Introduction
Philadelphia chromosome-negative myeloproliferative
neoplasms (MPNs), including polycythemia vera (PV), essential
thrombocytosis (ET) and primary myelofibrosis (PMF), are a group of
phenotypically related clonal hematopoietic diseases mainly
characterized by the overproduction of blood cells, apart from
cytopenia, particularly in PMF (1).
During the prolonged clinical course of MPNs, thrombosis, infection
due to leukocytopenia and transformation to acute myeloid leukemia
(AML) are the main causes of death (2–4).
The acquired Janus kinase 2 (JAK2) V617F mutation is
detected in >95% of patients with PV and 50–60% of patients with
ET or PMF (5). In JAK2V617F
mutation-negative MPNs, other gene mutations, such as those of the
myeloproliferative leukemia protein (MPL) and calreticulin (CALR)
genes, are often detected (6–9). In particular, it was reported that ET
and PMF patients with CALR mutations have a better prognosis
compared with patients with JAK2 or MPL mutations (6,7,9). However, the determination of gene
mutations is costly and not widely used. Apart from the JAK2V617F
mutation, gene mutation tests are not covered by insurance in
Japan.
The aim of our study was to determine the prognostic
factors obtainable from commonly used information, such as patient
characteristics, blood cell count, bone marrow examination and
imaging modalities.
Patients and methods
Patients
We reviewed 71 patients with Philadelphia
chromosome-negative MPNs diagnosed at the Hakodate Municipal
Hospital (Hakodate, Japan) between April, 2001 and April, 2014. The
diagnosis was based on the World Health Organization criteria
(10). Patients diagnosed at our
department but followed up in other hospitals were excluded from
the analysis. This study was approved by the Hakodate Municipal
Hospital Institutional Review Board. All the patients provided
written informed consent according to the Declaration of
Helsinki.
Definition of risk scoring
Abnormal values of white blood cell (WBC) count,
hemoglobin (Hb) concentration, hematocrit (Hct) and platelet (PLT)
count were defined as higher than the upper limits of normal values
(ULN), or lower than the lower limits of normal values (LLN).
Abnormal erythrocytic lineages were reflected in abnormal Hb and/or
Hct values. In addition to the number of abnormal blood cell
lineages, one point each was assigned for age >65 years and
splenomegaly. Each MPN patient was subsequently assigned a score of
1–6.
Cytogenetics and JAK2V617F
mutation
For the detection of the JAK2V617F mutation,
allele-specific polymerase chain reaction analysis was performed as
previously reported (11). We were
unable to investigate other JAK2 mutations, as this is not covered
by insurance in Japan. In our institute, detection of JAK2V617F
mutation became accessible in 2013.
Statistical analysis
The baseline characteristics of the patients were
described using median and range, and compared using the median
test, χ2 analysis and the Fisher's exact test, as
appropriate. Overall survival (OS) was defined as the time between
the first patient contact and death or the time of the last
follow-up. The endpoint of event-free survival (EFS) was defined as
transformation to AML, new appearance or aggravation of
splenomegaly, addition of an abnormal karyotype, new complication
of MF, increasing blood cell counts that required chemotherapy,
thrombosis, or patient death from any cause. OS and EFS were
evaluated by the Kaplan-Meier method and Mantel-Cox log-rank tests.
For univariate and multivariate analysis, logistic regression was
used to investigate the association of baseline characteristics
with OS and EFS. Statistical analysis was performed with StatMate V
software (ATMS Co., Ltd., Tokyo, Japan). P<0.05 was considered
to indicate statistically significant differences.
Results
Initial characteristics and clinical
course
Table I provides a
comparative presentation of the clinical and laboratory
characteristics of each type of MPN [PV, n=17; ET, n=34; PMF, n=16;
and MPN, unclassifiable (u), n=4]. Significant differences were
observed in Hb, Hct, PLT count and splenomegaly, but not in age,
gender distribution, WBC count, abnormal karyotype, or frequency of
the JAKV617F mutation.
 | Table I.Baseline characteristics of all MPN
patients (n=71). |
Table I.
Baseline characteristics of all MPN
patients (n=71).
|
| Types of MPN |
|
|---|
|
|
|
|
|---|
| Characteristics | PV (n=17) | ET (n=34) | PMF (n=16) | MPN, u (n=4) | P-value |
|---|
| Median age, years
(range) |
62 (44–76) |
69 (16–88) |
70 (30–81) |
75 (46–87) | 0.40028 |
| Gender, male, n
(%) | 11 (65) | 14 (41) | 11 (69) | 2
(50) | 0.23711 |
| Abnormal WBC count, n
(%) | 8
(47) | 13 (38) | 10 (62) | 4
(100) | 0.296929 |
|
>ULN | 8
(47) | 12 (35) | 9
(56) | 4
(100) |
|
|
<LLN | 0 (0) | 1 (3) | 1 (6) | 0 (0) |
|
| Abnormal Hb level, n
(%) | 15 (88) | 8
(24) | 10 (62) | 1
(25) | 1.51E-09 |
|
>ULN | 15 (88) | 1 (3) | 1
(6) | 1
(25) |
|
|
<LLN | 0 (0) | 7
(21) | 9
(56) | 0 (0) |
|
| Abnormal Hct, n
(%) | 17
(100) | 14 (41) | 13 (81) | 3
(75) | 1.98E-08 |
|
>ULN | 17
(100) | 11 (32) | 3
(19) | 3
(75) |
|
|
<LLN | 0 (0) | 3 (9) | 10 (63) | 0 (0) |
|
| Abnormal PLT count, n
(%) | 9
(53) | 34
(100) | 13 (81) | 3
(75) | 0.00013 |
|
>ULN | 9
(53) | 34
(100) | 9
(56) | 3
(75) |
|
|
<LLN | 0 (0) | 0 (0) | 4
(25) | 0 (0) |
|
| Splenomegaly, n
(%) | 9
(53) | 8
(24) | 11 (53) | 2
(50) | 0.018394 |
| Abnormal karyotype, n
(%) | 3
(18) | 6
(18) | 7
(44) | 2
(50) | 0.176625 |
| JAK2V617F mutation, n
(%) |
|
|
|
|
|
|
Tested | 13 (76) | 25 (74) | 7
(44) | 2
(50) |
|
|
Mutation-positive | 8
(62) | 17 (68) | 4
(57) | 1
(50) | 0.929494 |
| Follow-up duration,
months |
|
|
|
|
|
| Median
(range) |
43
(12–100) |
38 (2–151) |
41 (4–105) |
31 (22–36) | 0.10206 |
Causes of death and events
The causes of death and endpoints of EFS are
presented in Table II. A total of 3
patients succumbed due to transformation to AML, 2 patients due to
renal failure and 1 patient each due to pneumonia, hemophagocytic
syndrome, uncontrollable bleeding due to thrombocytopenia,
malignant pleural effusion, stroke, hepatic failure due to
hepatitis B virus infection and unexpected cardiopulmonary arrest.
All these patients had multilineage abnormalities in blood counts.
The numbers of events were as follows: Increasing blood cell counts
and chemotherapy, n=10; appearance or aggravation of splenomegaly,
n=6; complications of MF, n=3; transformation to AML, n=3; and
death, n=4 (2 from renal failure and 1 each from stroke and hepatic
failure). Addition of an abnormal karyotype was observed in 1
patient concurrently with complications of MF. Thrombosis was not
observed in this analysis.
| Table II.Causes of mortality and events. |
Table II.
Causes of mortality and events.
| Causes of
mortality |
| Events |
|
|---|
| Transformation to
AML | 3 | Increasing blood
cell counts and addition of chemotherapy | 10 |
| Renal failure | 2 | Appearance or
aggravation of splenomegaly | 6 |
| Pneumonia | 1 | Complication of
myelofibrosis | 3 |
| Hemophagocytic
syndrome | 1 | Transformation to
AML | 3 |
| Uncontrollable
bleeding | 1 | Death unrelated to
MPN |
|
| due to
thrombocytopenia |
| Renal
failure | 2 |
| Malignant pleural
effusion | 1 |
Stroke | 1 |
| Stroke | 1 | Hepatic
failure (HBV-positive) | 1 |
| Hepatic failure
(HBV-positive) | 1 |
|
|
| Unexpected
cardiopulmonary arrest | 1 |
|
|
The median follow-up duration for all MPN patients
was 37 months (range, 2–151 months). No significant differences
were observed among different types of MPN (P=0.10).
Association of OS and EFS with type of
MPN
The survival curves for each type of MPN (excluding
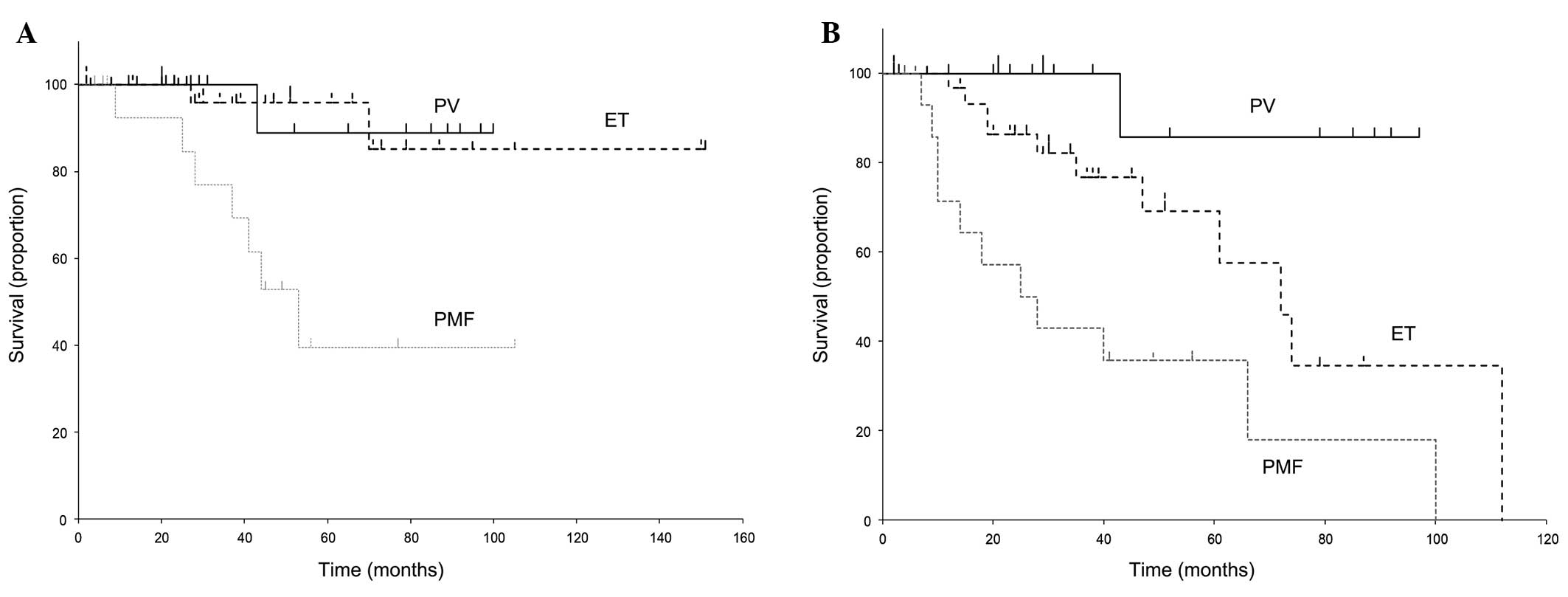
MPN, u) are presented in Fig. 1. The
median OS of PV, ET and PMF was 43 (range, 12–100), 38 (range,
2–151) and 41 (range, 4–105) months, respectively. The respective
median EFSs were 31 (12–97), 30 (2–112) and 22 (4–100) months. As
reported, the OS of PMF was worse compared with that of PV and ET
(P=0.0117). Similarly, EFS was significantly difference among the
different types of MPN (P=0.0210).
Logistic regression analysis and
prognostic factors for OS and EFS
The prognostic factors for OS and EFS on the
univariate and multivariate analysis are summarized in Tables III and IV. On univariate analysis, age (P=0.043),
gender (P=0.036), number of abnormal cell count lineages (P=0.020)
and splenomegaly (P=0.0055) were found to be significant prognostic
factors for OS. However, for EFS, only the number of abnormal cell
lineages (P=0.0065) was identified as a significant prognostic
factor. Our results suggest that abnormal karyotype and JAK2V167F
mutation did not significantly affect survival.
| Table III.Univariate logistic regression
analysis of prognostic factors for EFS and OS. |
Table III.
Univariate logistic regression
analysis of prognostic factors for EFS and OS.
| Factors | Odds ratio | P-value | 95% CI |
|---|
| OS |
|
|
|
| Age
(>65 years) | 1.07 | 0.043 | 1.00–1.15 |
| Gender
(male) | 0.18 | 0.036 |
0.036–0.90 |
| No. of
abnormal | 3.38 | 0.020 | 1.21–9.44 |
| blood
cell lineages |
|
|
|
|
Splenomegaly | 9.75 |
0.0055 | 1.95–48.8 |
|
Abnormal karyotype | 2.15 | 0.28 | 0.53–8.76 |
| EFS |
|
|
|
| Age
(>65 years) | 2.02 | 0.20 | 0.68–5.97 |
| Gender
(male) | 0.64 | 0.42 | 0.21–1.92 |
| No. of
abnormal | 2.73 |
0.0065 | 1.32–5.64 |
| blood
cell lineages |
|
|
|
|
Splenomegaly | 1.65 | 0.32 | 0.62–4.37 |
|
Abnormal karyotype | 1.23 | 0.73 | 0.38–3.93 |
|
JAK2V167F mutation | 1.39 | 0.63 | 0.36–5.46 |
| Table IV.Multivariate logistic regression
analysis of prognostic factors for EFS and OS. |
Table IV.
Multivariate logistic regression
analysis of prognostic factors for EFS and OS.
| Factors | Odds ratio | P-value | 95% CI |
|---|
| OS |
|
|
|
| Age
(>65 years) | 11.2 | 0.042 | 1.08–117.2 |
| Gender
(male) | 0.28 | 0.21 | 0.037–2.08 |
| No. of
abnormal | 2.55 | 0.13 | 0.75–8.67 |
| blood
cell lineages |
|
|
|
|
Splenomegaly | 11.8 | 0.016 | 1.56–90.0 |
|
Abnormal karyotype | 3.60 | 0.19 | 0.52–24.6 |
| EFS |
|
|
|
| Age
(>65 years) | 2.02 | 0.20 | 0.68–5.97 |
| Gender
(male) | 0.64 | 0.42 | 0.21–1.92 |
| No. of
abnormal | 2.54 | 0.014 | 1.20–5.36 |
| blood
cell lineages |
|
|
|
|
Splenomegaly | 1.34 | 0.59 | 0.44–4.05 |
|
Abnormal karyotype | 1.12 | 0.85 | 0.33–3.77 |
On multivariate analysis, a significantly worse
overall survival was observed for patients who were elderly
(P=0.042) and had splenomegaly (P=0.016). A significantly worse EFS
was observed with higher numbers of abnormal cell lineages
(P=0.014).
Scoring system based on age, number of
abnormal blood cell lineages and presence of splenomegaly
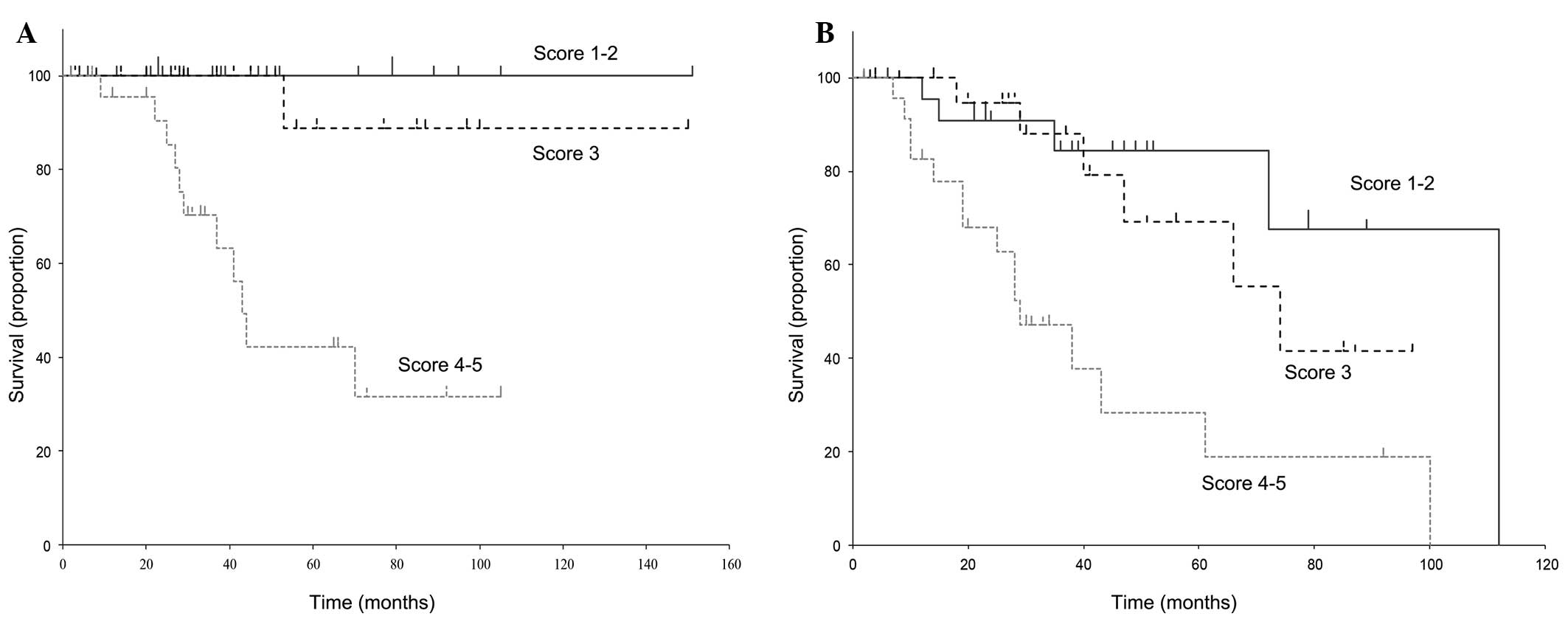
According to the previously described risk scoring
system based on age, abnormal blood cell lineages and splenomegaly,
each patient was assigned a sum score of 1–5 adverse points. The
five patient groups were then consolidated into three risk groups
based on the margin of intergroup survival differences (Fig. 2) as follows: i) Score 1–2 (n=23;
median OS, 45 months; median EFS, 38 months); ii) score 3 (n=24;
median OS, 39 months; median EFS, 29.5 months); and iii) score 4–5
(n=24; median OS, 32 months; median EFS, 26.5 months). Significant
differences were observed among these three groups in terms of OS
(P=0.011) and EFS (P=0.0059).
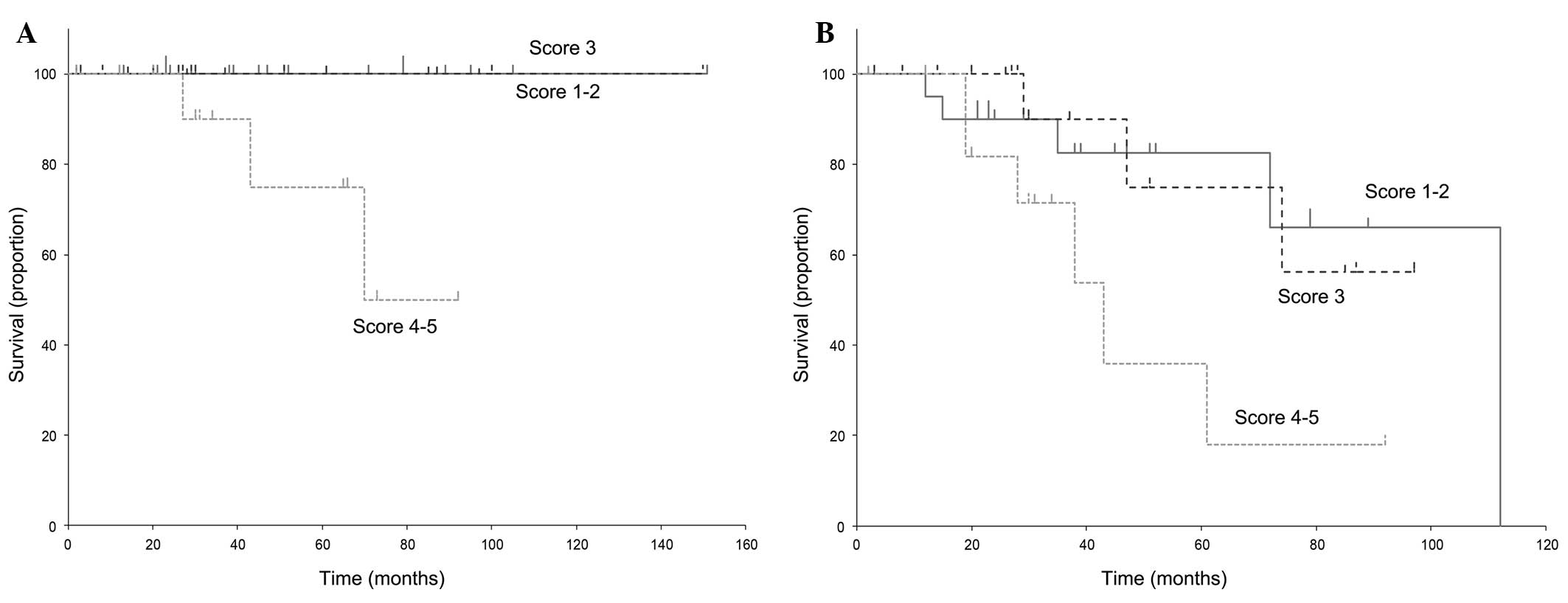
As illustrated in Fig.
1, PMF was associated with a worse prognosis compared with PV
and ET (17). Thus, we created two
categories of PV-ET analysis (Fig. 3)
and PMF analysis (Fig. 4). All the
PV/ET patients belonging to the score 1–2 and 3 groups remained
alive during the entire duration of the follow-up, with a median
survival of 46 months for the score 1–2 and 29.5 months for the
score 3 group. However, 3 patients succumbed to the disease in the
score 4–5 group (median survival, 34 months); this group had a
significantly poorer prognosis (P=0.0053). The respective median
EFSs of the 3 groups were 38.5, 29 and 30 months. Similarly, the
EFS of the score 4–5 group was significantly worse compared with
that of the other groups (P=0.022).
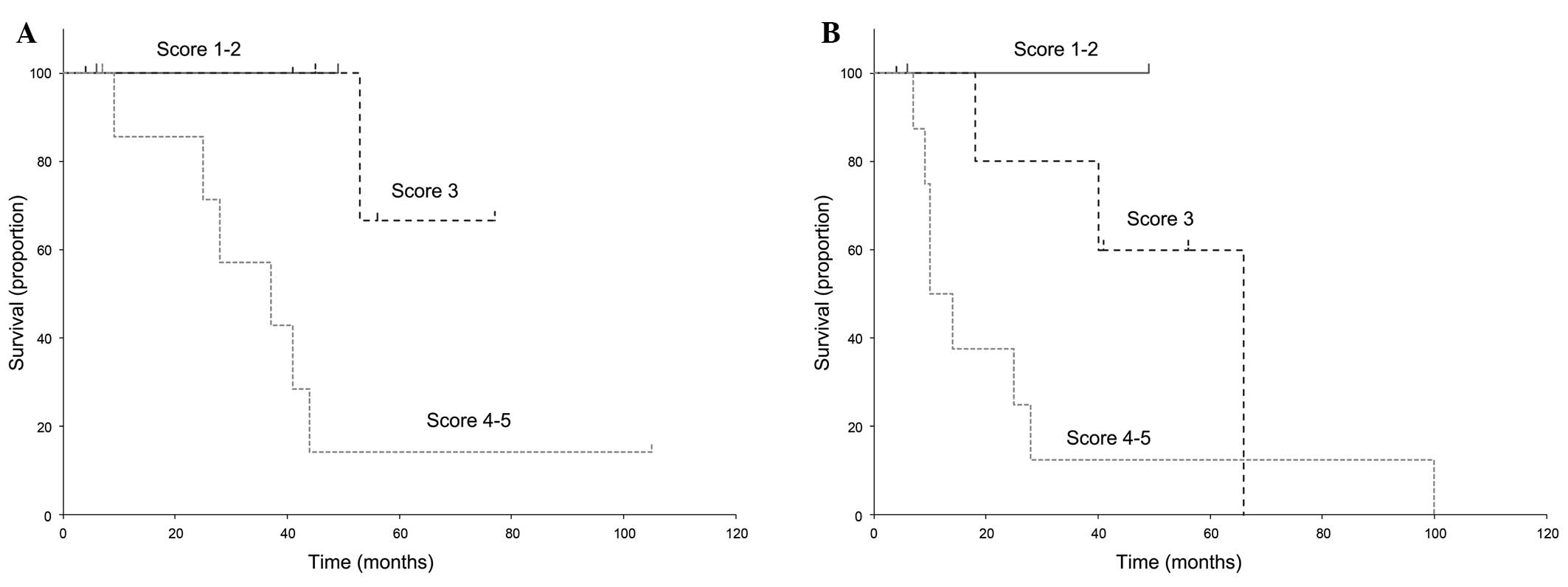
The OS and EFS curves of PMF patients are presented
in Fig. 4. Only 2 patients had a
score of 1–2, 6 patients had a score of 3, and 8 patients scored
4–5. No significant differences were observed within each group
(OS, P=0.177; EFS, P=0.520). However, our results suggested that
PMF patients with a score of 4–5 may have a worse outcome compared
with the lower scoring groups (OS, P=0.023 and EFS, P=0.142 in a
statistical analysis of the score 3 vs. the score 4–5 group).
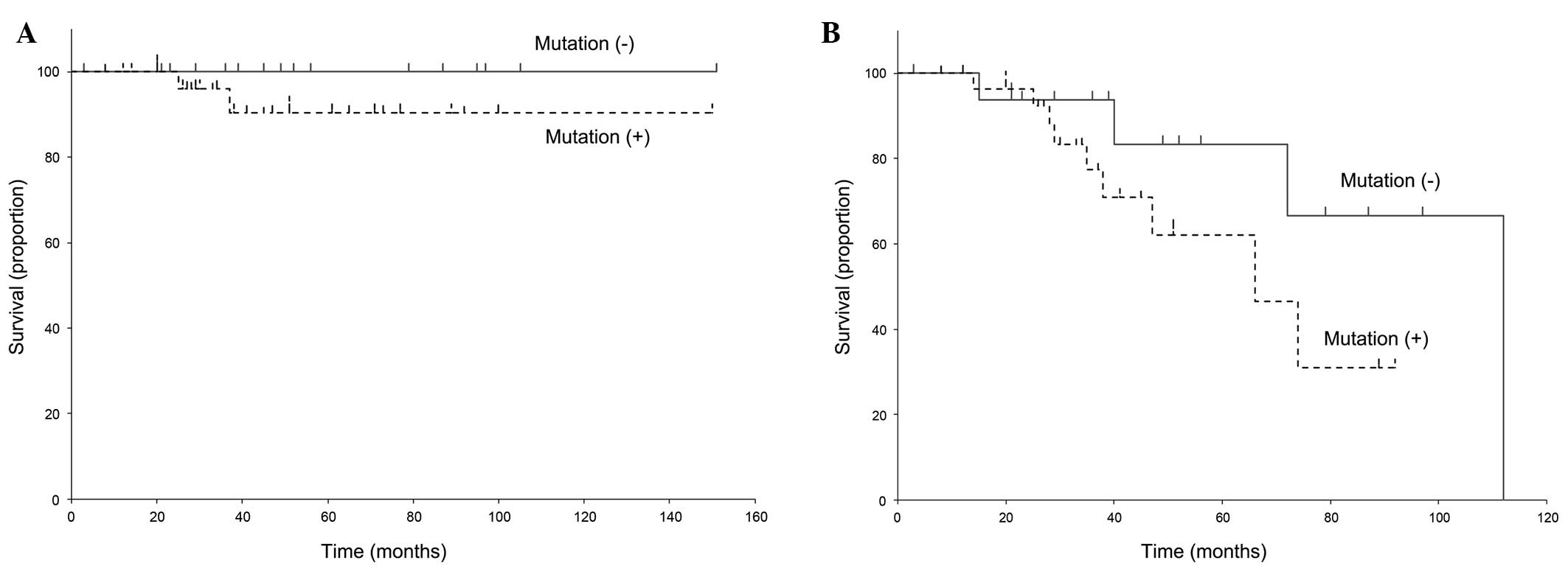
Prognosis and JAK2V617F mutation
A total of 47 patients were tested for the presence
of the JAK2V617F mutation. The survival curves are presented in
Fig. 5. We observed no statistical
association of OS and EFS with the JAK2V617F mutation in MPN
patients (OS, P=0.281; EFS, P=0.174).
Discussion
Several studies investigated the prognostic factors
for MPNs. In PV, the risk factors of transformation to MF or AML
were reported to be advanced age, duration of the disease and
leukocytosis at diagnosis (12) and
the prognostic factors of ET patients were reported to be advanced
age, anemia and thrombocytosis (PLT count
>1,000×109/l) (3). In
PMF, the International Working Group for Myeloproliferative
Neoplasms Research and Treatment propounds the risk stratification
categorized by advanced age (>65 years), anemia (Hb<10 gdl),
leukocytosis (leukocyte count >25×109/l), circulating
blasts, constitutional symptoms, unfavorable karyotype,
thrombocytopenia (PLT count <100×109/l) and required
red cell transfusion (DIPSS-plus) (4,13). By
contrast, the prognostic factors were evaluated as blood cell count
‘outside the normal range’, including not only anemia,
leukocytosis, and thrombocytopenia, but also polycythemia,
leukocytopenia and thrombocytosis. Our study proved that abnormal
blood cell count may be a prognostic index, even if the value
should be higher than ULN or lower than LLN.
Our results did not confirm the effect of abnormal
karyotype on prognosis. Previous studies demonstrated that the
prognosis may be poor in PMF patients with an unfavorable karyotype
that was defined as complex karyotype or single or double
abnormalities, including +8, −7/7q, i(17q), −5/5q, 12p-,
inv(3) or 11q23 rearrangement
(4,14,15). In
our study, only 3 patients out of 18 with an abnormal karyotype
fall into this unfavorable karyotype category (2 patients had
trisomy 8 and 1 patient had 11q23 rearrangement); thus, we were
unable to analyze the effect of that unfavorable karyotype on the
prognosis of MPNs. Gender is not a significant factor affecting the
survival of MPN patients according to a number of previous reports
(3,4,13).
Carobbio et al proved that male patients with ET are at
higher risk of venous thrombosis compared with female patients
(16). However, our results
demonstrated that men had a worse prognosis compared with women in
terms of OS.
The tyrosine kinase JAK2 is directly associated with
the pathogenesis of MPNs, with the identification of JAK2V617F as a
recurring gain-of-function mutation (17,18).
Almost all cases with PV, and ~50% of patients with ET and PMF,
carry this specific mutation; however, in our results, there was no
statistically significant difference in EFS between MPN patients
with and those without the JAK2V617F mutation. Similarly,
previously published results suggested that the JAK2V617F status
did not affect survival in MPNs (19,20). This
fact may suggest that the JAK2V617F mutation is a significant
underlying cause of MPNs, but additional gene mutations are
required to develop MPNs. Uncontrollable increasing blood cell
counts, additional MF and transformation to AML, which are
prognostic factors of MPNs, may be the consequences of additional
gene mutations. Other types of JAK2 mutations, e.g., JAK2 exon 12
mutations, may be detected in MPNs, particularly PV patients
(6,7,21), but it
remains unknown whether these JAK2 mutations, with the exception of
JAKV617F, affect prognosis. Among a number of gene mutations found
in MPN patients, it was recently demonstrated that ET and PMF
patients harboring CALR mutations may have a poor prognosis
(6,7,9).
The benefits of categorization of MPN patients by
our risk scoring are a cost-effective, ubiquitous and useful index
for general physicians. Apart from JAK2V167F, gene mutation
investigation is not covered by insurance in Japan and is only
accessible in university hospitals or research institutes. By
contrast, we may readily categorize MPN patients using a blood cell
counter. Additional methods, such as ultrasonography to evaluate
splenomegaly and bone marrow aspiration, are commonly used. These
ordinary examinations may be able to identify MPN patients with a
poor prognosis. Multilineage blood cell counts may indicate the
necessity of consulting with a hematologist.
References
|
1
|
Vardiman JW, Thiele J, Arber DA, Brunning
RD, Borowitz MJ, Porwit A, Harris NL, Le Beau MM,
Hellström-Lindberg E, Tefferi A, et al: The 2008 revision of the
World Health Organization (WHO) classification of myeloid neoplasms
and acute leukemia: Rationale and important changes. Blood.
114:937–951. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Streiff MB, Smith B and Spivak JL: The
diagnosis and management of polycythemia vera in the era since the
Polycythemia Vera Study Group: A survey of American Society of
Hematology members' practice patterns. Blood. 99:1144–1149. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tefferi A: Polycythemia vera and essential
thrombocythemia: 2013 update on diagnosis, risk-stratification, and
management. Am J Hematol. 88:507–516. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gangat N, Caramazza D, Vaidya R, George G,
Begna K, Schwager S, Van Dyke D, Hanson C, Wu W, Pardanani A, et
al: DIPSS plus: A refined Dynamic International Prognostic Scoring
System for primary myelofibrosis that incorporates prognostic
information from karyotype, platelet count, and transfusion status.
J Clin Oncol. 29:392–397. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Levine RL, Wadleigh M, Cools J, Ebert BL,
Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et
al: Activating mutation in the tyrosine kinase JAK2 in polycythemia
vera, essential thrombocythemia, and myeloid metaplasia with
myelofibrosis. Cancer Cell. 7:387–397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rumi E, Pietra D, Ferretti V, Klampfl T,
Harutyunyan AS, Milosevic JD, Them NC, Berg T, Elena C, Casetti IC,
et al: Associazione Italiana per la Ricerca sul Cancro Gruppo
Italiano Malattie Mieloproliferative Investigators: JAK2 or CALR
mutation status defines subtypes of essential thrombocythemia with
substantially different clinical course and outcomes. Blood.
123:1544–1551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rotunno G, Mannarelli C, Guglielmelli P,
Pacilli A, Pancrazzi A, Pieri L, Fanelli T, Bosi A and Vannucchi
AM: Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano
Malattie Mieloproliferative Investigators: Impact of calreticulin
mutations on clinical and hematological phenotype and outcome in
essential thrombocythemia. Blood. 123:1552–1555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nangalia J, Massie CE, Baxter EJ, Nice FL,
Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, et al:
Somatic CALR mutations in myeloproliferative neoplasms with
nonmutated JAK2. N Engl J Med. 369:2391–2405. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klampfl T, Gisslinger H, Harutyunyan AS,
Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B,
Pietra D, et al: Somatic mutations of calreticulin in
myeloproliferative neoplasms. N Engl J Med. 369:2379–2390. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Swerdlow SH, Campo E, Harris NL, Jaffe ES,
Pileri SA, Stein H, Thiele J and Vardiman JW: WHO classification of
tumours of haematopoietic and lymphoid tissues. World Health
Organization (4th). (Lyon). IARC Press. 2008.
|
|
11
|
Tanaka R, Kuroda J, Stevenson W, Ashihara
E, Ishikawa T, Taki T, Kobayashi Y, Kamitsuji Y, Kawata E,
Takeuchix M, et al: Fully automated and super-rapid system for the
detection of JAK2V617F mutation. Leuk Res. 32:1462–1467. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marchioli R, Finazzi G, Landolfi R, Kutti
J, Gisslinger H, Patrono C, Marilus R, Villegas A, Tognoni G and
Barbui T: Vascular and neoplastic risk in a large cohort of
patients with polycythemia vera. J Clin Oncol. 23:2224–2232. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Passamonti F, Cervantes F, Vannucchi AM,
Morra E, Rumi E, Pereira A, Guglielmelli P, Pungolino E, Caramella
M, Maffioli M, et al: A dynamic prognostic model to predict
survival in primary myelofibrosis: A study by the IWG-MRT
(International Working Group for Myeloproliferative Neoplasms
Research and Treatment). Blood. 115:1703–1708. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hussein K, Pardanani AD, Van Dyke DL,
Hanson CA and Tefferi A: International Prognostic Scoring
System-independent cytogenetic risk categorization in primary
myelofibrosis. Blood. 115:496–499. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tam CS, Abruzzo LV, Lin KI, Cortes J, Lynn
A, Keating MJ, Thomas DA, Pierce S, Kantarjian H and Verstovsek S:
The role of cytogenetic abnormalities as a prognostic marker in
primary myelofibrosis: Applicability at the time of diagnosis and
later during disease course. Blood. 113:4171–4178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Carobbio A, Thiele J, Passamonti F, Rumi
E, Ruggeri M, Rodeghiero F, Randi ML, Bertozzi I, Vannucchi AM,
Antonioli E, et al: Risk factors for arterial and venous thrombosis
in WHO-defined essential thrombocythemia: An international study of
891 patients. Blood. 117:5857–5859. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koppikar P and Levine RL: JAK2 and MPL
mutations in myeloproliferative neoplasms. Acta Haematol.
119:218–225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Levine RL and Gilliland DG:
Myeloproliferative disorders. Blood. 112:2190–2198. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thoennissen NH, Krug UO, Lee DH, Kawamata
N, Iwanski GB, Lasho T, Weiss T, Nowak D, Koren-Michowitz M, Kato
M, et al: Prevalence and prognostic impact of allelic imbalances
associated with leukemic transformation of Philadelphia
chromosome-negative myeloproliferative neoplasms. Blood.
115:2882–2890. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cervantes F, Passamonti F and Barosi G:
Life expectancy and prognostic factors in the classic
BCR/ABL-negative myeloproliferative disorders. Leukemia.
22:905–914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scott LM, Tong W, Levine RL, Scott MA,
Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison
CN, et al: JAK2 exon 12 mutations in polycythemia vera and
idiopathic erythrocytosis. N Engl J Med. 356:459–468. 2007.
View Article : Google Scholar : PubMed/NCBI
|