Introduction
Glioblastoma multiforme (GBM) is the deadliest
primary brain tumor. The standard-of-care for treating GBM includes
surgical resection followed by chemoradiation with temozolomide and
adjuvant temozolomide (1).
Invariably, these tumors progress after or during standard therapy,
and treatment at recurrence involves the use of the anti-angiogenic
antibody, bevacizumab. Once GBM progresses, survival is dismal and
there is an urgent need for effective therapies.
Recently, there has been significant interest and
some controversy regarding the use of the antiviral agent
valganciclovir for the treatment of newly diagnosed GBM patients.
Initially, Stragliotto et al (2) reported a randomized, double-blind,
placebo-controlled study in which newly diagnosed GBM patients
received either valganciclovir or placebo in addition to standard
therapy for 6 months; they reported no statistically significant
difference in overall survival (OS) in the valganciclovir arm
compared with placebo. However, this same group then published a
retrospective study with 50 patients who received valganciclovir,
reporting a median OS (mOS) of 25.0 months for all patients, 30.1
months in patients treated for at least 6 months, and 56.4 months
in patients under continuous valganciclovir therapy (3). The basis for these clinical studies were
data that indicated the protein and DNA for the cytomegalovirus
(CMV) could be detected in almost all GBMs (4–6), and that
survival was >18 months in GBMs with lower levels of CMV
infection (5). These clinical results
were very interesting to both neuro-oncology specialists and GBM
patients. However, there has been significant debate since these
publications, with certain groups suggesting CMV is not present in
GBM (7,8) and other groups contending it is
(4,9).
Moreover, the analysis of the retrospective study has come into
question, with concern raised over a type of selection bias
referred to as ‘immortal time bias’ (10). Immortal time bias is a prejudice, as
it refers to an amount of time in the follow-up period during which
the outcome cannot occur due to the exposure definition. Thus, in
the valganciclovir retrospective study, it would pertain to
patients that had ≥6 months of valganciclovir treatment, and within
this group death cannot have occurred in the first 6 months of
follow-up. However, Söderberg-Naucler et al repeated their
analysis using Cox regression, which may prevent such bias, and
still produced similar survival results in the valganciclovir group
(11).
With conflicting results regarding the presence of
CMV within GBMs, the mechanism underlying the beneficial effect of
valganciclovir on survival remains unknown; however, given the
impressive improvement in survival of newly diagnosed GBM patients,
the evaluation of valganciclovir in the recurrent setting is
warranted.
We herein report a single-institution, retrospective
analysis of GBM patients treated at recurrence with valganciclovir
plus bevacizumab. Progression-free survival (PFS) and OS were
compared to an institutional cohort for bevacizumab at recurrence
and a small survival advantage was observed in patients treated in
the recurrent setting with valganciclovir plus bevacizumab.
Materials and methods
Study design
Following approval by the Vanderbilt Institutional
Review Board, we performed a retrospective chart review of all the
patients treated for recurrent GBM at the Vanderbilt Neuro-Oncology
clinic. We identified 13 patients who received treatment with both
bevacizumab and off-label valganciclovir between August 1, 2013 and
December 31, 2014, and 8 patients who were started on
valganciclovir and bevacizumab concurrently. All patients but one
(gliosarcoma) had a pathological diagnosis of GBM. These patients
received bevacizumab at 10 mg/kg every 2 weeks and valganciclovir
starting with a loading dose of 900 mg b.i.d. for 21 days and then
450 mg b.i.d. as maintenance therapy.
For the control cohort, we searched the database for
all patients who were treated for first recurrence with GBM between
January 1, 2005 and December 31, 2014, and identified 50 patients
who met the following inclusion criteria: i) Had never received
valganciclovir, even in later recurrences; ii) only first
recurrence as to avoid any selection bias; iii) only a diagnosis of
GBM; iv) were treated with concurrent radiation therapy and
temozolomide followed by temozolomide therapy prior to recurrence;
iv) recurrence occurred after 8 weeks from completion of
chemoradiation; vi) had not had a second surgery for tumor
progression; vii) had not been on a clinical trial at any point
during their treatment; and viii) had signed consent forms to
participate in a database containing all clinical information and
tissue banking. A large portion of these control subjects received
a chemotherapy drug in addition to bevacizumab.
Statistical analysis
Continuous variables were summarized using the
median (range). Categorical variables were reported as frequencies
and percentages of treatment group. PFS was defined as the time
from therapy initiation to disease progression or death from any
cause. OS was defined as the time from therapy initiation to death
from any cause. All the patients had progressed as of the last
follow-up. Patients who remained alive at the last follow-up were
censored in the OS analysis. The distributions of PFS and OS were
estimated using the Kaplan-Meier method and compared between
treatment groups using the log-rank test.
Analysis of
O6-methylguanine-DNA methyltransferase gene (MGMT)
methylation and isocitrate dehydrogenase (IDH) mutation
Methylation of the MGMT promoter was detected
using DNA isolated from formalin-fixed, paraffin-embedded
specimens. Analysis was performed by methylation-specific
polymerase chain reaction (PCR) using an ABI7900 PCR system
(Applied Biosystems, Waltham, MA, USA). MGMT and
β-actin copy numbers were used to calculate the ratio of
MGMT/β-actin ×1,000, and samples with scores ≥2.00 were
considered to be methylated. Analysis for mutations in IDH1
and IDH2 was performed using multiplex PCR coupled with a
primer extension assay. Fluorescently labeled products were
analyzed using capillary electrophoresis. The mutations tested
included IDH1 R132H, R132 G, R132S, R132C, R132L, R132P and
the c.315C>T (G105G) polymorphism, as well as IDH2 R140G,
R140W, R140Q, R140L, R140P, R172G, R172W, R172K, R172M, R172T,
R172S and R172S.
Results
Patient characteristics
A total of 13 patients were identified, who had a
first recurrence of high-grade GBM and were exposed to both
bevacizumab and valganciclovir (Table
I). of these 13 patients, 8 were started concurrently on
bevacizumab plus valganciclovir at recurrence, whereas the
remaining 5 were started on bevacizumab at 3, 4, 6, 9 and 12 months
prior to valganciclovir initiation and prior to a second recurrence
(1st recurrence as the start date of bevacizumab administration).
The median exposure to valganciclovir was 9 months for all 13
patients (range, 3–16 months); the median exposure to
valganciclovir was 9.5 months for the 8 concurrently treated
patients (range, 3–16 months (Table
I). of the 13 patients, 12 had a pathological diagnosis of GBM,
whereas 1 patient was diagnosed with gliosarcoma. All 13 patients
were IDH wild-type and 29% had MGMT promoter
methylation. The median Karnofsky performance status score was 80%
and the median patient age was 61 years. Other chemotherapies used
during treatment (at further disease progression) included 2
patients treated with lomustine and 6 treated with vorinostat.
 | Table I.Patient characteristics. |
Table I.
Patient characteristics.
| Characteristics | Valganciclovir, total
exposed with bevacizumab (n=13) | Valganciclovir
started concurrently with bevacizumab (n=8) | Bevacizumab cohort
(n=50) |
|---|
| Age, median (range),
years | 61 (33–77) | 58.5 (49–77) | 58 (18–75) |
| Gender (%) |
|
|
|
| Male | 57 | 75 | 52 |
|
Female | 43 | 25 | 48 |
| KPS score, median
(range) | 80 (60–90) | 80 (60–90) | 80 (50–100) |
| Received
chemoradiation with temozolomide (%) | 100 | 100 | 100 |
| MGMT
methylated (%) | 29 | 37.5 | a |
| IDH mutated
(%) | 0 | 0 | a |
| Median ex posure to
valganciclovir (range), months | 9 (3–16) | 9.5 (3–16) | N/A |
Clinical results
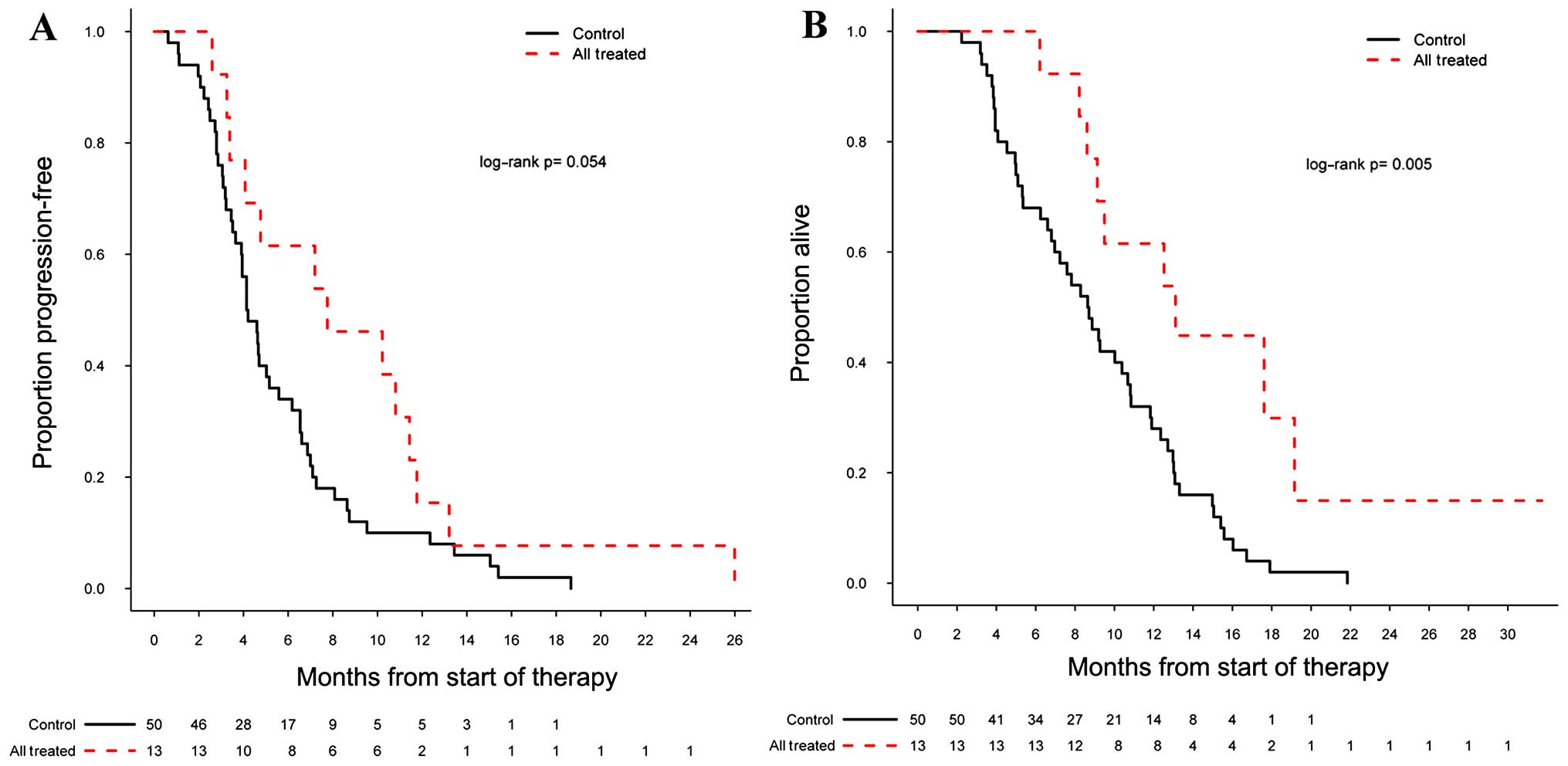
There was a significant difference in PFS (log-rank
P=0.054) and OS (log-rank P=0.005) in the 13 patients treated with
valganciclovir plus bevacizumab compared with the bevacizumab
control cohort, with a 6-month PFs (PF6) of 62% [95% confidence
interval (CI): 0.40–0.95] and a mOS of 13.1 months [95% CI:
9.13-not applicable (NA)] (Fig. 1A and
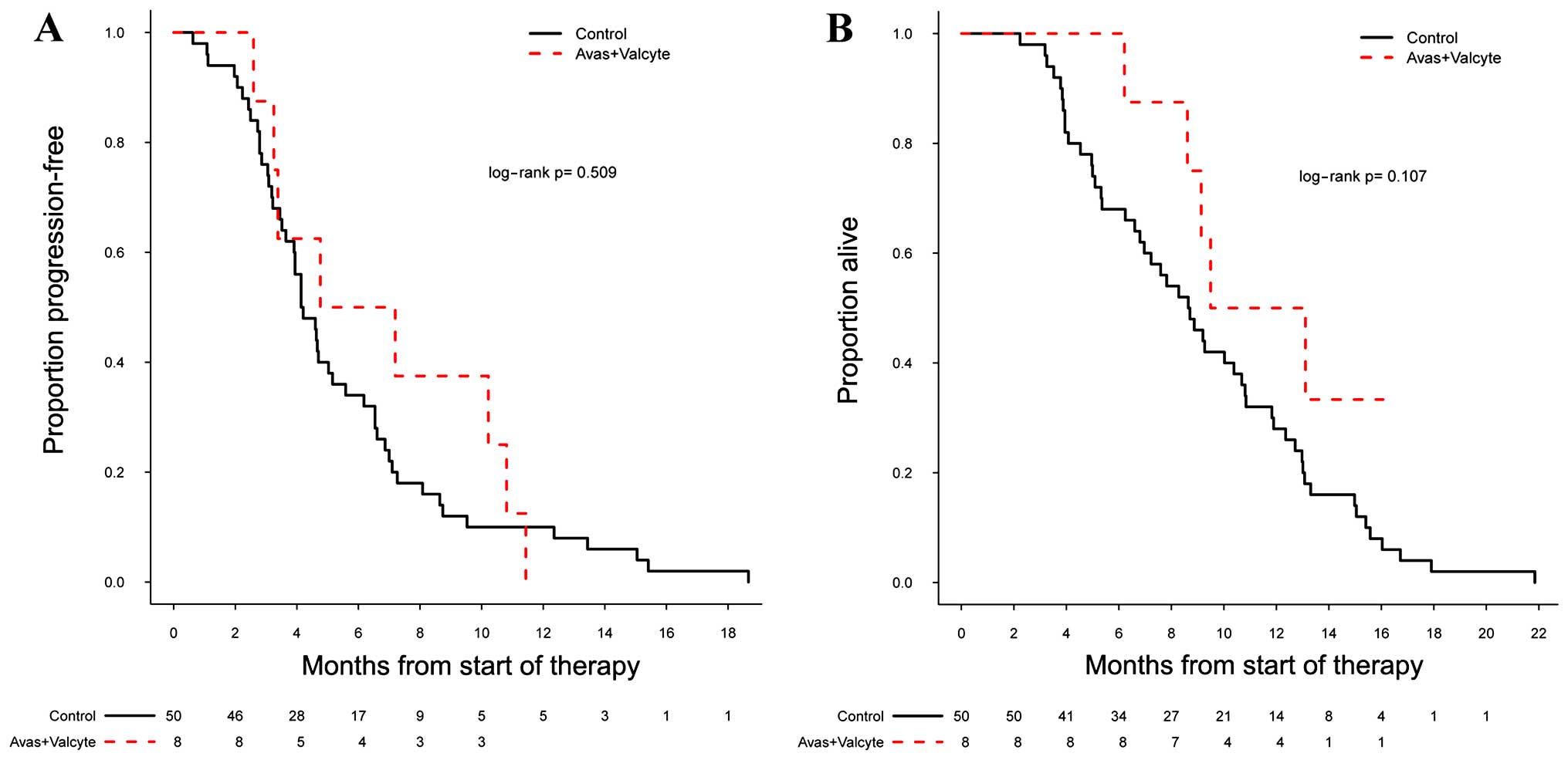
B). For 8 of the 13 patients who were started concurrently on
valganciclovir plus bevacizumab at first recurrence, the PF6 was
50% (95% CI: 0.25–1.0) and the mOS 11.3 months (95% CI: 9.1-NA);
neither of these reached significance compared with the control
group in terms of PFS (log-rank P=0.509) and OS (log-rank P=0.107).
For the institutional bevacizumab cohort, the PF6 was 34% (95% CI:
0.23–0.50) and the mOS 8.7 months (95% CI: 6.8–10.8) (Fig. 2A and B). Our institutional bevacizumab
cohort mOS (8.7 months) was similar to historical data with
bevacizumab, with a mOS ranging from 8.7 to 9.2 months (12,13).
Of the 13 patients who were administered
valganciclovir in our study, none developed leukopenia, skin
reactions or nausea; however, 4 patients developed thrombocytopenia
(platelet count <100,000/mcl), 3 of whom only had only one
laboratory value <100,000/mcl, whereas 1 patient had >1
laboratory readings <100,000/mcl.
Discussion
The treatment options for recurrent GBM are
extremely limited and the field of neuro-oncology urgently requires
additional therapies to supplement bevacizumab. The addition of
valganciclovir to the standard-of-care in newly diagnosed GBM
patients has shown promise in one retrospective study (3); however, this result and the presence of
CMV in GBM remains highly controversial.
At our institution, 13 patients with recurrent GBM
were placed on valganciclovir, of whom 8 were concurrently placed
on bevacizumab therapy at first recurrence, whereas the remaining 5
had valganciclovir added to their existing bevacizumab regimen. In
this study, we observed a small benefit in terms of os in patients
treated with valganciclovir plus bevacizumab compared with the
bevacizumab cohort. In addition, the use of valganciclovir with
bevacizumab appeared to be well tolerated in this setting.
We did not evaluate the presence of CMV protein in
these patients, due to the controversy regarding its detection.
Regardless, the question of whether valganciclovir prolongs
survival when combined with bevacizumab is a major issue and, to
the best of our knowledge, this study is the first to investigate
valganiclovir in the recurrent setting.
If CMV is indeed present in these tumors, as several
groups have reported, the mechanism of action of valganciclovir is
likely associated with its inhibition of CMV replication and, thus,
of CMV oncogene drivers (14).
However, if CMV is not present in GBM, or is present in amounts not
sufficient to affect tumor biology, the mechanism underlying the
beneficial effect of valganciclovir on survival remains unclear, as
other targets have not been identified. If indeed valganciclovir
affects survival in these tumors, it appears to require an extended
period of time (>6 months) to observe an appreciable clinical
difference (2); in the small group we
present in this study, the median exposure was 9 months. Such a
delayed effect is reminiscent of epigenetic modifiers, such as the
DNA methyltransferase inhibitors used in the treatment of
myelodysplastic syndromes, where ≥6 months of treatment are
required before observing a benefit (15). Thus, valganciclovir may inhibit GBM
growth in a CMV-independent manner.
We observed that the addition of valganciclovir to
bevacizumab led to a small increase in median OS in the recurrent
setting. However, this study is limited by its retrospective nature
and by the small number of patients that concurrently received
valganciclovir plus bevacizumab. As a group, there was a
significant increase in mOS, but in the 8 concurrently treated
patients the survival benefit did not reach significance. Moreover,
the 5 patients who had valganciclovir added to the existing
bevacizumab treatment, had not yet had a second recurrence; thus,
some selection bias is possible. Therefore, any conclusions drawn
by this study must be tempered and a larger prospective study is
required to further evaluate these data. Additional clinical
studies should be conducted in newly diagnosed patients where the
time-to-treat is usually >6 months. Given the significant
survival benefit reported in newly diagnosed GBM patients and the
small trend we present here, coupled with the great need for
improvement in this field, the risk-to-benefit ratio of such a
study would be small.
Acknowledgements
Dr Clark is supported by a Vanderbilt Clinical
Oncology Research Career Development Program K-12 Grant.
References
|
1
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stragliotto G, Rahbar A, Solberg NW, Lilja
A, Taher C, Orrego A, Bjurman B, Tammik C, Skarman P, Peredo I and
Söderberg-Nauclér C: Effects of valganciclovir as an add-on therapy
in patients with cytomegalovirus-positive glioblastoma A
randomized, double-blind, hypothesis-generating study. Int J
Cancer. 133:1204–1213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Söderberg-Nauclér C, Rahbar A and
Stragliotto G: Survival in patients with glioblastoma receiving
valganciclovir. N Engl J Med. 369:985–986. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cobbs CS, Harkins L, Samanta M, Gillespie
GY, Bharara S, King PH, Nabors LB, Cobbs CG and Britt WJ: Human
cytomegalovirus infection and expression in human malignant glioma.
Cancer Res. 62:3347–3350. 2002.PubMed/NCBI
|
|
5
|
Rahbar A, Stragliotto G, Orrego A, Peredo
I, Taher C, Willems J and Söderberg-Naucler C: Low levels of human
cytomegalovirus infection in glioblastoma multiforme associates
with patient survival; -a case-control study. Herpesviridae.
3:32012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bhattacharjee B, Renzette N and Kowalik
TF: Genetic analysis of cytomegalovirus in malignant gliomas. J
Virol. 86:6815–6824. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baumgarten P, Michaelis M, Rothweiler F,
Starzetz T, Rabenau HF, Berger A, Jennewein L, Braczynski AK, Franz
K, Seifert V, et al: Human cytomegalovirus infection in tumor cells
of the nervous system is not detectable with standardized
pathologico-virological diagnostics. Neuro Oncol. 16:1469–1477.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hellstrand K, Martner A and Bergström T:
Valganciclovir in patients with glioblastoma. N Engl J Med.
369:20662013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ranganathan P, Clark PA, Kuo JS, Salamat
MS and Kalejta RF: Significant association of multiple human
cytomegalovirus genomic loci with glioblastoma multiforme samples.
J Virol. 86:854–864. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu CJ and Hu YW: Immortal time bias in
retrospective analysis, Is there a survival benefit in patients
with glioblastoma who received prolonged treatment of adjuvant
valganciclovir? Int J Cancer. 135:250–251. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Söderberg-Naucler C, Peredo I, Rahbar A,
Hansson F, Nordlund A and Stragliotto G: Use of Cox regression with
treatment status as a time-dependent covariate to re-analyze
survival benefit excludes immortal time bias effect in patients
with glioblastoma who received prolonged adjuvant treatment with
valganciclovir. Int J Cancer. 135:248–249. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nghiemphu PL, Liu W, Lee Y, Than T, Graham
C, Lai A, Green RM, Pope WB, Liau LM, Mischel PS, et al:
Bevacizumab and chemotherapy for recurrent glioblastoma: A
single-institution experience. Neurology. 72:1217–1222. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Friedman HS, Prados MD, Wen PY, Mikkelsen
T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen
R, et al: Bevacizumab alone and in combination with irinotecan in
recurrent glioblastoma. J Clin Oncol. 27:4733–4740. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cobbs CS: Cytomegalovirus and brain tumor:
Epidemiology, biology and therapeutic aspects. Curr Opin Oncol.
25:682–688. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Garcia-Manero G and Fenaux P:
Hypomethylating agents and other novel strategies in
myelodysplastic syndromes. J Clin Oncol. 29:516–523. 2011.
View Article : Google Scholar : PubMed/NCBI
|