M, Kim M, Ullmer W, El-Ashry D,
et al: VEGF drives cancer-initiating stem cells through
VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene.
34:3107–3119. 2015. View Article : Google Scholar
|
|
38
|
Tanaka T, Ishiguro H, Kuwabara Y, Kimura
M, Mitsui A, Katada T, Shiozaki M, Naganawa Y, Fujii Y and Takeyama
H: Vascular endothelial growth factor C (VEGF-C) in esophageal
cancer correlates with lymph node metastasis and poor patient
prognosis. J Exp Clin Cancer Res. 29:832010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu H, Yang Y, Xiao J, Lv Y, Liu Y, Yang H
and Zhao L: COX-2-mediated regulation of VEGF-C in association with
lymphangiogenesis and lymph node metastasis in lung cancer. Anat
Rec (Hoboken). 293:1838–1846. 2010. View Article : Google Scholar
|
|
40
|
Wang TB, Wang J, Wei XQ, Wei B and Dong
WG: Serum vascular endothelial growth factor-C combined with
multi-detector CT in the preoperative diagnosis of lymph node
metastasis of gastric cancer. Asia Pac J Clin Oncol. 8:180–186.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gu J, Tang Y, Liu Y, Guo H, Wang Y, Cai L,
Li Y and Wang B: Murine double minute 2 siRNA and wild-type p53
gene therapy enhances sensitivity of the SKOV3/DDP ovarian cancer
cell line to cisplatin chemotherapy in vitro and in vivo. Cancer
Lett. 343:200–209. 2014. View Article : Google Scholar
|
|
42
|
Chakraborty S, Adhikary A, Mazumdar M,
Mukherjee S, Bhattacharjee P, Guha D, Choudhuri T, Chattopadhyay S,
Sa G, Sen A, et al: Capsaicin-induced activation of p53-SMAR1
auto-regulatory loop down-regulates VEGF in non-small cell lung
cancer to restrain angiogenesis. PLoS One. 9:e997432014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ravi R, Mookerjee B, Bhujwalla ZM, Sutter
CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenaz GL and Bedi A:
Regulation of tumor angiogenesis by p53-induced degradation of
hypoxia-inducible factor 1alpha. Genes Dev. 14:34–44.
2000.PubMed/NCBI
|
|
44
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sherr CJ and Roberts JM: Inhibitors of
mammalian G1 cyclin-dependent kinases. Genes Dev. 9:1149–1163.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qiao L, McKinstry R, Gupta S, Gilfor D,
Windle JJ, Hylemon PB, Grant S, Fisher PB and Dent P: Cyclin kinase
inhibitor p21 potentiates bile acid-induced apoptosis in
hepatocytes that is dependent on p53. Hepatology. 36:39–48. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kondo S, Barna BP, Kondo Y, Tanaka Y,
Casey G, Liu J, Morimura T, Kaakaji R, Peterson JW, Werbel B and
Barnett GH: WAF1/CIP1 increases the susceptibility of p53
non-functional malignant glioma cells to cisplatin-induced
apoptosis. Oncogene. 13:1279–1285. 1996.PubMed/NCBI
|
|
48
|
Coqueret O: New roles for p21 and p27
cell-cycle inhibitors: A function for each cell compartment? Trends
Cell Biol. 13:65–70. 2003. View Article : Google Scholar : PubMed/NCBI
|