Introduction
Ras and Akt are signaling proteins that mediate
fundamental aspects of normal growth and development in various
organs. However, when the Ras and Akt pathways become extremely
active, malignant transformation of normal tissue may occur
(1–3). For instance, Holland et al
(4) demonstrated that the combined
activation of Ras and Akt induces glioblastoma formation in mice.
Therefore, Ras and Akt signaling pathways have emerged as
attractive targets for the treatment of numerious types of tumors
including glioblastoma (3,5,6).
Glioblastoma is the most common type of brain tumor,
it is highly malignant and exhibits aggressive invasive growth.
Common genetic abnormalities, including Ras and phosphatase and
tensin homolog (PTEN) deleted on chromosome 10, in glioblastoma,
are associated with the aberrant activation or suppression of cell
signal transduction pathways and resistance to radiation and
chemotherapy (2,3). Notably, phosphodiesterase 4 (PDE4) is
widely expressed in brain tumors and promotes their growth
(7). Brain region-specific
differences in cyclic adenosine monophosphate (cAMP) levels have
been reported to account for the pattern of gliomagenesis, while
low levels of cAMP promote glioma formation in neurofibromatosis-1
genetically engineered mouse models (8,9).
PDE4 inhibitor has been shown to suppress glioblastoma growth in
vitro and in xenograft models (7,10,11).
cAMP is considered to be a ubiquitous regulator of
inflammatory and immunological reactions (12–14),
modulating several physiological processes via the activation of
protein kinase A (PKA) and the exchange protein directly activated
by cAMP (Epac). PKA and Epac are molecular players that are
downstream of cAMP (14,15). PDE inhibitors have been reported to
increase cellular cAMP levels and, in turn, prevent Akt activity in
rat glial and human osteosarcoma cells (15,16).
In the present study, the effects of cAMP on Ras and
Akt signaling pathways were examined in U87MG human malignant
glioma cells. cAMP was demonstrated to suppress cell growth and
p44/42 mitogen-activated protein kinase (MAPK) activity, which is
downstream of the Ras signaling pathway, but not Akt activity in a
PKA- and Epac-dependent manner. Findings of this study provide a
mechanistic basis for developing therapeutic approaches for
treating or preventing brain tumors.
Materials and methods
Chemicals
Forskolin and Dulbecco’s modified Eagle’s medium
(DMEM) were obtained from Wako Pure Chemical Industries, Ltd.
(Osaka, Japan). 8-Bromoadenosine 3′,5′-cAMP (8-Br-cAMP) and
8-(4-chlorophenylthio)-2′-O-methyladenosine cAMP
(8-CPT-cAMP, Epac activator) were obtained from Biaffin GmbH &
Co. KG (Kassel, Germany). Anti-phospho-specific
vasodilator-stimulated phosphoprotein (VASP) (Ser157) and H89 were
obtained from Calbiochem (La Jolla, CA, USA). Fetal bovine serum
(FBS) was obtained from Invitrogen Corporation (Carlsbad, CA, USA).
Anti-phospho-specific Akt (Ser473), anti-phospho-specific p44/42
MAPK (Thr202/Tyr204), anti-β-actin, horseradish peroxidase
(HRP)-linked anti-rabbit IgG, and anti-mouse IgG were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Cell culture
U87MG human glioblastoma cells were provided by Dr
Nakata (Kanazawa University, Kanazawa, Japan). Cells were
maintained in DMEM containing 10% FBS at 37°C in a 5%
CO2 incubator.
Western blot analysis
Western blot analysis was performed as described
previously (14,17).
Cell proliferation assay
Cell proliferation was analyzed using the Cell
Counting kit 8 (Wako Pure Chemical Industries, Ltd., Tokyo, Japan).
U87MG human malignant glioma cells were seeded in 96-well plates at
a density of 1×103 cells/well. Following a 24-h
incubation, the cells were treated with forskolin or 8-Br-cAMP for
24 h. The cells were then incubated with 10 μl WST-8 for 2
h. Absorbance of the colored formazan product produced by
mitochondrial dehydrogenases in metabolically active cells was
recorded at 450 nm as the background value. Cell proliferation was
expressed as a percentage of absorbance obtained in treated wells
compared with untreated (control) wells.
Statistical analysis
Data were presented as the means ± standard error of
the mean (SEM) from at least three independent experiments.
Statistical analysis was performed by analysis of variance (ANOVA),
followed by Dunnett’s test. P<0.05 or P<0.01 were considered
to indicate a statistically significant difference.
Results
Forskolin downregulates cell growth
Initially, we examined the effects of forskolin, an
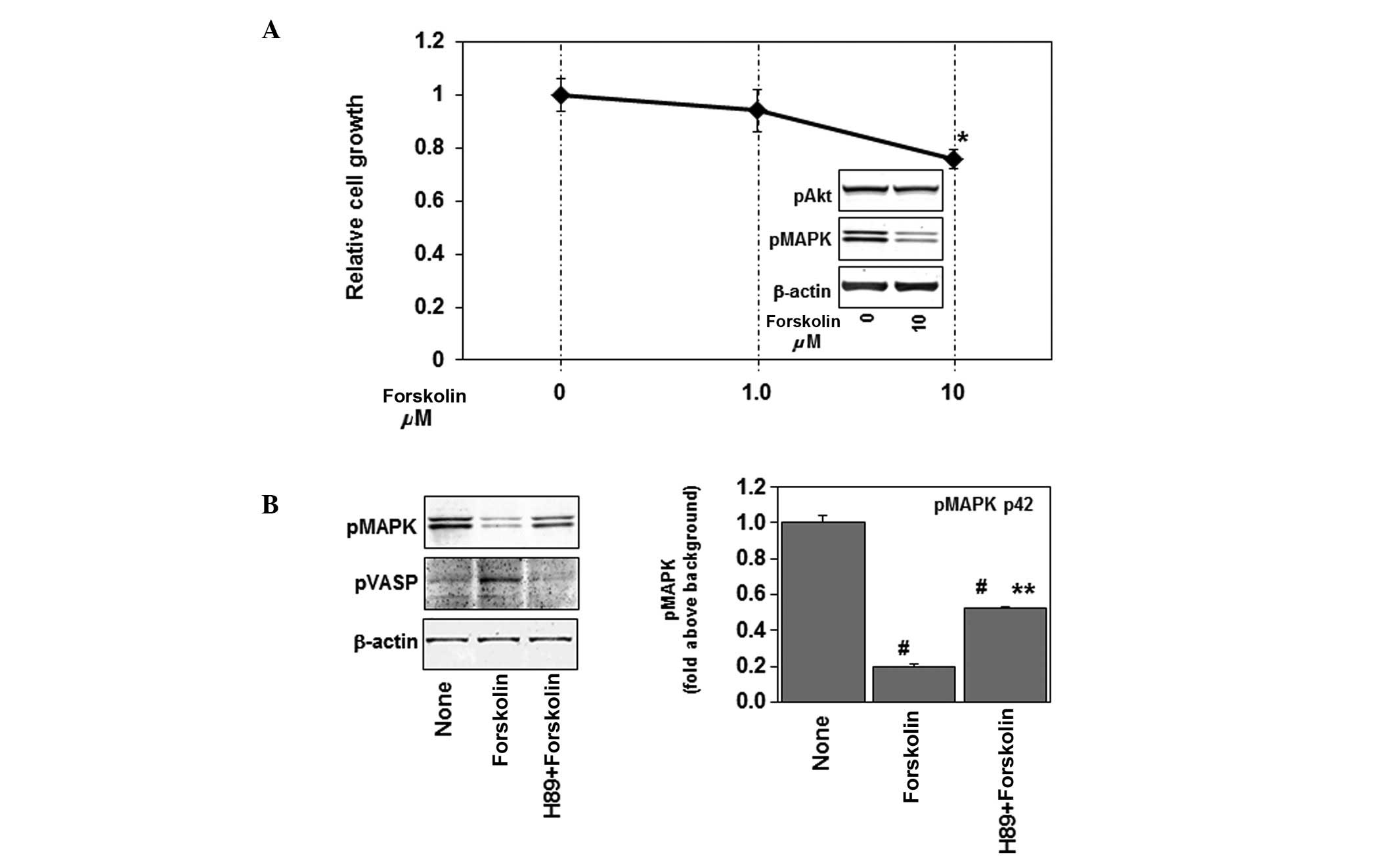
activator of adenylate cyclase, on cell growth. As shown in
Fig. 1A, 10 μM of
forskolin, but not 1 μM, slightly but markedly inhibited
cell growth (∼20%) in U87MG human glioblastoma cells.
Concomitantly, forskolin increased the expression of
phospho-vasodilator-stimulated phosphoprotein (phospho-VASP),
indicating an elevation of intracellular cAMP levels (Fig. 1B) (18,19).
Forskolin inhibits p44/42 MAPK but not
Akt activity
Ras signals to Raf, mitogen-induced extracellular
kinase (MEK), and MAPKs, especially p44/42 MAPK. The effects of
forskolin on p44/42 MAPK and Akt were examined. Treatment with 10
μM of forskolin inhibited phosphorylation of p44/42 MAPK but
not Akt in U87MG (Fig. 1A, inset
figure), indicating that the Ras-p44/42 MAPK signaling pathway
is inhibited by forskolin. PKA and Epac are molecular players
downstream of cAMP. As shown in Fig.
1B, the effects of the PKA inhibition completely by H89 blocked
the forskolin-induced phosphorylation of VASP, while partially
preventing the forskolin-induced inhibition of phosphorylation of
p44/42 MAPK. These results indicate that PKA is partially involved
in the effects of forskolin on the Ras-p44/42 MAPK signaling
pathway.
Cell-penetrating cAMP analog inhibits
cell growth and p44/42 MAPK, but not Akt activity
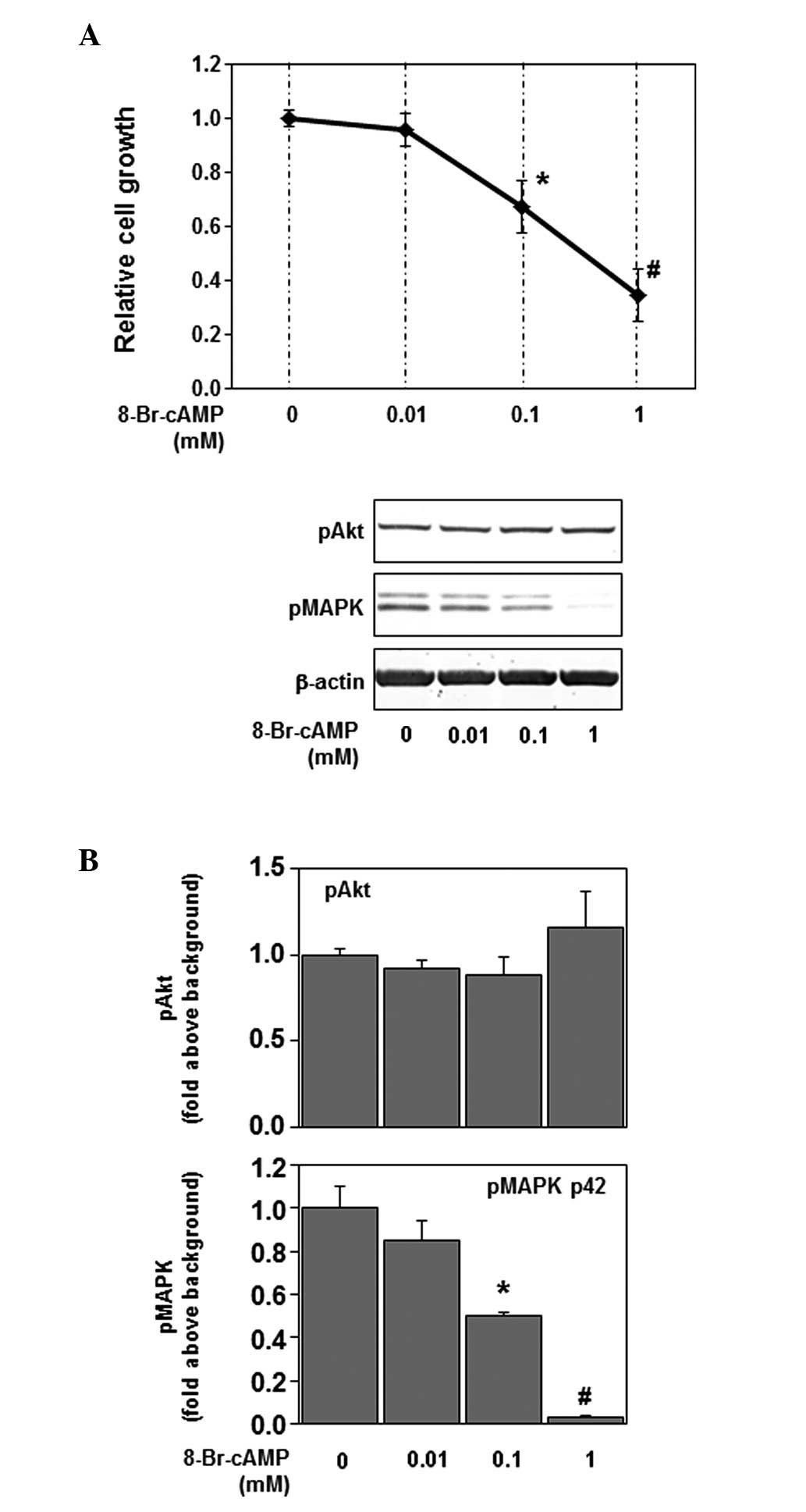
To determine whether intracellular cAMP is involved
in the inhibitory effect of forskolin, we examined the effects of
the non-hydrolyzable cAMP analog 8-Br-cAMP on cell growth and
phosphorylation of p44/42 MAPK and Akt. In U87MG, in which PDE4
expression is upregulated (20),
the non-hydrolyzable cAMP analog 8-Br-cAMP might be more effective
compared with forskolin stimulation. Therefore, 1 mM of 8-Br-cAMP
considerably suppressed cell growth and phosphorylation of p44/42
MAPK (Fig. 2A and B). As shown in
Fig. 2, 8-Br-cAMP inhibited cell
growth and phosphorylation of p44/42 MAPK in a dose-dependent
manner, mimicking the forskolin effect. However, 8-Br-cAMP did not
affect the phosphorylation of Akt (Fig. 2B). These results indicate that
intracellular cAMP is involved in the effects of forskolin on cell
growth and the Ras-p44/42 MAPK signaling pathway.
cA MP-induced p44/42 MAPK deactivation is
Epac-dependent
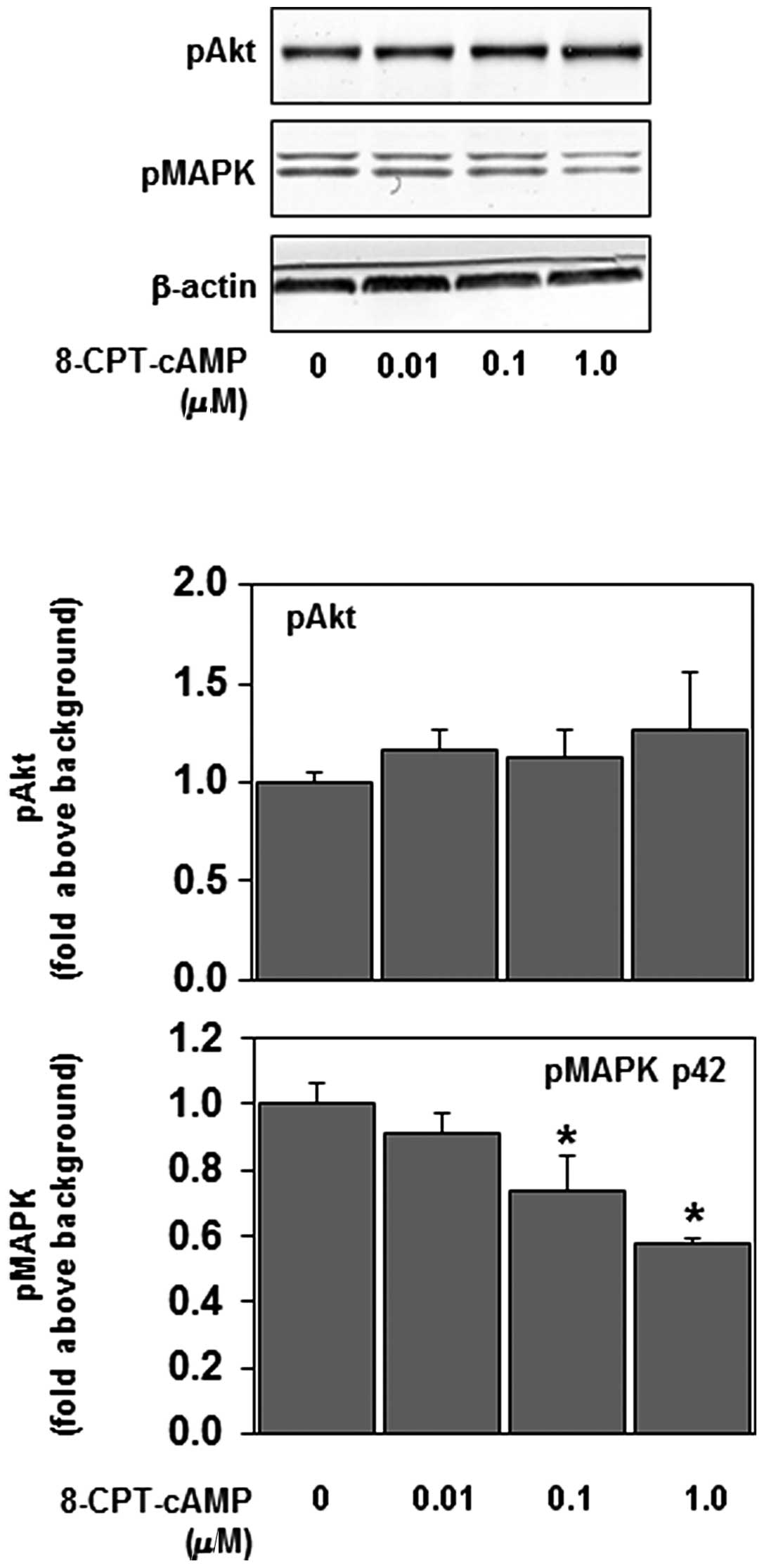
To examine the potential signaling role of Epac, the
ability of the Epac-selective agonist 8-CPT-cAMP to inhibit
phosphorylation of p44/42 MAPK was examined. 8-CPT-cAMP is a
non-hydrolyzable cAMP analog that activates Epac with greater
potency compared with PKA (21).
Recent studies have shown that concentrations of 8-CPT-cAMP as low
as 5 μM effectively activate Epac in studies using cultured
cells (14,15). In our experiments, 8-CPT-cAMP
inhibited the phosphorylation of p44/42 MAPK, but not that of Akt,
in a dose-dependent manner (Fig.
3). These results suggest that Epac is involved in the effect
of intracellular cAMP on the Ras-p44/42 MAPK signaling pathway.
Discussion
Ras-p44/42 MAPK signaling is a crucial signaling
pathway in the regulation of growth and development in normal
tissues and tumors (1–3). Increased activity of Ras-p44/42 MAPK
is found in several human glioblastoma (4,22).
In the present study, the upregulation of intracellular cAMP has
been demonstrated to inhibit p44/42 MAPK, but not Akt activity, and
proliferation in human glioblastoma cells in vitro.
Elevated levels of cAMP in the cell lead to the
activation of different cAMP targets, including PKA and Epac. The
two families of cAMP effectors provide a mechanism for the precise
and integrated control of cAMP signaling pathways in a spatial and
temporal manner. PKA and Epac may act independently, converge
synergistically or oppose each other in regulating a specific cell
function (18–21,23,24).
In this study, PKA and Epac have been demonstrated to be
responsible for cAMP-dependent p44/42 MAPK dephosphorylation
(Figs. 1 and 3), confirming that cAMP prevents cell
growth through PKA and Epac activation in U172 and U87MG human
glioblastoma cells (20).
In a recent study, cAMP was shown to inhibit the
activity of Akt through Epac-PTEN pathway activation in glial and
osteosarcoma cells (15,16). In this study, however, forskolin or
cAMP analogs failed to inhibit Akt activation in U87MG glioblastoma
cells (Figs. 1–3). One of the numerous functions of PTEN
is the regulation of Akt activity, thus, loss of PTEN function
induces Akt hyperactivation (3).
Since PTEN is not expressed in U87MG human glioblastoma cells
(15), cAMP is likely to fail to
inhibit Akt activation in PTEN-depleted U87MG glioblastoma
cells.
In their study, Holland et al (4) demonstrated that although activated
Ras or Akt alone is insufficient to induce glioblastoma formation,
their combination induces glioblastoma formation in mice.
Therefore, Ras and Akt signaling pathways have emerged as
attractive targets for the treatment of glioblastoma (2–5). PKA
and Epac activators as well as PDE4 inhibitors seem to be extremely
effective for the treatment of human glioblastoma.
In conclusion, we have shown that cAMP inhibits
p44/42 MAPK activity and proliferation in PTEN-depleted human
glioblastoma cells in vitro through PKA and Epac activation.
However, details regarding the mechanism underlying the PKA and
Epac suppression of p44/42 MAPK have yet to be elucidated. Further
studies are required to determine the mechanism of PKA and Epac
activation-dependent MAPK inhibition in glioblastoma cells.
Acknowledgements
This study was funded in part by
Grants-in-Aid for Science and Culture from the Ministry of
Education, Culture, Sports, Science and Technology of Japan.
References
|
1
|
Parsa AT and Holland EC: Cooperative
translational control of gene expression by Ras and Akt in cancer.
Trends Mol Med. 10:607–613. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McCubrey JA, Steelman LS, Abrams SL, Lee
JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA,
D’Assoro AB, Salisbury JL, Mazzarino MC, Stivala F and Libra M:
Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant
transformation and drug resistance. Adv Enzyme Regul. 46:249–279.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hers I, Vincent EE and Tavaré JM: Akt
signalling in health and disease. Cell Signal. 23:1515–1527. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Holland EC, Celestino J, Dai C, Schaefer
L, Sawaya RE and Fuller GN: Combined activation of Ras and Akt in
neural progenitors induces glioblastoma formation in mice. Nat
Genet. 25:55–57. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Höland K, Salm F and Arcaro A: The
phosphoinositide 3-kinase signaling pathway as a therapeutic target
in grade IV brain tumors. Curr Cancer Drug Targets. 11:894–918.
2011.PubMed/NCBI
|
|
6
|
McCubrey JA, Steelman LS, Abrams SL,
Bertrand FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M,
Tafuri A, Lunghi P, Bonati A and Martelli AM: Targeting survival
cascades induced by activation of Ras/Raf/MEK/ERK,
PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia
therapy. Leukemia. 22:708–722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldhoff P, Warrington NM, Limbrick DD Jr,
Hope A, Woerner BM, Jackson E, Perry A, Piwnica-Worms D and Rubin
JB: Targeted inhibition of cyclic AMP phosphodiesterase-4 promotes
brain tumor regression. Clin Cancer Res. 14:7717–7725. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brown JA, Gianino SM and Gutmann DH:
Defective cAMP generation underlies the sensitivity of CNS neurons
to neurofibromatosis-1 heterozygosity. J Neurosci. 30:5579–5589.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warrington NM, Gianino SM, Jackson E,
Goldhoff P, Garbow JR, Piwnica-Worms D, Gutmann DH and Rubin JB:
Cyclic AMP suppression is sufficient to induce gliomagenesis in a
mouse model of neurofibromatosis-1. Cancer Res. 70:5717–5727. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang L, Jackson E, Woerner BM, Perry A,
Piwnica-Worms D and Rubin JB: Blocking CXCR4-mediated cyclic AMP
suppression inhibits brain tumor growth in vivo. Cancer Res.
67:651–658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sengupta R, Sun T, Warrington NM and Rubin
JB: Treating brain tumors with PDE4 inhibitors. Trends Pharmacol
Sci. 32:337–344. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moore AR and Willoughby DA: The role of
cAMP regulation in controlling inflammation. Clin Exp Immunol.
101:387–389. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moon EY, Lee JH, Lee JW, Song JH and Pyo
S: ROS/Epac1-mediated Rap1/NF-kappaB activation is required for the
expression of BAFF in Raw264.7 murine macrophages. Cell Signal.
23:1479–1488. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saito T, Sugimoto N, Ohta K, Shimizu T,
Ohtani K, Nakayama Y, Nakamura T, Hitomi Y, Nakamura H, Koizumi S
and Yachie A: Phosphodiesterase inhibitors suppress
Lactobacillus casei cell-wall-induced NF-κB and MAPK
activations and cell proliferation through protein kinase A - or
exchange protein activated by cAMP-dependent signal pathway. Sci
World J. 2012:7485722012.PubMed/NCBI
|
|
15
|
Sugimoto N, Miwa S, Ohno-Shosaku T,
Tsuchiya H, Hitomi Y, Nakamura H, Tomita K, Yachie A and Koizumi S:
Activation of tumor suppressor protein PTEN and induction of
apoptosis are involved in cAMP-mediated inhibition of cell number
in B92 glial cells. Neurosci Lett. 497:55–59. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miwa S, Sugimoto N, Shirai T, Hayashi K,
Nishida H, Ohnari I, Takeuchi A, Yachie A and Tsuchiya H: Caffeine
activates tumor suppressor PTEN in sarcoma cells. Int J Oncol.
39:465–472. 2011.PubMed/NCBI
|
|
17
|
Sugimoto N, Shido O, Matsuzaki K,
Ohno-Shosaku T, Hitomi Y, Tanaka M, Sawaki T, Fujita Y, Kawanami T,
Masaki Y, Okazaki T, Nakamura H, Koizumi S, Yachie A and Umehara H:
Cellular heat acclimation regulates cell growth, cell morphology,
mitogen-activated protein kinase activation, and expression of
aquaporins in mouse fibroblast cells. Cell Physiol Biochem.
30:450–457. 2012. View Article : Google Scholar
|
|
18
|
Comerford KM, Lawrence DW, Synnestvedt K,
Levi BP and Colgan SP: Role of vasodilator-stimulated
phosphoprotein in PKA-induced changes in endothelial junctional
permeability. FASEB J. 16:583–585. 2002.PubMed/NCBI
|
|
19
|
Loza MJ, Foster S, Peters SP and Penn RB:
Beta-agonists modulate T-cell functions via direct actions on type
1 and type 2 cells. Blood. 107:2052–2060. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moon EY, Lee GH, Lee MS, Kim HM and Lee
JW: Phosphodiesterase inhibitors control A172 human glioblastoma
cell death through cAMP-mediated activation of protein kinase A and
Epac1/Rap1 pathways. Life Sci. 90:373–380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Enserink JM, Christensen AE, de Rooij J,
van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL and
Bos JL: A novel Epac-specific cAMP analogue demonstrates
independent regulation of Rap1 and ERK. Nat Cell Biol. 4:901–906.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alloussi SH, Alkassar M, Urbschat S, Graf
N and Gärtner B: All reovirus subtypes show oncolytic potential in
primary cells of human high-grade glioma. Oncol Rep. 26:645–649.
2011.PubMed/NCBI
|
|
23
|
Tamma G, Lasorsa D, Ranieri M,
Mastrofrancesco L, Valenti G and Svelto M: Integrin signaling
modulates AQP2 trafficking via Arg-Gly-Asp (RGD) motif. Cell
Physiol Biochem. 27:739–748. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gloerich M and Bos JL: Epac: defining a
new mechanism for cAMP action. Annu Rev Pharmacol Toxicol.
50:355–375. 2010. View Article : Google Scholar : PubMed/NCBI
|