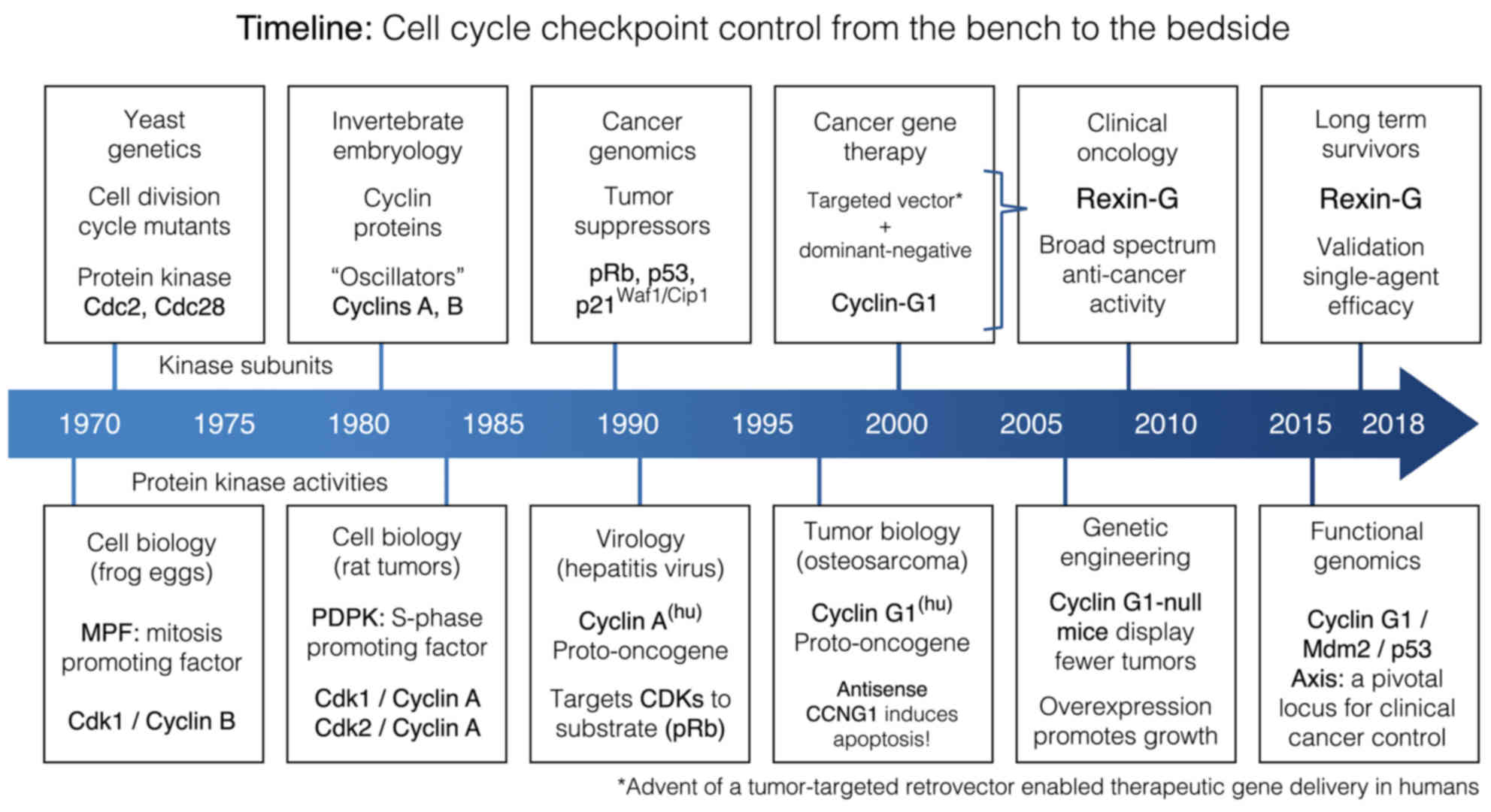
Introduction
Decades of research in the fields of biochemistry,
cell biology, molecular genetics, virology, genetic engineering,
functional genomics and cancer gene therapy, have converged in
identifying the executive components of a commanding regulatory
axis of mammalian cell cycle control: Providing new mechanistic
insights, biochemical pathways, unifying concepts and checkpoint
control elements with profound implications in the prevention,
diagnosis and treatment of cancer (Fig.
1). The challenge remains, however, to successfully integrate
these celebrated epochs of gene discovery and biochemical pathway
characterization into a practical understanding of cell cycle
control befitting the actual praxis and applied pharmacology of
contemporary clinical oncologists. This focused review, prepared by
contributing scientists and clinical practitioners in the field of
genetic medicine, is intended to present the current
state-of-the-art in applied cell cycle checkpoint control as it
relates to cancer management.
Tumor initiation, tumor promotion and
Ser/Thr protein phosphorylation: Then and now
From a clinical perspective, the molecular
mechanisms of chemical co-carcinogenesis first came to light in the
1940s with the pioneering studies of croton oil (i.e., phorbol
esters) and 3,4-benzpyrene in a now-classic mouse skin model
(1), in which increased tumor
production was only observed when the inflammatory phorbol ester
followed, rather than preceded, the application of the DNA-damaging
carcinogen, thereby defining the separable and discernible stages
of tumor initiation, promotion and progression (2). The subsequent discovery that protein
kinase C (PKC), which plays a major role in signal transduction and
cell proliferation, is the cellular receptor for the
tumor-promoting phorbol esters (3)
ushered in a wave of pharmaceutical interest in selective PKC
inhibitors, only to be thwarted by the general multifunctionality
of PKC, the multiplicity of PKC isoenzymes, the limited specificity
of PKC modulators and the remaining unanswered questions and
intricacies of PKC function, which stifled the promise of targeting
PKCs for cancer therapeutics (4).
Nevertheless, the importance of discrete Ser/Thr kinase activities,
which operate via recognition-site-specific protein phosphorylation
events, in the control of biochemical pathways governing mammalian
cell growth and proliferation, as well as tumorigenesis, invasion
and metastasis, has emerged as a major regulatory theme.
Focus on cyclin-dependent targeting of
proline-directed protein phosphorylation
Basic research in yeast genetics characterized a
number of cell division cycle (Cdc) mutants, thereby identifying
important genes, notably Cdc28 in baker's yeast (S.
cerevisiae) and its homologue Cdc2 in fission yeast (S.
pombe), which encode a unique serine/threonine protein kinase
that is not only critical for mitosis in yeast, but is highly
conserved in both structure and function throughout the evolution
of all eukaryotes, including Homo sapiens (5). The molecular cloning and
characterization of the Cdc2/Cdc28 kinase (CDK1 in mammals) and its
implicit role in governing the defined stages and checkpoints of
the eukaryotic cell division cycle supported by the independent
discovery of cyclins A and B as prominent oscillating proteins of
unknown function in sea urchin embryos (A. punctata), which
were eventually cloned, biochemically characterized and determined
to be positive-acting regulatory subunits of the executive
Cdc2/Cdc28 kinase, thereby linking nascent protein synthesis (i.e.,
cyclin proteins) to the ordered stepwise progression of the cell
division cycle through its defined phases (6). From this point onwards, the term
‘cyclin-dependent’ kinase (CDK) has been used to characterize the
vertebrate homologs of this key regulatory enzyme. Recognized as a
triumph of basic research in simple model systems, as well as a
major advance in terms of elaborating the executive enzymology
regulating cell cycle transitions, the principal investigators,
Hartwell, Nurse and Hunt, were awarded the Nobel Prize for Medicine
in 2001 (7). Armed with the DNA
coding sequences for both the catalytic subunit (kinase) and the
putative regulatory subunits (cyclins) of this executive protein
kinase from primitive eukaryotic model systems, the molecular
cloning and characterization of homologous coding sequences in
higher animals began in earnest (8,9),
enabling scientists to solve a lingering paradox in modern cell
biology, i.e., the molecular basis for deconstructing cell cycle
regulation in higher animals into two separable and distinct
protein kinase activities, each with very different substrate
specificities: The purified mitosis-promoting factor (MPF), which
controls the G2-to-M phase transition (10) vis-à-vis the S phase-promoting factor
(SPF), which orchestrates the G1-to-S phase transition (11,12).
Mechanistic insights were derived from international
studies of site-specific protein phosphorylation in rat
pheochromocytoma, which led to the identification and
characterization of the growth factor-sensitive proline-directed
protein kinase (PDPK), based on its ability to phosphorylate a
unique site on tyrosine hydroxylase, a key regulatory enzyme in the
synthesis of adrenal catecholamines (13). These collaborative studies led to the
discovery that the preferred amino acid sequence recognized by this
unique interphase kinase activity, in terms of substrate
specificity, is X-Ser/Thr-Pro-X-X, thereby predicating the
requirement for a single orienting proline residue immediately
adjacent to the actual phosphorylation site (14), affirming the appellation
‘proline-directed’ protein kinase, and presenting this unique
Ser/Thr kinase activity (which is biochemically distinguishable
from the M phase-specific histone H1 kinase activity of purified
MPF) as a likely candidate for the illusive SPF in mammals
(15). Structurally, the high
frequency of S-P-X-X and T-P-X-X motifs (presumably type-1 β-turns)
found in gene regulatory proteins is not only indicative of a new
DNA-binding unit (16), but also of
proline-directed protein phosphorylation sites that have been
canalized by natural selection, along with this site-specific
protein kinase activity.
Molecular cloning and characterization of homologous
kinase subunits in vertebrates revealed that the biochemical and
physiological activities of MPF are attributable to CDK1/cyclin B
complexes, the Ser/Thr kinase activity of which regulates the
cytoskeletal dynamics associated with mitosis (10,12). In
1991, Hall et al characterized the subunits of the purified
PDPK as a complex of CDK1 and cyclin A (17); when CDK2, a second homologue of the
yeast Cdc2/Cdc28 kinase, was identified in humans, this homologous
kinase, which is expressed somewhat earlier in the cell cycle
compared with CDK1, was also found to partner with cyclin A and is
enzymatically active as a CDK2/cyclin A heterodimer (18). Moreover, in addressing the paradox of
differential substrate specificities, it was determined that the
cyclin A subunit of these CDK complexes not only acts as a positive
regulatory subunit, in terms of kinase activation, but it is the
inducible ‘cyclin’ subunit that determines the substrate
specificity of the active protein kinase. In this case, the cyclin
A subunit physically targets the cyclin A/CDK holoenzymes to the
Retinoblastoma (Rb) tumor suppressor protein (19), where progressive site-specific
phosphorylation of pRb serves to inactivate the tumor suppressor
(i.e., transcription/E2F repressor) (20), thereby linking the molecular
activation of G1-phase transcription in humans to the expression of
specific cyclin proteins (21). The
cyclin-targeted CDK activities serve to overcome the suppressive
function of Rb-related ‘pocket’ proteins (pRb, p107 and p130) that
govern the feed-forward mechanics of the cell cycle, i.e., the
coupling of protein phosphorylation and gene transcription, which
drives cell cycle progression (22,23).
Focus on G1-phase regulation: Oncogenic
cyclins vis-à-vis tumor suppressive gatekeepers
A fundamental characteristic of cancer genetics is
the molecular dysregulation of cell cycle checkpoint control
elements, which normally ensures the orderly progression of cell
growth, DNA synthesis and mitotic cell division, while actively
ensuring genomic fidelity. Among the manifold genetic alterations
known to contribute to the pathogenesis of cancer in humans,
including the molecular genetic disruptions of tumor viruses, the
majority of these mutations are observed in genes that regulate
progression through the G1 phase of the cell division cycle,
including pRb-related tumor-suppressor proteins, which govern cell
cycle progression, and the much-studied p53 tumor suppressor
(mutated in >50% of human cancers), which serves as a molecular
‘guardian’ of DNA fidelity and an ‘executioner’ via its
pro-apoptotic function (24).
Alterations in the enzymatic machinery that controls the decisions
to progress from a resting state (G0) into the cell cycle (G0-to-G1
transition) and/or to progress from the G1 to the S phase led to
the identification of a growing family of human cyclins and their
CDK partners that are directly implicated in the mechanisms of
tumorigenesis and cancer (25–27).
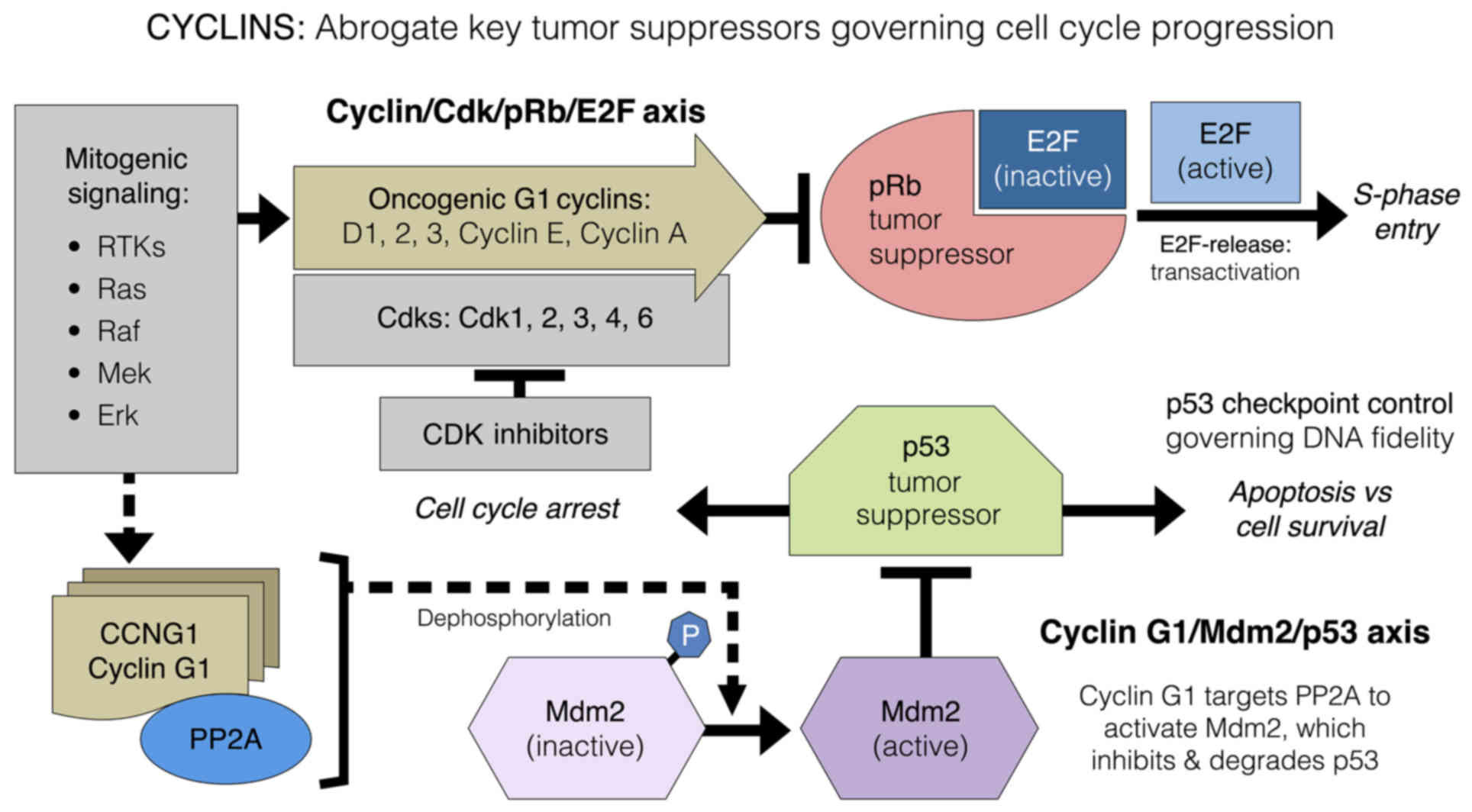
Thus, it is becoming abundantly clear that the major tumor
suppressive elements (i.e., pRb and p53) that control progression
through the mammalian cell cycle (28) are themselves a target for molecular
inactivation by the expression and growth-promoting activities of
two distinct types of potentially oncogenic cyclin proteins: The G1
cyclins (D-, E- and A-type cyclins) targeting CDKs to the Rb-Axis,
and the perplexing cyclin G1, which disables the functions of p53
(Fig. 2).
The regulatory importance of human G1 cyclin
expression in the promotion of cell proliferation and cancer is
indicated by the telltale manner of their discovery and cloning in
humans, which is directly linked to the pathogenesis of neoplastic
disease. Human cyclin A was first identified biochemically as a
co-precipitate, along with the pRb tumor suppressor, in a screen
for proteins that bind with high affinity to the cell-transforming
adenoviral E1A oncoprotein (29);
soon thereafter, the molecular cloning of the human cyclin A gene
was achieved by tracking the various insertion sites where the
hepatitis B virus (HBV) physically integrates into the human genome
(30). Importantly, it was
determined that the HBV-induced dysregulation of the human cyclin A
locus not only enforces a generalized overexpression of the gene,
but it also physically mutated/truncated the N-terminal segment of
the cyclin A protein, removing signal sequences necessary for its
cyclical proteolytic degradation and producing a hybrid HBV-cyclin
A transcript encoding a ‘stabilized’ cyclin A protein (hence
interphase PDPK activity), which may indeed participate in the
development and progression of hepatocellular carcinoma (HCC)
(31). In subverting the cyclin A
locus, a key component of the mammalian cell cycle machinery, the
transforming HBV had apparently created a ‘dominant-positive’
mutant gene product to essentially commandeer cell cycle control by
maintaining a critical growth-promoting function of the cyclin A
subunit (i.e., kinase activation via the retained ‘cyclin box’),
while deleting the natural proteolytic processing, which normally
eliminates cyclin A entirely, upon completion of each cell division
cycle. Such viral subversion and oncogenic activation of a
growth-regulatory element identifies human cyclin A as a bona fide
proto-oncogene (15,32), and hence a useful biomarker for
characterizing tumor cell proliferation in histochemical detail
(33).
The identification of the D-type cyclins (D1, D2 and
D3) as oncogenes forged new and important links between mitogenic
signal transduction, D-type cyclin gene expression and
tumorigenesis (34): The growth
factor-sensitive cyclin D1 was initially identified as a
PRAD1/bcl-1 proto-oncogene, which is subject to gene amplification
and/or rearrangement, resulting in cyclin D1 overexpression in a
wide spectrum of human cancers, including B-cell neoplasms,
carcinomas of the head and neck, various sarcomas and human breast
cancers (>50%); cyclin D2 was identified as the integration site
of a murine leukemia virus, while the cyclin D2 and D3 genes are
amplified and overexpressed in numerous human cancers, including
leukemias, lymphomas, glioblastomas, colorectal, renal and
pancreatic carcinomas (26,35). Partnering promiscuously as
heterodimers with a number of CDK subunits (CKD1, 2, 3, 4 and 6),
D-type cyclins function in early G1 to target the CDK complexes to
the pRb-related tumor suppressor proteins, where site-specific
inhibitory phosphorylation of pRb, to a limited extent, primes pRb
(36) and advances cell cycle
progression to a critical control point in the late G1 phase, known
as the restriction point, or R-point (37): This is where the induction of cyclin
E-dependent kinase activity contributes to the hyperphosphorylation
and inactivation of Rb, thereby releasing the E2F transcription
factors and driving cells irreversibly through the G1-to-S phase
transition, beyond which additional extracellular signals in the
form of mitogenic growth factors are no longer required (38,39).
Taken together as an important class of growth-promoting
proto-oncogenes, the orderly and progressive expression of specific
G1 cyclins (D-type cyclins first, followed by cyclin E and cyclin
A) are now viewed as inducible rate-limiting activators of G1 phase
progression, the dysregulation of which is potentially
oncogenic.
Just as the executive CDKs of G1 progression are
engaged by positive regulatory subunits with profound oncogenic
potential (i.e., G1 cyclins), G1 progression is also negatively
controlled by polypeptide CDK inhibitors (CDKIs) whose expression
is linked with the classical tumor suppressor p53. The finding that
p53, a sequence-specific DNA-binding transcription factor,
selectively induces p21WAF1/CIP1 (a universal CDKI) as a
mediator of p53-initiated cell cycle arrest, has attracted interest
in CDKIs as a strategic locus for prospective therapeutic
interventions (40). Indeed, two
families of endogenous CDKIs were found to function operationally
as tumor suppressors: i) The WAF1/CIP/KIP family of CDKIs (p21, p27
and p57) appear to inhibit the activity of all major CDKs, while
the INK4 family of CDKIs (p16, p15, p18 and p19) more specifically
inhibit the cyclin D-dependent kinases CDK4 and CDK6, which
phosphorylate/inactivate the pRb suppressor in early G1 (35,41). Of
note, genetic alterations involving the p16INK4a locus
(chromosome 9p21) have been identified as germline mutations in
melanoma-prone patients, and as deletions or mutations in a large
percentage of primary human tumors, including sarcomas, lymphomas,
leukemias, squamous cell carcinomas and pancreatic adenocarcinomas.
In addition, genetic engineering of homologous deletions in mice
confirmed the overall importance of this locus in the suppression
of tumorigenesis (41,42).
Targeting CDKs and CDK inhibitors (CDKIs)
for cancer therapy: Current status, issues
Elucidation of the ‘executor’ roles of G1 cyclins,
CDKs and CDKIs, as they are critical regulatory components of the
cell cycle machinery that are frequently altered in human cancers,
prompted renewed interest in the development of specific kinase
inhibitors expected to block cell cycle progression and/or induce
growth arrest, thereby providing a prospective abundance of targets
for novel antineoplastics (43).
Unfortunately, but not unexpectedly (see PKC issues above), the
first generations of pharmaceutical pan-CDKIs failed to meet such
expectations, due to the general lack of target specificity
(44). Even the p21WAF1/Cip1
protein, which is the classic pan-CDKI that mediates p53-dependent
cell cycle arrest, allowing for DNA repair, is sufficiently
complex, in terms of the regulation of its expression, binding
activities and subcellular localization, to function operationally,
either as a tumor suppressor under certain circumstances (cell
cycle block) or as a tumor-promoting oncogene (by preventing
apoptosis), depending on the cellular context (45).
Over the past 20 years, a substantial number of
prospective pan-CDKIs have been developed as potential cancer
therapeutics and tested in numerous clinical trials in a variety of
different tumor types, only to fail upon final analysis to meet
objective clinical endpoints with an acceptable profile of systemic
toxicities (46,47). The principal reasons provided to
explain the general failure of non-selective pan-CDKIs in the
clinical setting are as follows: i) Lack of clear understanding of
the physiological mechanisms of action as to which CDKs are
actually being inhibited in vivo; ii) lack of patient
selection and stratification on the basis of pertinent companion
biomarkers that may help identify a subset of good clinical
responders; and iii) lack of a therapeutic window, meaning the
inability to achieve therapeutic levels of the drugs due to their
intrinsic inability to discriminate between cancerous and healthy
tissues (47). Despite the
disappointing early attempts to develop pharmaceutical CDKIs for
cancer therapy, a more specific focus on the cellular activities of
CDK4 and CDK6, which are activated by the oncogenic D-type cyclins,
has recently achieved limited success (47,48).
Nonetheless, the major challenges of precise target specificity,
rapidly acquired drug resistance, untoward toxicities in normal
tissues, optimal patient selection (companion biomarkers) and
efficient drug delivery remain.
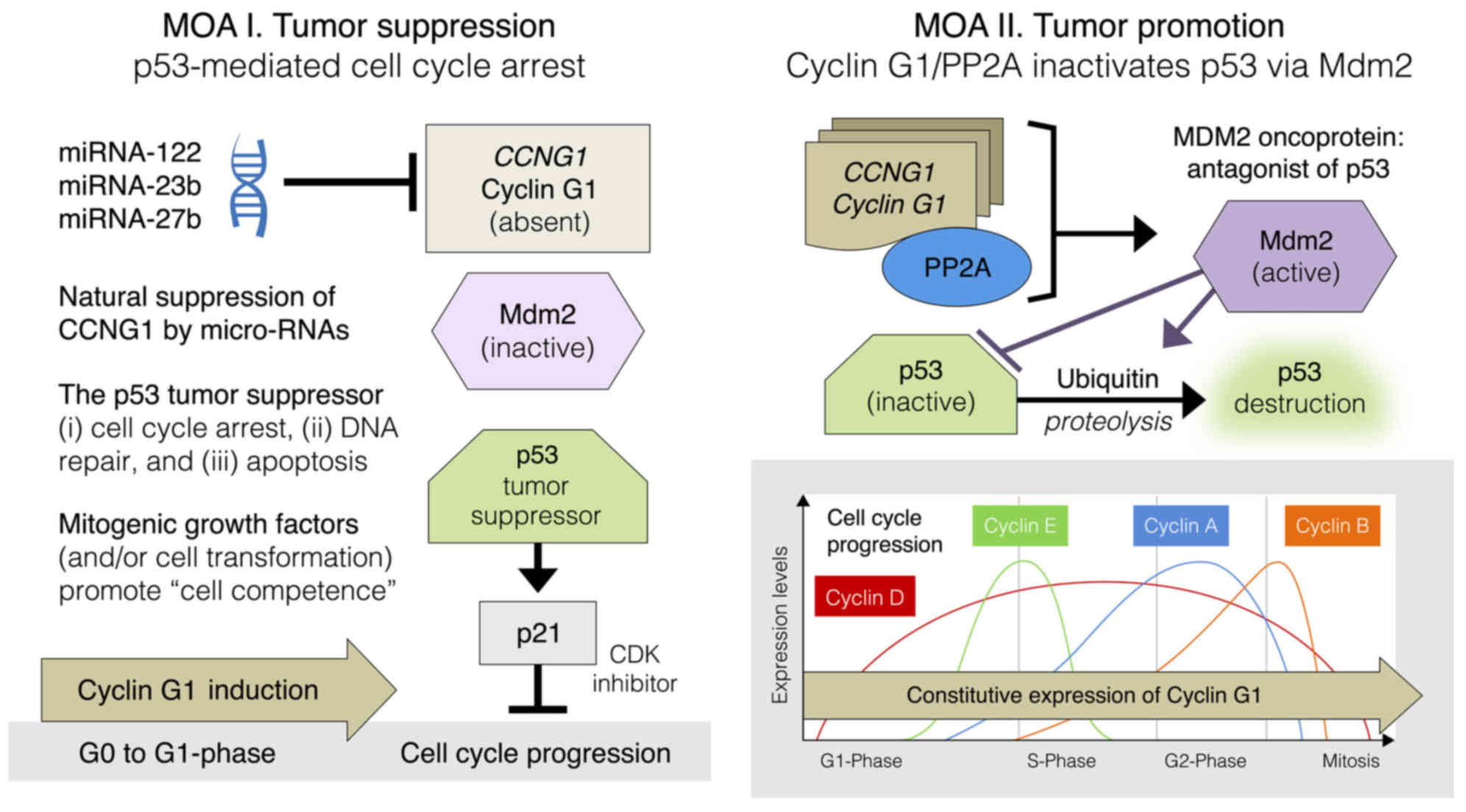
Cyclin G1, a non-canonical cyclin, opposes
p53, the fragile ‘guardian of the genome’
Cyclin G1 is a non-canonical cyclin that i)
structurally looks like a cyclin (cyclin box), ii) behaves like the
earliest of all G1 cyclins (induction: G0-to-G1 boundary) and iii)
physically targets a key regulatory enzyme (Mdm2) to a critical
checkpoint substrate (p53) in the executive regulation of the cell
division cycle. While the observed growth-promoting function of
human cyclin G1 is somewhat analogous to the progressive cyclin D,
E, A/CDK/pRb/E2F axis controlling cell cycle progression (Fig. 2), it is well-known that cyclin G1
operates via a separable and distinct regulatory pathway, namely
the commanding cyclin G1/Mdm2/p53 axis, where precision targeted
Ser/Thr phosphatase activity (i.e., cyclin G1-dependent Ser/Thr
phosphatase activity) is intimately linked, through biochemical
activation of the transforming protein of the MDM2 oncogene,
to the commanding checkpoint control function(s) of the p53 tumor
suppressor protein (Fig. 3).
The human cyclin G gene, now cyclin G1 or
CCNG1 (49), was initially
cloned by Hall et al at Children's Hospital Los Angeles (Los
Angeles, CA, USA), where it was determined to be overexpressed in
human osteosarcoma cells (50),
providing the first of several important links to cancer. The
observed overexpression of the cyclin G1 gene in human
osteosarcoma, which remained constitutive throughout the cell
division cycle in synchronized MG-63 cells, was rather perplexing,
since murine cyclin G was identified in mice in a molecular screen
for transcriptional targets of the p53 tumor suppressor, thereby
(mistakenly) suggesting a growth-inhibitory function rather than a
growth-promoting function for cyclin G1 (51). However, it was experimentally
determined that the enforced overexpression of cyclin G1 in either
normal or neoplastic cell lines did not cause the anticipated cell
cycle arrest, nor did experimental overexpression of cyclin G1
induce apoptosis (51), as the p53
tumor suppressor (referred to as the molecular guardian of the
genome and policeman of the oncogenes) is known to do under various
conditions (52). By contrast, the
ectopic overexpression of cyclin G1 is reported to promote the
clonal expansion of normal human fibroblasts and to accelerate cell
growth in RKO (p53+) colon carcinoma cells (53), whereas the molecular suppression (or
knockout) of cyclin G1 expression by antisense strategies was found
to be uniformly lethal (54). These
studies revealed an essential growth-promoting function for cyclin
G1, thus identifying CCNG1 as an accessible gene locus that
may, through its suppression, demonstrably inhibit the growth of
human tumor xenografts (upon intra-tumoral injection) in a nude
mouse model of cancer (55,56).
In 1995, Hall et al, in collaboration with
E.M. Gordon, Medical Director of the Vector Production Unit, USC
Gene Therapy Laboratories (Los Angeles, CA, USA) altered the focus
of his research laboratory from the biochemistry of
proline-directed protein kinases, CDK pathway characterization and
related gene discovery to work together in translational research:
Their goal was to develop a targeted and injectable gene delivery
vehicle (vector) that would be efficient, effective and safe for
repeated use in clinical practice. While their pioneering work on a
tumor-targeted retrovector was progressing, Hall and Gordon
utilized available retroviral vector-mediated gene transfer
technologies to investigate functional knockouts of cyclin D1
(antisense CycD1) and cyclin G1 (antisense CycG1) in MG-63
osteosarcoma cells, in comparison with the enforced expression of
p21WAF1/CIP1, a universal CDKI expressed in sense
orientation (54). After examining
the comparative experimental results in terms of both cytostatic
(all three constructs inhibited cell proliferation) and cytocidal
activities (i.e., TUNEL staining for overt apoptosis), Hall and
Gordon focused on cyclin G1 knockouts for potential clinical
development. At that time, this was a ‘road less traveled’ in terms
of regulatory biology, which did indeed make a difference in the
clinical setting.
The commanding cyclin G1/Mdm2/p53 axis
operating in normal and neoplastic cells
A critical ‘executor’ element in the cyclin
G1/Mdm2/p53 axis is the human homologue of the murine double minute
gene 2 (Mdm2) oncogene. Originally characterized as an
amplified gene locus encoding a potent transforming (i.e.,
tumorigenic) oncoprotein in mice, the human Mdm2 gene was
found to be amplified in numerous human cancers, including soft
tissue sarcomas (20%), osteosarcomas (16%) and esophageal
carcinomas (13%) (57,58). More recently, links to the
pathogenesis of breast cancer confirm that abnormally high levels
of the Mdm2 oncoprotein are detected in at least one-third (38%) of
human breast cancers, which cannot be explained by gene
amplification alone (59). Shortly
after its characterization, it was discovered that the Mdm2
oncoprotein forms a tight physical complex with the p53 tumor
suppressor, thereby inhibiting p53-mediated transactivation events
(60). In addition to inhibiting
p53-dependent transcription, it was determined that the Mdm2
oncoprotein functions biochemically as a specific E3 ubiquitin
ligase that is responsible for the ubiquitination and degradation
of the p53 tumor suppressor protein (61). Thus, the Mdm2 oncoprotein antagonizes
the p53 tumor suppressor on two levels: By ubiquitin-independent
repression of p53 transactivation, and by ubiquitin-dependent
proteolysis of the p53 protein (62), as seen in Fig. 3 (MOA II, tumor promotion).
This antagonistic association of the Mdm2
oncoprotein with p53 function and stability suggested that
overexpression of the Mdm2 gene is yet another molecular
mechanism, in addition to deletion or inactivating mutations in the
TP53 gene per se, that may functionally inactivate
the p53 tumor suppressor in the acquisitive process of neoplastic
transformation. Therefore, it was reasoned that pharmaceutical
agents targeting the Mdm2/p53 interaction in transformed cells
retaining wild-type TP53 status may be a promising new
approach to cancer therapy (63),
which is considered particularly important in the context of
hematological malignancies, where the frequency of TP53
mutations is relatively low (in comparison with solid tumors),
while Mdm2 is frequently amplified and overexpressed
(64). Pharmacological targeting of
the pivotal Mdm2/p53 interaction with natural compounds, small
molecules, and even stapled (helix-linked) peptides, has been
extensively investigated in an effort to restore the tumor
suppressor activity of wild-type p53 (65). Years of preclinical experimentation
have identified promising drug candidates for further clinical
evaluation; however, issues of drug-related toxicity and acquired
resistance persist, and it was concluded that this particular class
of compounds would definitely benefit from rational combinations
with a second, potentially synergistic, pharmacological strategy
(64).
In the elaboration of a pivotal Mdm2/p53 axis
controlling both DNA-fidelity and cell fate, the oncogenic function
of the non-canonical cyclin G1 was revealed: From a biochemical
perspective, the critical functions of the Mdm2 oncoprotein, which
is the major-negative regulator of p53, are intricately controlled
by multiple site-specific protein phosphorylation events (66), thereby necessitating a site-specific
Ser/Thr dephosphorylation event in order for the Mdm2 oncoprotein
to be conformationally activated to carry out the destabilization
of the p53. Moreover, this crucial Mdm2-activating
dephosphorylation event was found to be cyclin G1-dependent. It was
determined that the cyclin G1 protein forms a stable complex with
the Ser/Thr protein phosphatase designated 2A (PP2A) (67,68),
specifically with the B'-class of regulatory subunits that
determine both the substrate specificity and the subcellular
localization of PP2A complexes (69). Conceptually, cyclin G1 binds tightly
to the Mdm2 oncoprotein in vitro and in vivo, where
it recruits PP2A to the Mdm2 oncoprotein, thereby stimulating the
ability of the PP2A to dephosphorylate and activate Mdm2 at the
critical regulatory site (68). In
this capacity, cyclin G1 acts both as a targeting subunit and as a
selectivity factor that stimulates and propels PP2A catalytic
activity toward Mdm2. The discovery that cyclin G1 associates
physically with both PP2A and Mdm2 and, furthermore, that this
physical association regulates the accumulation and degradation of
the p53 protein (70), provides new
and important insights into the oncogenic function of cyclin G1 and
suggests that the main role of cyclin G1 may be to activate the
Mdm2 oncoprotein to override the cell cycle checkpoint control
functions of p53 (71). These
findings provide a mechanistic explanation for the observation that
cyclin G1 expression is associated with growth promotion rather
than with growth arrest (54,72).
This conclusion is further supported by studies of genetic
engineering in mice, where it was discovered that cyclin
G1-deficient mice not only survive, but exhibit a reduced incidence
of hepatic tumors upon exposure to hepatocarcinogens followed by
partial hepatectomy (73). This
decrease in tumor susceptibility associated with loss of cyclin G1
function is attributed to a resulting increase in p53 levels and a
corresponding increase in p53 tumor suppressor activity (73). Taken together, these findings raise
the possibility that the strategic modulation of cyclin G1 function
in the commanding cyclin G1/Mdm2/p53 axis may be targeted at the
molecular level for the development of novel anticancer agents.
Targeting cyclin G1 function in experimental
hyperplasia: Applications to cancer control
Upon examining the role of cyclin G1 in normal
proliferative cell populations, which exhibit normal levels of
wild-type TP53 and Mdm2 gene expression, it was
determined that downregulation of cyclin G1 (CCNG1) by
retrovirus-mediated antisense gene transfer inhibits vascular
smooth muscle cell proliferation and subsequent neointima formation
(a form of hyperplasia) seen in rat carotid arteries following
balloon catheter injury (74).
Similar cytostatic and cytocidal effects of antisense cyclin G1
treatment were observed in proliferative cell populations of both
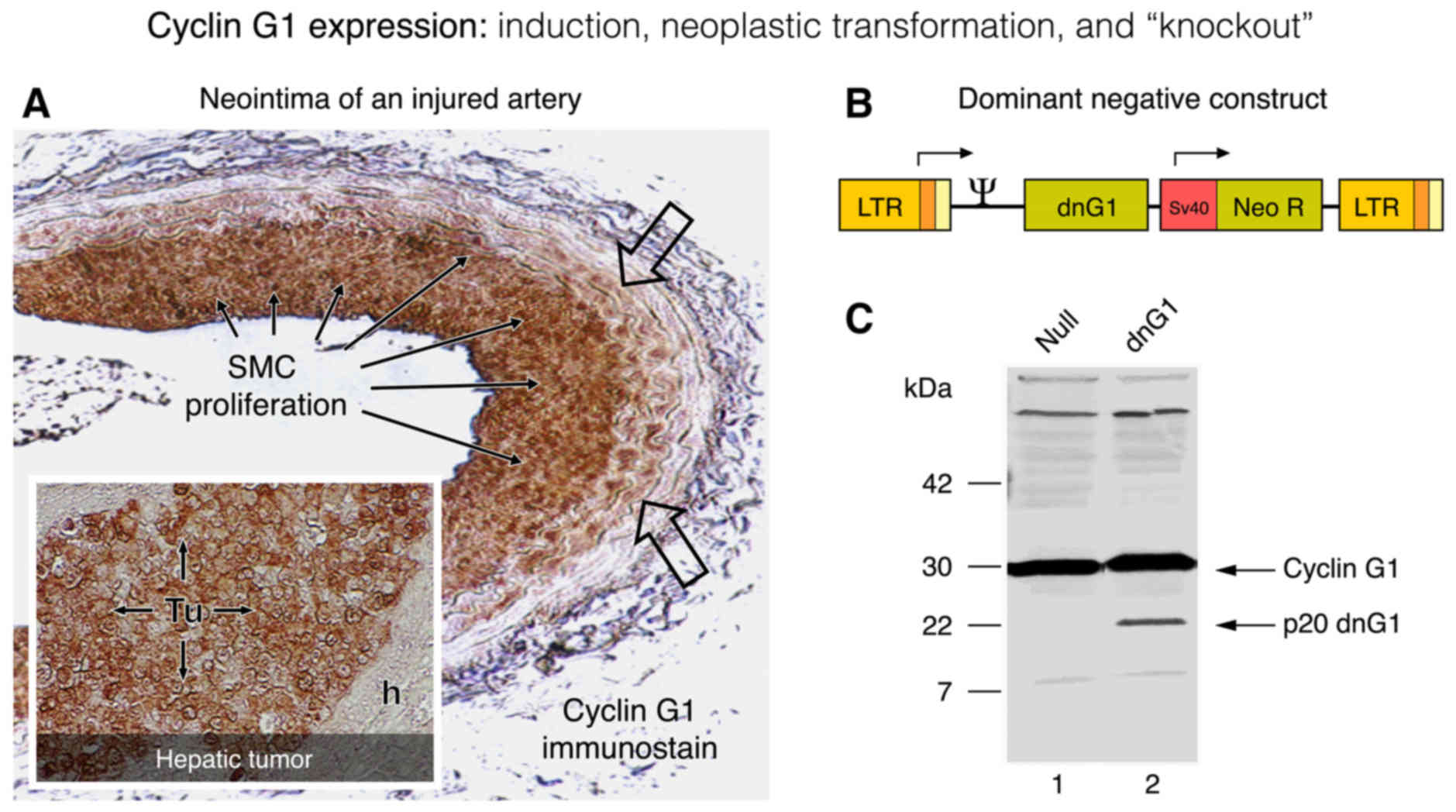
rabbit keratocytes and human fetal lens epithelial cells (75). Immunohistochemical staining for
cyclin G1 expression in balloon-injured arteries revealed a marked
induction of gene expression, from non-detectable levels in
quiescent arteries to relatively high levels of immunoreactivity in
the proliferative neointimal masses (Fig. 4A). The inducible expression of cyclin
G1 observed in vascular injury/restenosis is reminiscent of the
marked induction of cyclin G1 in the regenerating liver following
partial hepatectomy (76), rising
rapidly from exceedingly low basal levels measured in the quiescent
liver to appreciable steady-state levels. In contrast to the
normally low levels seen in the liver, the expression of cyclin G1
in human tumor xenografts appears to be constitutively elevated
(Fig. 4A).
Targeting cyclin G1 function with a
dominant-negative (dnG1) mutant construct. After realizing that
antisense ‘knockdown’ of cyclin G1 expression in normal and
neoplastic cells was indeed informative and promising, yet
reasoning that partial or incomplete suppression of CCNG1 gene
expression may still be problematic under physiological conditions
(where enzymatic phosphotransferase rates are measured in msec and
compensatory changes in gene expression are likely to occur), it
was considered preferable to develop and deploy a
‘dominant-negative mutant’ construct of the cyclin G1 protein, the
enforced expression of which would serve to counteract vital
aspects of the cyclin G1 structure, binding and/or metabolism,
thereby acting as a cyclin G1 antagonist, even in the presence of
an abundance of the wild-type protein (Fig. 4B). Working with structural deletions
and point mutations in the human cyclin G1 coding sequences, based
on the homology of the repeating helical domains comprising what is
referred to as the cyclin box, and the conserved amino acid
residues predicted from crystallography to serve as contact points
of the cyclin A/CDK2 complex (49,50), an
N-terminal deletion mutant was selected for development as the most
potent negative-acting mutant of cyclin G1 (77). This dominant-negative mutant
construct of human cyclin G1 (designated dnG1) is devoid of the
extended N-terminal domain proximal to the cyclin box domain, and
is missing the first two helical segments (α1 and α2 helices) of
the cyclin box itself (Fig. 9).
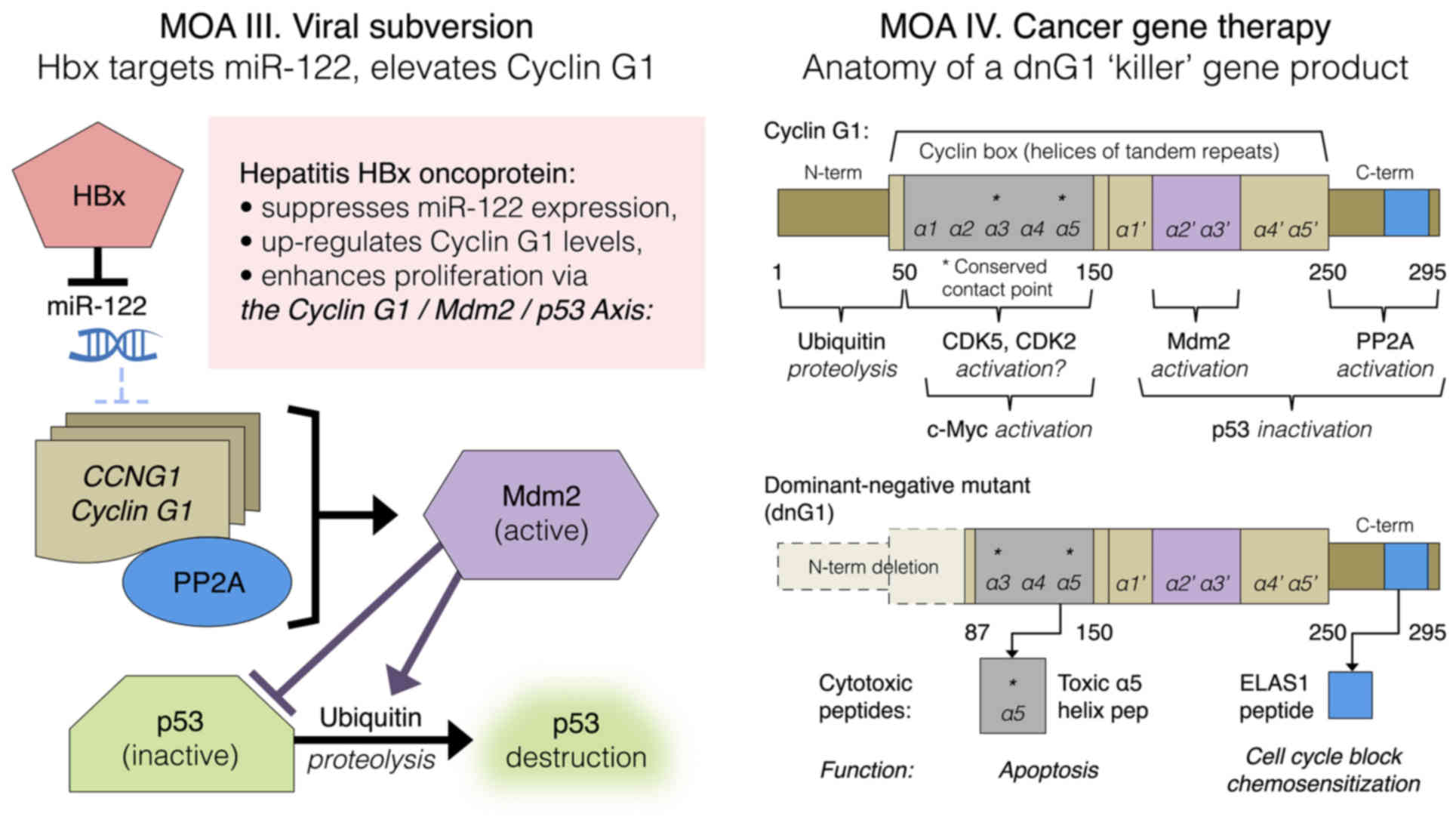 | Figure 9.Left panel: Mechanism-of-action (MOA)
III. HBx-mediated viral subversion of the hepatocellular division
cycle operates by suppression of miR-122, hence de-repression of
CCNG1 expression. Right panel: MOA IV. Cancer gene therapy,
dng1 structure. The cytocidal dnG1 protein, a dominant-negative
mutant construct of cyclin G1, is devoid of the ‘ubiquitinated’
N-terminus (proteolytic processing), as well as the first two
helical segments (α1 and α2) of the definitive cyclin box,
characteristically arrayed in cyclins as a tandem set of helical
segments, including two highly-conserved residues (asterisks)
essential for cyclin-dependent kinase (CDK) binding. The cytocidal
dnG1 protein, which induces apoptosis in proliferative cells,
retains the presumptive (?) CDK contact points (Helix α3*, α5*) and
the structural domains attributed to PP2A, β' and Mdm2 binding.
Remarkably, new therapeutic peptides (e.g., ELAS1 and α5 Helix
peptides) derived from structures or homologous interfaces
contained within the dnG1 protein are reported to induce cell cycle
blockade and apoptosis, respectively. |
Development of a targeted retroviral vector
to efficiently deliver the dnG1 ‘designer gene’
The turn of the 21st century brought major
biotechnological advances in the design and clinical applications
of actively-targeted gene delivery vehicles, specifically
pathotropic (disease-seeking) gene therapy vectors, which enabled
the first systemically administered, tumor-targeted retroviral
vectors to be validated in clinical medicine (78). While the gristly pathobiology of the
tumor extracellular matrix (ECM), as well as the molecular
mechanisms of retroviral vector-mediated gene transfer and
expression, are beyond the scope of this focused review, the
potential clinical implications of an ‘active’ tumor-targeted gene
delivery vehicle are wide-ranging. Focusing on the abnormal
collagenous proteins (i.e., lesion signature proteins) that are
pathologically exposed during the process of tissue injury (such as
balloon angioplasty), Hall and Gordon adaptively engineered a
physiological surveillance function embodied within the complex
structure of von Willebrand coagulation Factor (vWF), which
normally guides platelets to sites of significant tissue injury,
into the surface envelope protein (gp70) of the Moloney murine
leukemia virus, thereby creating a desirable ‘gain-of-function’
(pathological-matrix-targeting), without impairing the natural
receptor-mediated binding and entry of the targeted viral particles
into target cells, thus preserving the efficiency of gene transfer
(79,80). Eventually, in anticipation of human
gene therapy applications, the enabling tumor-targeting
gain-of-function, first established in rodents, was genetically
engineered into the 4070A ‘amphotropic’ murine leukemia virus
envelope protein that is capable of transducing human cells
(81,82).
Armed with two powerful enabling biotechnologies, i)
the matrix/lesion-targeted retrovector and ii) the cytocidal
dominant-negative mutant construct of cyclin G1 (dnG1), validation
of both targeted gene delivery and single-agent efficacy was
demonstrated in preclinical studies of vascular restenosis, where
significant long-term inhibition of neointima formation in
balloon-injured rat arteries was observed (77,83).
This initial proof-of-concept was followed by preclinical studies
on the safety and efficacy of the matrix/lesion-targeted vector
bearing the inhibitory dnG1 construct (Mx-dnG1) applied as simple
eye drops for the prevention of corneal haze induced by excimer
laser-injury in rabbits (84). The
encouraging results of this preclinical study prompted a formal
evaluation of potential applications of anti-proliferative eye
drops in humans by the NIH Recombinant DNA Advisory Committee
(RAC), for a phase I/II intervention for superficial
opacity/corneal scarring that occurs after phototherapeutic
keratectomy (85). With these
critical proofs-of-principle in hand (i.e., the efficacious
preclinical management of experimental hyperplasia with Mx-dnG1),
Hall and Gordon shifted their pioneering studies of lesion-targeted
gene delivery to include the always pertinent and challenging
models of metastatic cancer (86,87)
(Figs. 5–7), where the physiological obstacles to
tumor-targeted gene delivery (dilution, filtration, turbulence,
sheer forces, inactivation) remain paramount, and the clinical
management of cancer cell growth, as well as the clinical
management of tumor-angiogenesis, represents a major unmet medical
need.
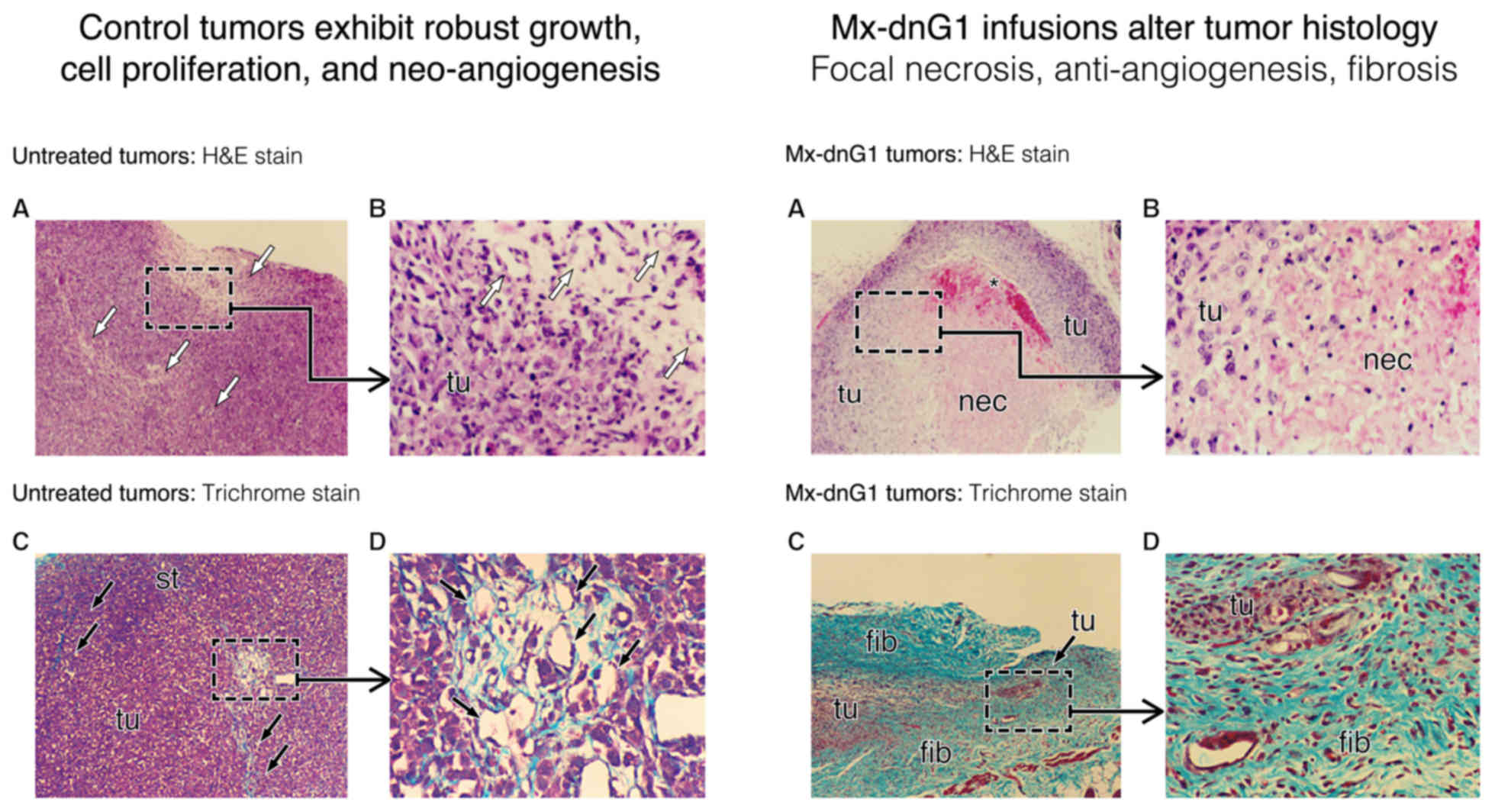 | Figure 7.Characteristic changes in tumor
histology following intravenous Mx-dnG1 treatment. Untreated tumor
xenografts (left panel) exhibit robust tumor (tu) cell
proliferation with active zones of neo-angiogenesis (A, open
arrows; B, magnified); Masson's trichrome stain (C, D magnified)
reveals exposed collagenous proteins (solid arrows, blue stain)
associated primarily with angiogenesis and reactive stroma
formation (st). By contrast, Mx-dnG1 treatment (right panel)
reduces the flagrant population of proliferative tumor cells (tu)
significantly (A, boxed; B, magnified), producing large regions of
focal necrosis (nec) accompanied by overt anti-angiogenesis (*).
Residual tumors are ultimately reduced to besieged clusters of
proliferative tumor (tu) cells (C, boxed; D, magnified), as
reactive/reparative fibrosis (fib), including nascent (i.e.,
targetable) ECM deposition (blue stain), dominates the histology of
tumor regression observed in Mx-dnG1 treated animals. |
Important insights into the mechanisms and
efficiencies of tumor-targeted gene delivery were gained from a
liver model of metastatic cancer, wherein human MiaPaca2 pancreatic
cancer cells were infused into the portal vein to establish tumors
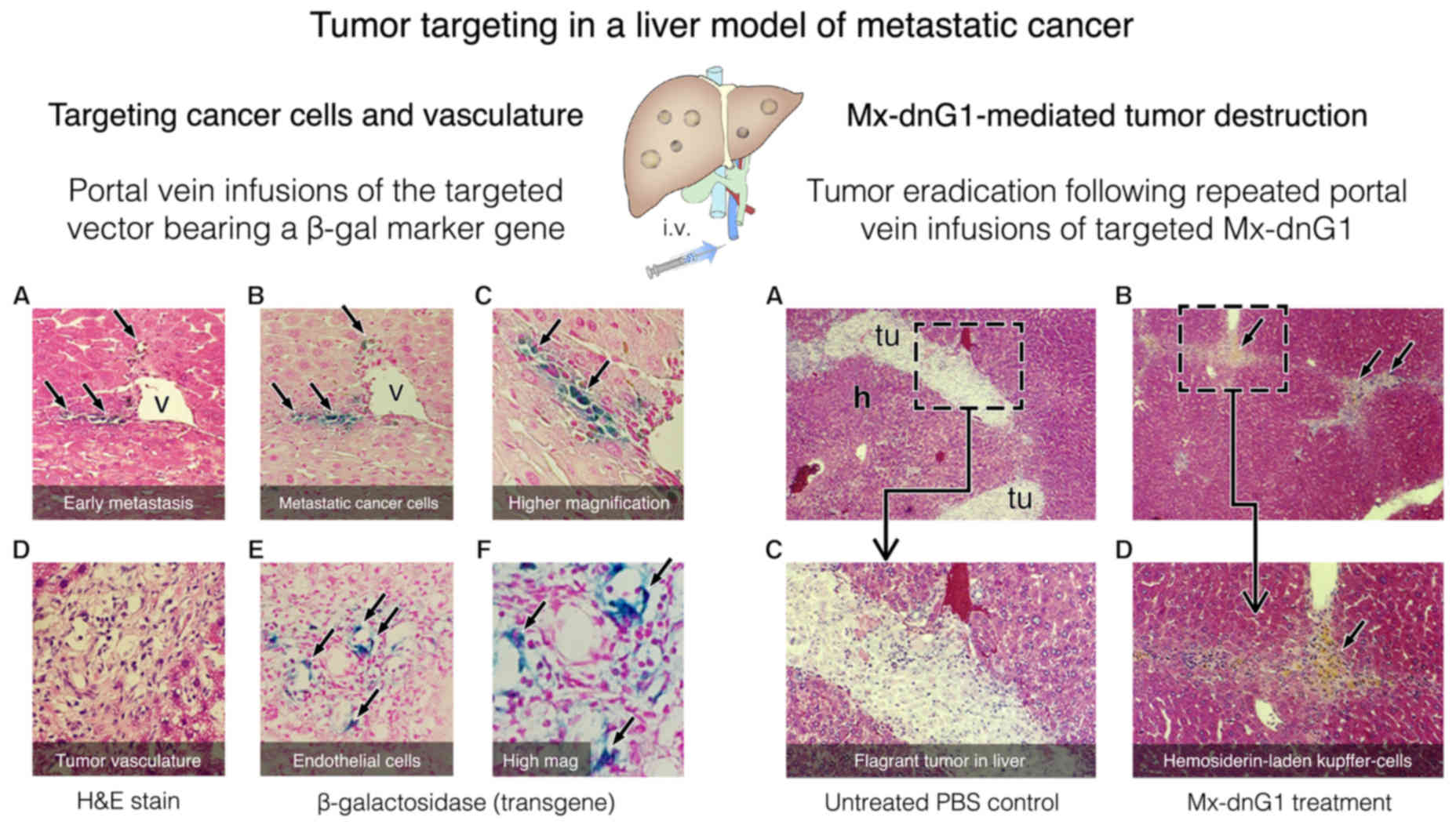
in nude mice (Fig. 5). In this model
of regional gene delivery to established liver metastases, marker
studies demonstrated an informative and selective targeting of
cancer cells within the liver during the earliest stages of the
metastatic process (Fig. 5A and C;
left panel). Of note, this exquisite targeting of collagenous
matrix signature proteins (i.e., biochemical footprints) left by
invasive cancer cells, long before a discernable tumor is formed
within the liver, has considerable implications in the diagnosis
and treatment of human cancers. Later on, when flagrant tumors are
formed within the liver and robust tumor angiogenesis is apparent,
the tumor targeting extended to the tumor vasculature, where the
proliferative endothelial cells are transduced by the vector, as
demonstrated by β-galactosidase transgene expression (Fig. 5D-F; left panel). First and foremost,
when the marker gene was replaced by dnG1, as in the cancer gene
therapy vector (Mx-dnG1), dose-dependent tumor eradication was
observed upon repeated intravenous infusions (86) (Fig. 5;
right panel).
Prospective clinical trials for this regional
approach to cancer gene therapy underwent formal review (and
approval) for use in humans by the Food and Drug Administration
(FDA)/RAC, a tumor site-specific phase I/II evaluation of the
safety and efficacy of hepatic arterial infusion of a
matrix-targeted retroviral vector bearing a dnG1 construct as
treatment for colorectal carcinoma metastatic to the liver
(88). However, at this point, Hall
and Gordon opted instead to explore the even greater potential for
systemic gene delivery (87), in
light of the potential for more wide-ranging clinical applications
in the treatment of metastatic cancers (Figs. 6–7).
In a classic nude mouse model of metastatic cancer
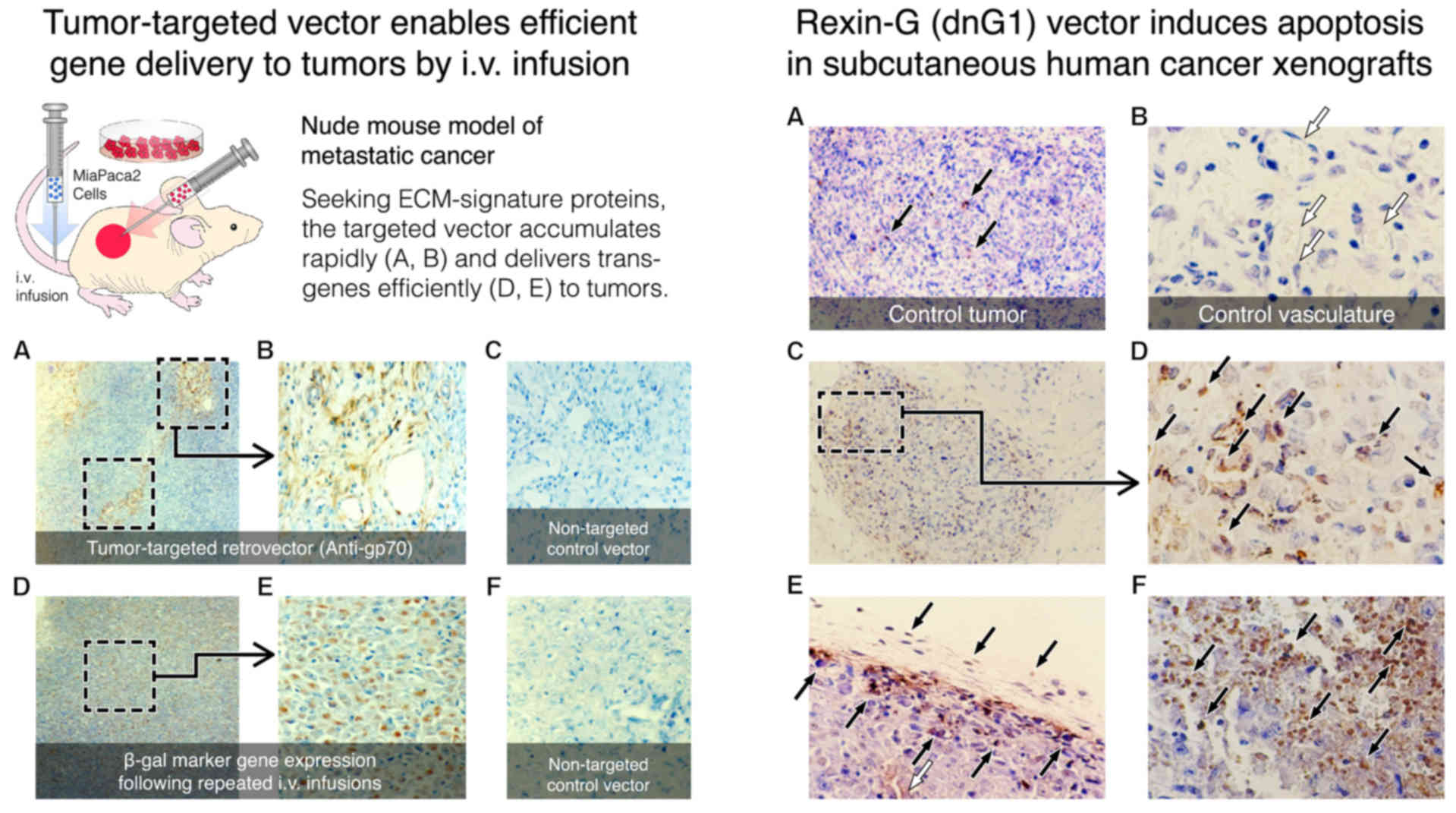
(Fig. 6; left panel), it was
observed that tumor-targeted gene delivery is not only conceivable
(A and B vs. C), it is apparent (D and E vs. F); in addition, it is
evident that the physiological partitioning of the circulating
vector to tumors is dependent on the high-affinity matrix
(Mx)-binding domain that was genetically transposed from the D2
module of the vWF propeptide into the primary structure of the
retroviral envelope protein (79–83,87).
Notably, in order to accomplish tumor-targeted gene delivery within
1 h, the matrix/lesion-targeted vector infused into the tail vein
of the mice necessarily transited the heart, the lungs, and the
heart once more, prior to entering the systemic circulation,
including the blood vessels feeding the tumor xenografts (Fig. 6A and B; left panel), where it appears
to spread out, similar to Coomassie blue dye in a natural sponge,
as it physically contacts and effectively transduces proliferative
tumor cells with a β-galactosidase marker gene (Fig. 6D and E; left panel).
When the marker gene was replaced with the
inhibitory dnG1 construct (Fig. 6;
right panel), it was confirmed that the cytocidal mechanism of
action of dnG1, as observed in both hyperplastic lesions and
cancerous xenografts, is attributed to the activation of DNA
fragmentation via the process of apoptosis. This pioneering study
demonstrated that the Mx/lesion-targeted dnG1 retroviral vector
(Mx-dnG1) deployed by peripheral vein injection i) accumulates
rapidly in tumor vasculature, ii) transduces tumor cells with
exceedingly high-level efficiency and iii) enables demonstrable
therapeutic gene delivery and long-term efficacy of dnG1 without
eliciting appreciable toxicity (87). In addition to the marked apoptosis
observed in Mx-dnG1 treated tumors (Fig.
6; right panel), characteristic histological changes in tumors
after repeated systemic administration includes: Zones of focal
necrosis, overt anti-angiogenesis and reparative fibrosis
accompanied by ECM deposition, which occupies an increasingly
greater proportion of the residual tumor, as the cancer cells and
the proliferative tumor vasculature are eradicated progressively by
the repeated intravenous infusions of Mx-dnG1 (Fig. 7; left panel A-D control tumors vs.
right panel A-D treated tumors).
Following the establishment of three preclinical
proofs of principle, namely that i) tumor-targeted gene therapy is
feasible via simple intravenous infusions, ii) tumor-targeted
delivery of the dnG1 ‘killer gene’, which exhibits both
anti-angiogenic and anti-tumor activities, is capable of altering
the course of metastatic pancreatic cancer, an otherwise fatal
disease and iii) the design features and general safety profile of
the first targeted, injectable gene therapy vector had been
critically evaluated by the FDA/RAC and formally approved for
clinical use in humans, the first clinical studies were
initiated.
Clinical studies demonstrate broad-spectrum
anticancer activity and single-agent efficacy
In 2003, a clinical-grade retroviral expression
vector bearing an inhibitory dnG1 construct of the cyclin G1 gene
(designated Rexin-G) became the world's first targeted, injectable
vector to be approved for clinical trials in the treatment of
intractable metastatic disease. Uniquely suited, by design, to
perform this tumor-targeting function within the context of the
human circulatory system, Mx-dnG1 (aka Rexin-G) is a pathotropic
(disease-seeking) gene delivery vehicle bearing the dnG1 ‘killer’
gene, a therapeutic tumor-targeted nanoparticle that selectively
seeks out and accumulates in metastatic lesions upon simple
intravenous infusion. Preclinical validation of tumor-targeted gene
delivery, accompanied by critical demonstrations of dose-dependent
efficacy in pertinent cancer models, was followed by the first
clinical studies, which aimed to demonstrate the potential for
achieving broad-spectrum, single-agent, anticancer efficacy in the
clinic. A summary of completed clinical studies is shown in
Table I.
 | Table I.Clinical studies evaluating repeated
intravenous infusions of a pathotropic Mx-dnG1 retrovector
(Rexin-G) as monotherapy for chemoresistant metastatic cancer. |
Table I.
Clinical studies evaluating repeated
intravenous infusions of a pathotropic Mx-dnG1 retrovector
(Rexin-G) as monotherapy for chemoresistant metastatic cancer.
| Completed
studies | Types of cancer
treated | Description of
results, conclusions | (Refs.) |
|---|
| International phase
I/II protocols for pancreatic cancer are expanded to include all
solid tumors | Advanced pancreatic
cancer, expanded to include metastatic melanoma, breast, uterus,
colon cancer, and leiomyosarcoma | Demonstrations of
overall safety, lack of toxicity, and potential for clinical
efficacy is established; a dose-response calculus is advanced | (90,91) |
| Phase I (USA)
low-dose safety study in advanced or metastatic pancreatic Ca | Advanced pancreatic
cancer | General safety is
confirmed; no RCR no inactivating antibodies; no DLT, enabled
further dose escalation(s) | (92) |
| Phase I/II dose
escalation studies of safety and efficacy |
Chemotherapy-resistant sarcoma, breast
cancer and pancreatic cancer | Demonstrates
significant anticancer activity (efficacy) without toxicity | (93,94) |
| Phase I/II with
advanced phase II study of resistant sarcoma and osteosarcoma |
Chemotherapy-resistant sarcoma and
osteosarcoma | Safety and
dose-dependent clinical efficacy is demonstrated; significant
improvements in overall survival | (95,96) |
| Advanced phase I/II
study of chemotherapy-resistant metastatic pancreatic Ca | Advanced pancreatic
cancer | Safety and
dose-dependent clinical efficacy is demonstrated; significant
improvements in overall survival | (97) |
As with all targeted biologics, a major objective
was to assess and establish the clinical safety of the vector
system, while progressively seeking efficacy within the rule-based
designs and dose escalation protocols used in phase I cancer
clinical trials (89). Cooperative
(US FDA sanctioned/BFAD-approved) international studies conducted
in the Philippines enabled stepwise intra-patient dose escalations,
thereby achieving clinical dose-dependent efficacy in an expedient
manner (90,91), while the US FDA required more
traditional studies of potential dose-limiting toxicities (DLT)
and/or cumulative toxicities to be conducted at sub-therapeutic
doses (92) before a formal
determination of adequate safety would enable a progressive
stepwise dose escalation. When clinical significant dose-dependent
efficacy was demonstrated in US-based phase I/II studies of
intravenous Rexin-G for advanced sarcoma and osteosarcoma (93–95),
including significant improvements in both progression-free and
overall survival, the FDA approved an ‘across-the-board’ dose
escalation allowance (applicable to all ongoing trials), thereby
enabling these higher, more efficacious doses (i.e., optimal
biological doses) to also be administered to breast and pancreatic
cancer patients (96–98).
From these clinical studies, including the
physiological dose-response kinetics, a predictive pharmacology of
tumor-targeted genetic medicine began to emerge: A calculus of
parity (or therapeutic equivalence) was introduced, in which the
metastatic tumor burden of a given patient is evaluated and used in
calculations, including vector targeting efficiencies and
performance, in determining the effective intravenous dose(s)
required to achieve tumor eradication (91). Advancements in bioprocess development
enabled high-potency clinical grade vectors, which further
simplified intravenous infusions and led to the accelerated
approval of Rexin-G for all solid tumors by the Philippine FDA in
2007. This was followed by a rapid progression of clinical studies
in the US, where Rexin-G received several Orphan Drug designations,
including FDA Fast-Track status for metastatic pancreatic cancer,
which stands at the cusp of phase III clinical trials (97,98).
Long-term follow-up of an advanced phase I/II study using Rexin-G
for chemotherapy-resistant sarcomas revealed the clinical value of
the observed Rexin-G (dnG1) dose-response relationships in terms of
overall patient survival: The 1-year survival rate increased from
0% at dose level I to 28.5% at dose level II–III, to an impressive
38.5% at dose level IV–V (with 31% of the patients alive at 2
years) following initiation of Rexin-G monotherapy, which may be
considered as a gold standard in terms of objective clinical
responses (96–98).
At the time of this review, Rexin-G had been
administered safely to >270 patients worldwide, including
>3,000 total intravenous infusions without serious side effects,
vector issues, inactivating antibodies, or DLT. Documentation of
significant anticancer activity, single-agent efficacy and
objective clinical responses (including evidentiary histology),
have been reported in a broad spectrum of cancers derived from all
three germ layers (98–100). Clinical confirmation of i)
tumor-targeted gene delivery, ii) a pro-apoptotic dnG1 mechanism of
action and iii) characteristic changes in tumor histology
(originally observed in preclinical models) are clearly
recapitulated in the clinical setting, as is clearly seen in an
opportunistic surgical (liver) biopsy obtained from a metastatic
pancreatic cancer patient undergoing intravenous Rexin-G treatment
(Fig. 8). In addition to
statistically significant increases in patient survival, a
considerable number of advanced-stage, chemotherapy-resistant
cancer patients treated with repeated infusions of Rexin-G as
monotherapy, including metastatic breast cancer, pancreatic cancer,
malignant melanoma, osteosarcoma and soft tissue sarcoma patients,
remain cancer-free or without active disease progression ≥9-12
years after the initiation of Rexin-G treatment (101).
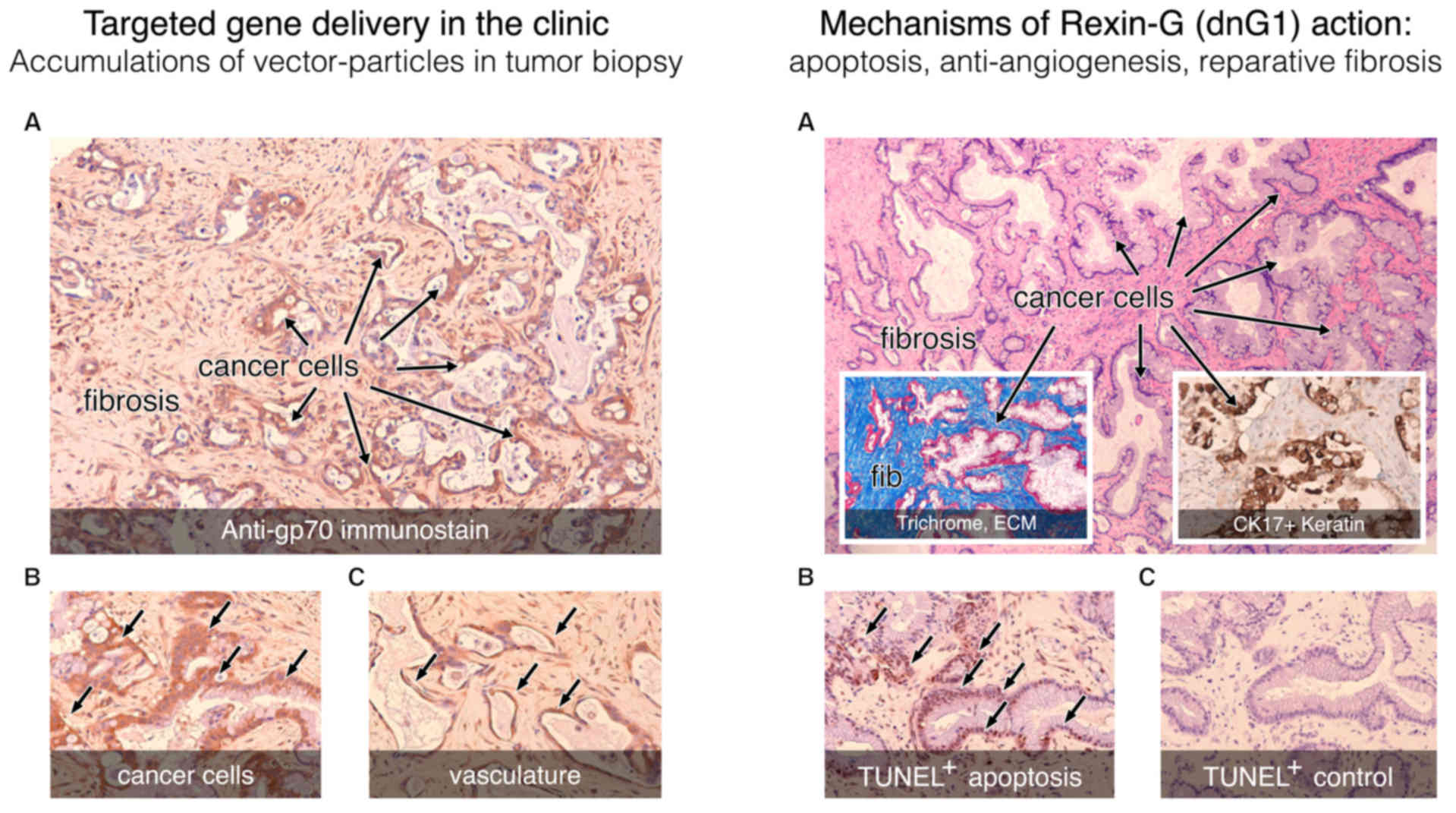 | Figure 8.Histochemical analysis of a tumor
biopsy: Pancreatic cancer, metastatic to the liver, following
intravenous infusions of tumor-targeted Mx-dnG1 (Rexin-G). Left
panel, tumor targeting: Immunohistochemical staining for the
retrovector gp70 envelope protein (brown staining) demonstrates
widespread vector penetration and accumulation within the tumor
(A), with particularly high levels of immunostaining appearing in
the cancer cells (A and B, arrows) and associated vasculature (C,
arrows). Right panel, mechanisms of action: (A) Clusters of
residual (CK17+) pancreatic cancer cells (A, inset of
western blot) are distinguished from the reactive fibrosis (fib; A,
inset) seen with Masson's trichrome stain, which stains the
collagenous extracellular matrix proteins bright blue. Application
of the TUNEL method for detecting apoptotic DNA fragmentation
reveals massive levels of apoptosis (arrows) in cancer cells (B vs.
C, control) as the actual mechanism of dnG1 action. |
Restoring long-lost tumor suppression with a
dominant-negative cyclin-G1 antagonist
Strong support for the proposed growth-promoting
function of cyclin G1 in the commanding cyclin G1/Mdm2/P53 axis
came from functional genomics, which identified microRNAs (miRNAs)
as a new class of endogenously expressed, small non-coding
gene-regulatory RNA molecules that interact with the p53
transcription factor and its network of effectors at multiple
levels (102,103), thereby impacting both the mechanics
of cell cycle progression and the fate of proliferative cells.
High-throughput screens investigating the role of miRNAs in the
pathogenesis of human HCC identified miR-122 as the most
predominant species of hepatic regulatory miRNA, which is either
lost or severely downregulated in ~70% of HCC cancers and in all
HCC-derived cell lines (104).
These studies identified cyclin G1 as a primary target of miR-122
suppression, which accounts for the inverse correlation between
miR-122 and high levels of cyclin G1 expression observed in primary
liver carcinomas. Loss of miR-122 expression in liver cancer
correlates with a poor prognosis indicative of tumor progression,
including a notable gain in invasive metastatic properties
(105). As depicted in Fig. 3, miR-122 regulates p53 protein
through cyclin G1 in HCC cells (106): The miR-122/cyclin G1 interaction
modulates p53 protein stability and transcriptional activity.
Accordingly, enforced/restored expression of miR-122 decreases cell
viability (107) and induces cell
cycle arrest and apoptosis in HCC cell lines (108).
Important insights into the natural tumor suppressor
function of miR-122 were gained from genetic engineering, where it
was determined that miR-122 gene knockout mice are prone to hepatic
tumor development, while AAV-mediated delivery of miR-122 to the
livers of such mice inhibits tumorigenesis (109). By repressing the expression of
cyclin G1 (CCNG1 transcription), miR-122 increases p53
protein levels and suppressor activity and inhibits tumorigenesis
in liver cancer models. Insights into the proto-oncogenic function
of cyclin G1 were gleaned from studies of HBV subversion in HCC
(110), where it was determined
that the HBx-protein directly mediates suppression of miR-122
expression and enhances hepatoblastoma cell proliferation through
the functional modulation of the cyclin G1/Mdm2/p53 axis; i.e.,
downregulation of the expression of the tumor suppressive miR-122
increases cyclin G1 expression, which in turn abolishes
p53-mediated suppression of HBV replication and promotes
hepatocellular proliferation (Fig.
9; MOA III viral subversion). Thus, it appears that the
inhibitory dnG1 expression construct described herein, by
counteracting the oncogenic function(s) of cyclin G1, is capable of
restoring a lost, missing, or deregulated axis of tumor
growth/suppression, which operates by triggering an endogenous
apoptotic mechanism of action, in the presence or absence of
p53.
Cyclin G1 proto-oncogene promotes cell
survival and progression over DNA fidelity
The establishment of cyclin G1 (CCNG1) as a
bona fide proto-oncogene and a powerful growth-promoting protein
has profound implications for the clinical staging, prognosis and
management of human cancers. Normally induced in response to tissue
injury (G0/G1 boundary), as in hyperplasia (Fig. 4), cyclin G1 is frequently
overexpressed in human breast and prostate cancers (111), osteosarcomas, colorectal cancers
(112) and HCC (113), wherein CCNG1 overexpression
is clearly correlated with a poor prognosis. As mentioned above,
CCNG1-null mice survive and even exhibit decreased
incidence, size and malignancy of tumors due to the resulting
dominance of DNA-sensing p53-dependent tumor suppressor functions
(73); however, in proliferative
populations of tumor cells, including tumor neovasculature, the
blockade of cyclin G1 expression or function is uniformly lethal.
This apparent dependence of cancer cells on sustained cyclin G1
expression for cell survival, which is acquired during the course
of diverse multistage carcinogenesis, is referred to as ‘oncogene
addiction’ (114); indeed, this
acquired state of dependency on cyclin G1 identifies CCNG1
as a vulnerable locus in human cancers, which can be targeted on
several levels to gain a positive therapeutic advantage.
Examination of the structure of cyclin G1 provides
conceptual insights as to its multiplex biochemical functions
(Fig. 9; MOA IV, right panel). The
cyclin G1 protein is an unusually unstable protein, exhibiting a
half-life of ~20 min; this instability is attributed to
ubiquitin-mediated proteolysis involving the extreme N-terminus of
the protein (115). The homology
between cyclin G1 and known regulatory cyclins suggests that cyclin
G1 may activate one or more CDK partners under certain conditions.
Indeed, cyclin G1 is capable of physically interacting with CDK1,
2, 4, 5 and 6, the kinase activity of which appears to contribute
physiologically to the short half-life of the cyclin G1 protein
(116). However, neither Histone
H1- nor Rb-kinase activity was detected in cyclin G1/CDK2
immunoprecipitates (116),
suggesting that cyclin G1 association with a specific CDK partner,
as well as the resulting CDK activity, may be more tightly
regulated by additional factors and/or is uniquely targeted to one
or more undetermined protein substrates. Subsequently, convincing
aspects of cyclin G1 function in direct association with CDK5 have
been revealed: Activation of CDK5 by cyclin G1 binding physically
targets the active kinase complex to the c-Myc oncoprotein, the
overexpression of which immortalizes cells, reduces growth factor
requirements, promotes cell cycle progression and inhibits cell
differentiation (117). The
resulting cyclin G1/CDK5 phosphorylation of c-Myc on Ser-62
(-X-Ser62-Pro-X-) stabilizes the multifunctional
transcription factor, resulting in transcriptional activation of a
defined (E-Box-containing) set of growth-promoting genes, including
both cyclins and CDKs, which are capable of driving cell
proliferation (118).
Mechanistically, c-Myc is a critical platelet-derived growth
factor-inducible ‘competence gene’ that activates diverse cellular
processes associated with entry and progression through the cell
cycle, including the synthesis of cellular components in
preparation for cell division. In this manner, by activating and
selectively targeting CDK5 kinase activity to activate and
stabilize the c-Myc oncoprotein, the overexpression of cyclin G1
enables cancer cells to overcome radiation-induced (i.e.,
DNA-damage-induced) cell cycle arrest (117). Although the transcriptional targets
of c-Myc include a number of DNA repair genes, thereby coupling DNA
replication to the pathways and processes that preserve the
integrity of the genome (118), the
net effect of cyclin G1 function in association with CDK5 (or CDK2)
is to abrogate DNA-fidelity checkpoint controls to promote cell
survival, cell competence and cell cycle progression at the peril
of increasing error-prone DNA synthesis, as is often found in
cancers.
Significant clues into the function of cyclin G1/CDK
complexes as a ‘survival factor’ were obtained in the nervous
system, where cyclin G1 expression was induced by nerve injury in
mature (post-mitotic) motor neurons and remained elevated during
the early stages of nerve regeneration along with other
‘immediate-early’ genes associated with cell survival (119), suggesting that cyclin G1 functions
as a survival factor in post-mitotic neurons, maintaining cell
viability during the process of tissue repair. Following this
mechanistic insight, we find that mild cognitive impairment, a
transitional disease state that precedes more serious
neurodegenerative problems associated with Alzheimer's disease
(AD), is accompanied by increased co-expression of cyclin G1, CDK2
and CDK5 in afflicted brain regions (120). This is in keeping with the theory
that neuronal cell death associated with AD has, as its root cause,
an ectopic re-entrance into the cell cycle (121), which results in the
hyperphosphorylation of microtubule-associated tau proteins
characteristic of AD neurofibrillary tangles. Various neurotoxic
insults induce hyperactivation of CDK5, with fatal consequences for
the cell (122). On the other hand,
lack of CDK5 activity results in neuronal cell death (123), suggesting that precise control of
CDK5 activity is crucial for neuron survival via the site-specific
phosphorylation of the apoptosis inhibitor Bcl-2 (124). Apart from cyclin G1/CDK5 complexes,
which target and activate the c-Myc competence gene in
cancer cells (117), a
brain-specific regulatory subunit of CDK5 has been identified as
p35, the N-terminal truncation of which (from p35 to p25) causes
prolonged activation of CDK5, mis-localization and pathological
phosphorylation of substrates; i.e., the p25/Cdk5 kinase
hyperphosphorylates tau, disrupts the cytoskeleton and promotes
cell death by apoptosis in primary neurons (122,125).
In this same manner in which the proteolytic truncation (N-terminal
deletion) of this p35 CDK5-activator protein converts the p35/CDK5
kinase complex from a survival factor to a cytotoxic p25/CDK5
kinase activity that promotes cell death by apoptosis in neurons,
the ‘designer’ N-terminal truncation of cyclin G1 into a dnG1
mutant protein (Fig. 9) converts the
survival functions (i.e., anti-apoptotic, pro-survival) of the
CCNG1 oncogene (mediated via cyclin G1/CDK5-dependent c-Myc
activation) into the pro-apoptotic, cytocidal functions of the
truncated mutant dnG1 ‘killer’ gene. In this case, a potent
proteolytically stable antagonist of wild-type CCNG1 gene
function is generated.
A note to future biomedical researchers. While the
functionality of cyclin G1 in the nervous system is beyond the
scope of this oncological review (cancer cells are constitutively
competent), this mechanistic link to cell survival and
proliferative competence has important implications in terms of
artificially/therapeutically promoting nerve regeneration and
associated tissue repair, in light of the striking potentiality for
tail and/or limb regeneration that is evident in lower
vertebrates.
Whereas the early induction of cyclin G1 observed
as cells exit from quiescence (G0-to-G1 transition) in experimental
models of hyperplasia and liver regeneration is indicative of its
role in establishing cellular competence to proliferate, the
putative CDK kinase partner would thereby represent a G0/G1
competence-promoting factor (CPF), perfectly analogous to SPF and
MPF. The target substrate for this cyclin G1-dependent kinase
activity, CDK5 and/or possibly CDK2, (116,120)
is the c-Myc oncogene, the activation of which is associated
with the start of cellular proliferation, cell cycle progression,
increased genomic instability, reduced adhesion, EMT, tumor
invasion and metastasis. The overexpression of c-Myc in ~50%
of human cancers correlates with poor patient survival (126), while c-Myc is often considered to
be among the most desirable cancer targets, although it appears to
be among the most ‘undruggable’ of molecular targets in all of
cancer therapy (127).
In addition to i) promoting cell growth and
survival as an enzymatic cyclin G1/CDK5 (or cyclin G1/CDK2) CPF in
reparative tissues, and ii) disabling p53 checkpoint control via
the cyclin-G1/Mdm2/p53 axis, resulting in error-prone DNA-synthesis
that is characteristic of an advanced metastatic state, a
cyclin-G1-dependent pathway also appears to mediate the troublesome
EMT, which characteristically results in a more aggressive,
invasive cancer phenotype (observed in distant metastases) and,
hence, a worsening prognosis (113). Recently, CCNG1 was found to
be amplified in a particularly aggressive subset of triple-negative
breast cancer (TNBC) cells and in patients with
chemotherapy-resistant TNBC, in which cyclin G1 overexpression was
directly linked to the molecular mechanisms regulating both
polyploidization and chemotherapy resistance (128): Upon paclitaxel exposure, the
pro-survival function of cyclin G1 promotes breast cancer cell
survival by inducing polyploidy, thereby limiting treatment options
and outcomes involving taxanes, yet further validating cyclin G1 as
a strategic target for medical intervention in the treatment of
advanced metastatic breast cancer (Table
I).
Combinatorial approaches and companion
diagnostics for CCNG1 targeted therapies
Dysregulation of cyclin G1 is the driving oncogenic
event in a large number of human cancers. By restoring a critical
locus of CCNG1 suppression, the enforced expression of
miR-122, as well as cyclin G1 silencing, suppresses tumor growth
and increases the sensitivity of HCC cells to doxorubicin (DOX)
(106); thus, exciting new
possibilities for combinatorial therapies are emerging. Indeed,
cyclin G1 expression regulates and determines the outcome of taxane
chemotherapy: Elevated cyclin G1 expression accompanies paclitaxel
(PTX) -induced mitotic arrest and promotes cancer cell survival
after PTX exposure, whereas cyclin G1 depletion by RNA interference
delays mitotic slippage and enhances paclitaxel-induced apoptosis
(129), providing molecular
insights to guide future combinatorial treatment options. Moreover,
additional insights into the molecular mechanisms of
chemotherapeutic drug resistance have recently been uncovered: It
was recently reported that miR-27b, a novel miRNA that regulates
multidrug resistance (MDR) in gastric cancer, targets and
suppresses CCNG1 expression and restores p53-dependent
checkpoint activities. Ectopic expression of miR-27b increases the
chemosensitivity of gastric cancer cells to several
chemotherapeutic drugs by inhibiting CCNG1 (130). Likewise, ectopic expression of the
tumor-suppressive miR-23b, which is downregulated in colon cancer
and potently mediates multiple steps of metastasis, including tumor
growth, invasion and angiogenesis, selectively targets CCNG1
for suppression (131): By binding
with the 3′ untranslated region of CCNG1, miR-23b
downregulates cyclin G1 mRNA and protein expression, thereby
inhibiting ovarian tumorigenesis and cancer progression (132). A variety of molecular approaches
targeting cyclin G1 expression and/or function for cancer control
are shown in Table II.
| Table II.Medical intervention via targeting
cyclin G1 (CCNG1) for cancer therapy. |
Table II.
Medical intervention via targeting
cyclin G1 (CCNG1) for cancer therapy.
| Modes of medical
intervention | Description of
mechanisms, efficacy, conclusions | (Refs.) |
|---|
| Mx-dnG1 (Rexin-G):
Dominant-negative | √ Apoptosis of
transduced cancer cells | (90–101) |
| mutant construct of
cyclin G1 (dnG1) | √ Potent tumor
anti-angiogenic activity |
|
| delivered
intravenously by means of a | √ Broad spectrum
antitumor activity |
|
| tumor-targeted
retroviral expression vector | √ Single-agent
efficacy, long-term survivors |
|
|
CCNG1-suppressive
oligonucleotides: | Suppression of
cyclin G1 protein expression |
|
| • CCNG1
antisense fragments: | √ Induces apoptosis
in cancer cells, tumors | (54–56) |
| • Suppressive RNA,
miR 122: | √ Induces apoptosis
in cancer cells, tumors; | (105–109) |
| • siRNA-mediated
CCNG1 knockdown: | √ Increases
sensitivity to DOX via apoptosis; |
|
| • Suppressive RNA,
miR-27b | √ Inhibits cell
invasion/metastatic phenotype |
|
| • Suppressive RNA,
miR-23b | √ CCNG1
depletion enhances taxane toxicity | (129) |
|
| √ Regulates
multidrug resistance in gastric Ca. | (130) |
|
| √ Induces
apoptosis, suppresses tumorigenesis, cancer progression and
metastasis | (131,132) |
| Cytotoxic peptide
drugs: |
|
|
| • ELAS1 peptide
(cyclin G1 C-terminus): | √ Induces
apoptosis, CPT chemosensitization | (133,140) |
| • Toxic a5 Helix
peptides (cyclin box): | √ Induces
apoptosis, necrosis, antitumor action | (136–138) |
Establishment of the once-paradoxical cyclin G1
proto-oncogene as a dominant, growth-promoting, potentially
cancer-causing component of the commanding cyclin G1/Mdm2/p53 axis
has enormous implications in terms of the future of clinical
medicine, including cancer diagnostics, prognostics, and the
potential for rational combinatorial therapies. The frequency of
cyclin G1 overexpression observed in human cancers, as well as the
newfound association to the molecular mechanisms of cancer
metastasis (EMT) and chemotherapy resistance, provide new avenues
for patient profiling, staging and treatment, including
chemosensitization by strategic targeting of cyclin G1 as a locus
of diagnostic evaluation and clinical control. The advent of the
injectable, tumor-targeted vector and its demonstrated safety in
clinical trials, enables physicians of the future to think and to
reach beyond previous limitations in clinical trial designs. The
development of Mx-dnG1 (Rexin-G), which, by itself, induces
apoptosis in cancer cells and tumor-associated vasculature (in the
presence or absence of p53), is a powerful clinical tool in terms
of applied cell cycle checkpoint control, which merits
conscientious clinical development. Indeed, the functional
characterization of discrete structural domains that are present
within the cytocidal dnG1 have recently identified the ELAS1
peptide, which is based on a conserved sequence motif within the
extended C-terminal domain of cyclin G1, as a therapeutically
useful peptide drug (133).
Reportedly, the ELAS1 peptide, which competitively blocks the
physical association of cyclin G1 with the B’γ subunit of PP2A,
sensitizes osteosarcoma cells to camptothecin and irinotecan, and
induces apoptotic death in both prostate adenocarcinoma and
squamous cell carcinoma, albeit in a wild-type
TS53-dependent manner. By comparison, the ability of dnG1 to
activate apoptosis in the presence (hyperplasia, neoangiogenesis)
or absence (most metastatic cancer cells) of wild-type p53
predicates structure-function relationships involving programmed
cell death, which remained to be identified within the truncated
α-helices (molecular interfaces) of the dnG1 cyclin box domain.
Further elaboration of the structure-function
relationships of the cyclin box (134), which provide a mechanistic link to
cell death/signaling mechanisms, came from screening studies of the
ubiquitin-proteasome system, which has profound implications in
cancer biology. In addition to degrading specific cyclin proteins
during progression of the cell cycle, and generating peptide
antigens that are bound and presented to the immune system by the
major histocompatibility complex, the ubiquitin-proteasome system
was also found to generate intracellular peptides with profound
regulatory activity and importance (135,136).
Accordingly, a comprehensive screen for such intracellular peptides
in synchronized HeLa cells identified a small peptide derived from
the proteolysis of cyclin D2, specifically the α5 helix of the
cyclin box, which induces cell death via apoptosis and/or necrosis
in all cell lines tested (i.e., broad-spectrum efficacy), including
human cervical cancer, breast cancer, melanoma and thyroid
tumor-derived lines (136).
Moreover, when this cyclin D2 α5 helix-derived peptide is fused to
a cell-penetrating peptide, the resulting construct, infused in
vivo in a rat brain tumor model, reduced the volume of rat C6
glioblastomas by ~50% (136). Using
a fluorescently labeled peptide monitored by real-time confocal
microscopy in MDA-MB-231 breast cancer cells, the α5 helix-derived
peptide entered the cells within min, localized to the nucleus as
well as the cytoplasm, and induced cell death in breast cancer
cells, specifically in G1/S-arrested cells and in cells that were
actively progressing through S phase (137).
Reasoning that the protein/protein interface and
conformational changes required in the molecular mechanism of
cyclin-dependent CDK activation would be potential targets for the
design of specific inhibitors of cell cycle progression, a small
peptide inhibitor (C4) derived from the α5 helix (aa 285-306) of
cyclin A was found to be capable of binding to cyclin A/CDK2
complexes, thereby inhibiting the kinase activity of kinase
complexes harboring CDK2 in a competitive manner (138). Intricate structural studies
indicate that this C4 (α5 helix) peptide binds to the active site
of CDK2 to interfere with the interaction of target phosphoacceptor
substrates, independently of ATP binding in the catalytic site,
thus overcoming one of the major drawbacks of inhibitors that
target the ATP-binding site of protein kinases (which tend to
result in poor selectivity) and introducing a novel class of
designer peptide inhibitors of cyclin-dependent kinase activation,
with the potential to treat a wide range of tumor types (138). The cytotoxic efficacy of this C4
(α5 helix) peptide was validated in cellulo by demonstrating
that proliferation of all the human tumor cell lines tested (breast
cancer, liver cancer and T-cell leukemia) is blocked in a
dose-dependent manner when the C4 (α5 helix) peptide is coupled to
a cell-penetrating carrier peptide (138).
As both the crystal structure of cyclin A and the
mechanism of cyclin A/CDK2 interaction have been extensively
characterized (134), and the major
contact points with the catalytic CDK subunit have been determined
to be the α3, α4 and α5 helices of the cognate cyclin subunit, the
demonstration that the ‘designer’ peptide inhibitor C4 (α5 helix)
does not compete with cyclin A in terms of cyclin A/CDK2 complex
formation, nor does it simply interact with monomeric CDK2; rather,
the binding of cyclin A to CDK2 promotes a conformational change in
CDK2 that exposes the substrate docking site on the CDK, thereby
facilitating the binding of the C4 (α5 helix) across the catalytic
cleft of CDK2 and blocking the phosphorylation of target substrates
(138). Based on the profound
mechanistic insights provided by this detailed structure-activity
analysis, as well as the broad-spectrum anticancer efficacy
demonstrated by the cyclin D1-derived α5 helix peptide, recently
identified by functional proteomics (136,137),
we may now present a clearer and more comprehensive picture of the
molecular mechanisms by which the dnG1 construct, developed by
direct experimentation (77) and
embodied by Rexin-G in clinical medicine (98), is able to produce such broad-spectrum
anticancer efficacy when administered repeatedly as a single
therapeutic agent (Tables I and
II).
Clearly, dnG1, which is structurally truncated, and
yet retains the α3, α4 and α5 helices of the predicted substrate
(c-Myc) docking site (Fig. 9),
presents a rational, stable and competitive intracellular inhibitor
of cyclin G1/CKD5 activity (and possibly cyclin G1/CDK2 activity),
which blocks the phosphorylation/activation of c-Myc, thwarts
proliferative competence and leads to cell death via apoptosis and
necrosis in cancer cells derived from all three germ layers, save
for artificially-immortalized ‘vector producer’ cell lines (e.g.,
293T cells) that have been purposefully transformed by the
strategic integration of the SV40 large T antigen, a
dominant-acting oncoprotein, which coincidently targets pRb, p53
and PP2A for subversion (139) and
often leads to malignant transformation. Retaining the structural
interaction domains that bind Mdm2 and PP2A, the C-terminus of
cyclin G1 not only exhibits the PP2A-B’γ association domain, it
apparently ‘displays’ this domain at least an order of magnitude
more efficiently as a protein than the ELAS1 peptide drug, which
was designed to mimic, thus compete with, this interaction
(133). Of note, this ELAS1 peptide
(thus dnG1) was found to sensitize U2OS osteosarcoma cells to
radiation therapy (RT), as well as camptothecan/irinotecan
chemotherapy (140), thereby
providing a new avenue for dnG1 (Rexin-G) to further improve the
efficacy of chemoradiotherapy (CRT).
While the p53 tumor suppressor normally guards DNA
fidelity and serves as the executioner (by inducing apoptosis) of
aberrant cells, this locus of control is frequently lost in the
process of cell transformation, tumorigenesis, invasion, EMT and/or
metastasis, as the balance of power, i.e., the dominance of the
commanding axis, shifts from p53 tumor suppression to the oncogenic
cyclin G1/Mdm2 phenotype. From a therapeutic aspect, dnG1 functions
mechanistically, hence clinically, to counteract the root
proliferative competence of cancer cells, that being the
pro-survival, anti-apoptotic, tumor-promoting actions of cyclin
G1/CDK activity associated with the phosphorylation/activation of
the c-Myc oncoprotein. In retaining the structural elements and
interfaces of the commanding cyclin G1/Mdm2/p53 axis that interact
with Mdm2 and PP2A, the dnG1 protein provides an additional means
of sensitizing cancer cells to RT, cytotoxic chemotherapies and
CRT. In the revealing crucible of clinical medicine, wherein
investigational new drugs, such as Rexin-G, are initially tested
under rather extreme conditions (e.g., late-stage, metastatic
and/or chemotherapy-resistant cancers) wherein standard therapies
have failed, the repeated clinical demonstrations of tumor
eradication (in the presence or absence of p53) in multiform cancer
types, including eradication of primary tumors, metastatic lesions,
and even lymphatic metastasis (98,99),
resulting in long-term dose-dependent survival benefits (101), provides a compelling molecular
exemplar for the future of cancer gene therapy.
Conclusions along with a note concerning the
future of clinical oncology
The commanding cyclin G1/Mdm2/p53 axis is presented
herein with collegial appreciation for the multidisciplinary nature
of the task: The decades of basic, translational and clinical
cancer research that has brought this ‘accessible’ locus of
checkpoint control to light. The model is purposefully simple yet
powerful in its applications, intended to provide a logical
framework for new information, including the development of
pertinent companion diagnostics, and to serve as a guide for
clinical oncologists to better understand the executive regulatory
components that determine the efficacy and outcomes of cancer
therapies through the increasingly complex molecular mechanisms of
cell cycle checkpoint control. This review is presented in
appreciation of the numerous cancer patients whose informed consent
has served to teach and tell, and whose long-term, cancer-free
survival stands as a meaningful milestone in clinical oncology.
The broad-spectrum, single-agent efficacy of
Mx-dnG1 (Rexin-G) demonstrated in end-stage cancers, where standard
therapies had failed, is an excellent step forward; and one can
only imagine what improved clinical outcomes may arise when the
physiological surveillance properties of the tumor-targeted Mx-dnG1
vector is eventually permitted to be administered worldwide at an
earlier, less intractable stage of metastatic cancer (Figs. 5 and 8). Furthermore, these first pioneering
demonstrations of clinical safety, efficacy and long-term survival
of Mx-dnG1 (Rexin-G)-treated cancer patients serve as a challenge
to those who may otherwise distort or obscure what has been
historically accomplished, and to serve as a guidepost for the next
generation of clinical oncologists who will assuredly be more
knowledgeable and skilled in applied molecular medicine. Finally,
this review article is presented well in advance of the time when
genetic medicine will no longer be viewed as a competitive (or
disruptive) biotechnology, but will rather be considered a rational
and beneficial addition to the combinatorial practice of the
postmodern medical oncologist.
Acknowledgements
The authors gratefully acknowledge Heather C.
Gordon for graphic illustrations and previous funding from the
National Science Foundation, National Institutes of Health, NATO
Scientific Exchange Program, American Heart Association, Whittier
Family Foundation, and the FDA Orphan Drug Program in support of
this pioneering biomedical research.
Funding
Funded in part by the Sarcoma Oncology Center, 2811
Wilshire Blvd., Suite 414, Santa Monica CA 90403, USA, and in part
by DELTA Next-Gen, LLC, 2700 Neilson Way, Suite 1423, Santa Monica
CA 90405, USA.
Availability of data and materials
The materials included in this manuscript,
including all relevant raw data, will be made freely available to
any researchers who wish to use this for non-commercial purposes,
while preserving any necessary confidentiality and anonymity.
Authors' contributions
EG reviewed the published literature, wrote the
manuscript, analyzed the critical stages in Rexin-G drug
development, reviewed and edited the final manuscript. JR reviewed
the published literature, wrote sections of the manuscript and the
reference section, and edited the final manuscript. SL reviewed the
published literature, wrote and edited the reference section, and
edited the final manuscript. SC reviewed the published literature,
analyzed the clinical trials using Rexin-G, reviewed and edited the
final manuscript. FH reviewed the published literature, analyzed
the molecular mechanisms of action of cyclin G1 in the cell cycle
control pathway, wrote the manuscript, reviewed and edited the
final manuscript. All authors approve the submission of this
manuscript for publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
Drs Gordon and Hall are co-inventors of Rexin-G
targeted gene delivery system, which was developed at the
University of Southern California Keck School of Medicine, and are
co-founders of DELTA Next-Gen, LLC. Dr Chawla, Mr. Ravicz and Mr.
Liu have no competing interests to disclose.
References
|
1
|
Goel G, Makkar HP, Francis G and Becker K:
Phorbol esters: Structure, biological activity, and toxicity in
animals. Int J Toxicol. 26:279–288. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weinstein IB: The origins of human cancer:
Molecular mechanisms of carcinogenesis and their implications for
cancer prevention and treatment-twenty-seventh G.H.A. Clowes
memorial award lecture. Cancer Res. 48:4135–4143. 1988.PubMed/NCBI
|
|
3
|
Kikkawa U, Takai Y, Tanaka Y, Miyake R and
Nishizuka Y: Protein kinase C as a possible receptor protein of
tumor-promoting phorbol esters. J Biol Chem. 258:11442–11445.
1983.PubMed/NCBI
|
|
4
|
Cooke M, Magimaidas A, Casado-Medrano V
and Kazanietz MG: Protein kinase C in cancer: The top five
unanswered questions. Mol Carcinog. 56:1531–1542. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reid BJ, Culotti JG, Nash RS and Pringle
JR: Forty-five years of cell-cycle genetics. Mol Biol Cell.
26:4307–4312. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yasutis KM and Kozminski KG: Cell cycle
checkpoint regulators reach a zillion. Cell Cycle. 12:1501–1509.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jackson PK: The hunt for cyclin. Cell.
134:199–202. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reed SI: G1 specific cyclins: In search
for an S-phase promoting factor. Trends Genet. 7:95–99. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pines J and Hunter T: Cyclin-dependent
kinases: A new cell cycle motif? Trends Cell Biol. 1:117–121. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kishimoto T: Entry into mitosis: A
solution to the decades-long enigma of MPF. Chromosoma.
124:417–428. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nasmyth K: Viewpoint: Putting the cell
cycle in order. Science. 274:1643–1645. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Strausfeld UP, Howell M, Descombes P,
Chevalier S, Rempel RE, Adamczewski J, Maller JL, Hunt T and Blow
JJ: Both cyclin A and cyclin E have S-phase promoting (SPF)
activity in Xenopus egg extracts. J Cell Sci. 109:1555–1563.
1996.PubMed/NCBI
|
|
13
|
Vulliet PR, Hall FL, Mitchell JP and
Hardie DG: Identification of a novel proline-directed
serine/threonine protein kinase in rat pheochromocytoma. J Biol
Chem. 264:16292–16298. 1989.PubMed/NCBI
|
|
14
|
Hall FL, Mitchell JP and Vulliet PR:
Phosphorylation of synapsin I at a novel site by proline-directed
protein kinase. J Biol Chem. 265:6944–6948. 1990.PubMed/NCBI
|
|
15
|
Hall FL and Vulliet PR: Proline-directed
protein phosphorylation and cell cycle regulation. Curr Opin Cell
Biol. 3:176–184. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suzuki M: SPXX, a frequent sequence motif
in gene regulatory proteins. J Mol Biol. 207:61–84. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hall FL, Braun RK, Mihara K, Fung YT,
Berndt N, Carbonaro-Hall DA and Vulliet PR: Characterization of the
cytoplasmic proline-directed protein kinase in proliferative cells
and tissues as a heterodimer comprised of p34cdc2 and p58cyclin A.
J Biol Chem. 266:17430–17440. 1991.PubMed/NCBI
|
|
18
|
Elledge SJ, Richman R, Hall FL, Williams
RT, Lodgson N and Harper JW: CDK2 encodes a 33-kDa cyclin
A-associated protein kinase and is expressed before CDC2 in the
cell cycle. Proc Natl Acad Sci USA. 89:2907–2911. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peeper DS, Parker LL, Ewen ME, Toebes M,
Hall FL, Xu M, Zantema A, van der Eb AJ and Piwnica-Worms H: A- and
B-type cyclins differentially modulate substrate specificity of
cyclin-cdk complexes. EMBO J. 12:1947–1954. 1995.
|
|
20
|
Giacinti C and Giordano A: RB and cell
cycle progression. Oncogene. 25:5220–5227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bertoli C, Skotheim JM and de Bruin RA:
Control of cell cycle transcription during G1 and S phases. Nat Rev
Mol Cell Biol. 14:518–528. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Foster DA, Yellen P, Xu L and Saqcena M:
Regulation of G1 cell cycle progression: Distinguishing the
restriction point from a nutrient-sensing cell growth
checkpoint(s). Genes Cancer. 1:1124–1131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Csikász-Nagy A, Kapuy O, Tóth A, Pál C,
Jensen LJ, Uhlmann F, Tyson JJ and Novák B: Cell cycle regulation
by feed-forward loops coupling transcription and phosphorylation.
Mol Syst Biol. 5:2362009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weinberg RA: The biology of cancer, 2nd
edition, Chapter 9: p53 and apoptosis: Master guardian and
executioner. Garland Sci; New York: 2014
|
|
25
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deshpande A, Sicinski P and Hinds PW:
Cyclins and cdks in development and cancer: A perspective.
Oncogene. 24:2909–2915. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Malumbres M: Cyclin-dependent kinases.
Genome Biol. 15:1222014. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sherr CJ and McCormick F: The RB and p53
pathways in cancer. Cancer Cell. 2:103–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Giordano A, McCall C, Whyte P and Franza
BR Jr: Human cyclin A and the retinoblastoma protein interact with
similar but distinguishable sequences in the adenovirus E1A gene
product. Oncogene. 6:481–485. 1991.PubMed/NCBI
|
|
30
|
Wang J, Chenivesse X, Henglein B and
Bréchot C: Hepatitis B virus integration in a cyclin A gene in a
hepatocellular carcinoma. Nature. 343:555–557. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang J, Zindy F, Chenivesse X, Lamas E,
Henglein B and Bréchot C: Modification of cyclin A expression by
hepatitis B virus DNA integration in a hepatocellular carcinoma.
Oncogene. 7:1653–1656. 1992.PubMed/NCBI
|
|
32
|
Bréchot C: Oncogenic activation of cyclin
A. Curr Opin Genet Dev. 3:11–18. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bodey B, Williams RT, Carbonaro-Hall DA,
Horvath A, Tolo VT, Luck JV Jr, Taylor CR and Hall FL:
Immunocytochemical detection of cyclin A and cyclin D in
formalin-fixed, paraffin-embedded tissues: Novel, pertinent markers
of cell proliferation. Mod Pathol. 7:846–852. 1994.PubMed/NCBI
|
|
34
|
Motokura T and Arnold A: Cyclin D and
oncogenesis. Curr Opin Genet Dev. 3:5–10. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hunter T and Pines J: Cyclins and Cancer
II: Cyclin D and CDK inhibitors come of age. Cell. 79:573–582.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Santamaria D, Barrière C, Cerqueira A,
Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M and
Barbacid M: Cdk1 is sufficient to drive the mammalian cell cycle.
Nature. 448:811–815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Blagosklonny MV and Pardee AB: The
restriction point of the cell cycle. Cell Cycle. 1:103–110. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hwang HC and Clurman BE: Cyclin E in
normal and neoplastic cell cycles. Oncogene. 24:2776–2786. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Narasimha AM, Kaulich M, Shapiro GS, Choi
YJ, Sicinski P and Dowdy SF: Cyclin D activates the Rb tumor
suppressor by mono-phosphorylation. Elife. 3:2014.doi:
10.7554/eLife.02872. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
El-Deiry WS: p21(WAF1) mediates cell cycle
inhibition, relevant to cancer suppression and therapy. Cancer Res.
76:5189–5191. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roussel MF: The INK4 family of cell cycle
inhibitors in cancer. Oncogene. 18:5311–5317. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Serrano M, Lee H, Chin L, Cordon-Cardo C,
Beach D and DePinho RA: Role of the INK4a locus in tumor
suppression and cell mortality. Cell. 85:27–37. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shapiro GI and Harper JW: Anticancer drug
targets: Cell cycle and checkpoint control. J Clin Invest.
104:1645–1653. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Casimiro MC, Velasco-Velázquez M,
Aguirre-Alvarado C and Pestell RG: Overview of cyclins D1 function
in cancer and the CDK inhibitor landscape: Past and present. Expert
Opin Investig Drugs. 23:295–304. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Peyressatre M, Prével C, Pellerano M and
Morris MC: Targeting cyclin-dependent kinases in human cancers:
From small molecules to Peptide inhibitors. Cancers (Basel).
7:179–237. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sherr CJ, Beach D and Shapiro GI:
Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov.
6:353–367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Horne MC, Goolsby GL, Donaldson KL, Tran
D, Neubauer M and Wahl AF: Cyclin G1 and Cyclin G2 comprise a new
family of cyclins with contrasting tissue-specific and cell
cycle-regulated expression. J Biol Chem. 271:6050–6061. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wu L, Liu L, Yee A, Carbonarohall D, Tolo
V and Hall F: Molecular-cloning of the human CYCG1 gene encoding a
G-type cyclin-overexpression in human osteosarcoma cells. Oncol
Rep. 1:705–711. 1994.PubMed/NCBI
|
|
51
|
Okamoto K and Beach D: Cyclin G is a
transcriptional target of the p53 tumor suppressor protein. EMBO J.
13:4816–4822. 1994.PubMed/NCBI
|
|
52
|
Efeyan A and Serrano M: p53: Guardian of
the genome and policeman of the oncogenes. Cell Cycle. 6:1006–1010.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Smith ML, Kontny HU, Bortnick R and
Fornace AJ Jr: The p53-regulated cyclin G gene promotes cell
growth: p53 downstream effectors cyclin G and Gadd45 exert
different effects on cisplatin chemosensitivity. Exp Cell Res.
230:61–68. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Skotzko M, Wu L, Anderson WF, Gordon EM
and Hall FL: Retroviral vector-mediated gene transfer of antisense
Cyclin G1 (CYCG1) inhibits proliferation of human osteogenic
sarcoma cells. Cancer Res. 55:5493–5498. 1995.PubMed/NCBI
|
|
55
|
Chen DS, Zhu NL, Hung G, Skotzko MJ,
Hinton DR, Tolo V, Hall FL, Anderson WF and Gordon EM: Retroviral
vector-mediated transfer of an antisense cyclin G1 construct
inhibits osteosarcoma tumor growth in nude mice. Hum Gene Ther.
8:1667–1674. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hung G, Skotzko MJ, Chang M, Zhu NL,
Parekh D, Hall FL, Gordon EM and Anderson WF: Intratumoral
injection of an antisense cyclin G1 retroviral vector inhibits
growth of undifferentiated carcinoma xenografts in nude mice.
Pediatr Hematol Oncol. 4:317–325. 1997.
|
|
57
|
Piette J, Neel H and Maréchal V: Mdm2:
Keeping p53 under control. Oncogene. 15:1001–1010. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Momand J, Jung D, Wilczynski S and Niland
J: The MDM2 gene amplification database. Nucleic Acids Res.
26:3453–3459. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Haupt S, Vijayakumaran R, Miranda PJ,
Burgess A, Lim E and Haupt Y: The role of MDM2 and MDM4 in breast
cancer development and prevention. J Mol Cell Biol. 9:53–61.
2017.PubMed/NCBI
|
|
60
|
Momand J, Zambetti GP, Olson DC, George D
and Levine AJ: The mdm-2 oncogene product forms a complex with the
p53 protein and inhibits p53-mediated transactivation. Cell.
69:1237–1245. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Iwakuma T and Lozano G: DM2, an
introduction. Mol Cancer Res. 1:993–1000. 2003.PubMed/NCBI
|
|
62
|
Shi D and Gu W: Dual roles of MDM2 in the
regulation of p53: Ubiquitination dependent and ubiquitination
independent mechanisms of MDM2 repression of p53 activity. Genes
Cancer. 3:240–248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shangary S and Wang S: Targeting the
MDM2-p53 interaction for cancer therapy. Clin Cancer Res.
14:5318–5324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tisato V, Voltan R, Gonelli A, Secchiero P
and Zauli G: MDM2/X inhibitors under clinical evaluation:
Perspectives for the management of hematological malignancies and
pediatric cancer. J Hematol Oncol. 10:1332017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Estrada-Ortiza N, Neochoritisa CG and
Dömlinga A: How to design a successful p53-MDM2/X interaction
inhibitor: A thorough overview based on crystal structures. Chem
Med Chem. 11:757–772. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Meek DW and Knippschild U:
Posttranslational modification of MDM2. Mol Cancer Res.
1:1017–1026. 2003.PubMed/NCBI
|
|
67
|
Okamoto K, Kamibayashi C, Serrano M,
Prives C, Mumby MC and Beach D: p53-dependent association between
cyclin G and the B' subunit of protein phosphatase 2A. Mol Cell
Biol. 16:6593–6602. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Okamoto K, Li H, Jensen MR, Zhang T, Taya
Y, Thorgeirsson SS and Prives C: Cyclin G recruits PP2A to
dephosphorylate Mdm2. Mol Cell. 9:761–771. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Westermarck J and Hahn WC: Multiple
pathways regulated by the tumor suppressor PP2A in transformation.
Trends Mol Med. 14:152–160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kimura SH and Nojima H: Cyclin G1
associates with MDM2 and regulates accumulation and degradation of
p53 protein. Genes Cells. 7:869–880. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Giono LE and Manfredi JJ: The p53 tumor
suppressor participates in multiple cell cycle checkpoints. J Cell
Physiol. 209:13–20. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chen X: Cyclin G: A regulator of the
p53-Mdm2 network. Dev Cell. 2:518–519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Jensen MR, Factor VM, Fantozzi A, Helin K,
Huh CG and Thorgeirsson SS: Reduced hepatic tumor incidence in
cyclin G1-deficient mice. Hepatology. 37:862–870. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhu NL, Wu L, Liu PX, Gordon EM, Anderson
WF, Starnes VA and Hall FL: Down-regulation of cyclin G1 expression
by retrovirus-mediated antisense gene transfer inhibits vascular
smooth muscle cell proliferation and neointima formation.
Circulation. 96:628–635. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kampmeier J, Behrens A, Wang Y, Yee A,
Anderson WF, Hall FL, Gordon EM and McDonnell PJ: Inhibition of
rabbit keratocyte and human fetal lens epithelial cell
proliferation by retroviral-mediated transfer of antisense cyclin
G1 and antisense MAT1 constructs. Hum Gene Ther. 11:1–8. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Jensen MR, Factor VM and Thorgeirsson SS:
Regulation of Cyclin G1 during murine hepatic regeneration
following Dipin-induced DNA damage. Hepatology. 28:537–546. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Xu F, Prescott MF, Liu PX, Chen ZH, Liau
G, Gordon EM and Hall FL: Long term inhibition of neointima
formation in balloon-injured rat arteries by intraluminal
instillation of a matrix-targeted retroviral vector bearing an
improved cytocidal Cyclin G1 construct. Int J Mol Med. 8:19–30.
2001.PubMed/NCBI
|
|
78
|
Waehler R, Russell SJ and Curiel DT:
Engineering targeted viral vectors for gene therapy. Nature Rev
Genet. 8:573–587. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hall FL, Gordon EM, Wu L, Zhu NL, Skotzko
MJ, Starnes VA and Anderson WF: Targeting retroviral vectors to
vascular lesions by genetic engineering of the MoMLV gp70 envelope
protein. Hum Gene Ther. 8:2183–2192. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Weimin Wu B, Cannon PM, Gordon EM, Hall FL
and Anderson WF: Characterization of the proline-rich region of
murine leukemia virus envelope protein. J Virol. 72:5383–5391.
1998.PubMed/NCBI
|
|
81
|
Hall FL, Liu L, Zhu NL, Stapfer M,
Anderson WF, Beart RW and Gordon EM: Molecular engineering of
matrix-targeted retroviral vectors incorporating a surveillance
function inherent in von Willebrand factor. Hum Gene Ther.
11:983–993. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhu NL, Gordon EM, Liu L, Terramani T,
Anderson WF and Hall FL: Collagen-targeted retroviral vectors
displaying domain D2 of von Willebrand factor (vWF-D2) enhance gene
transfer to human tissue explants. Int J Pediatr Hematol Oncol.
7:325–335. 2001.
|
|
83
|
Gordon EM, Zhu NL, Forney Prescott M, Chen
ZH, Anderson WF and Hall FL: Lesion-targeted injectable vectors for
vascular restenosis. Hum Gene Ther. 12:1277–1287. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Behrens A, Gordon EM, Li L, Liu PX, Chen
Z, Peng H, La Bree L, Anderson WF, Hall FL and McDonnell PJ:
Retroviral gene therapy vectors for prevention of excimer
laser-induced corneal haze. Invest Ophthalmol Vis Sci. 43:968–977.
2002.PubMed/NCBI
|
|
85
|
Song JC, McDonnell PJ, Gordon EM, Hall FL
and Anderson WF: Phase I/II evaluation of safety and efficacy and a
matrix-targeted retroviral vector bearing a dominant negative
cyclin G1 construct (Mx-dnG1) as adjunctive intervention for
superficial corneal opacity/corneal scarring. Hum Gene Ther.
14:306–309. 2003.PubMed/NCBI
|
|
86
|
Gordon EM, Liu PX, Chen ZH, Liu L, Whitley
MD, Gee C, Groshen S, Hinton DR, Beart RW and Hall FL: Inhibition
of metastatic tumor growth in nude mice by portal vein infusions of
matrix-targeted retroviral vectors bearing a cytocidal cyclin G1
construct. Cancer Res. 60:3343–3347. 2000.PubMed/NCBI
|
|
87
|
Gordon EM, Liu PX, Chen ZH, Liu L, Whitley
M, Liu L, Wei D, Groshen S, Hinton DR, Anderson WF, Beart RW Jr and
Hall FL: Systemic administration of a matrix-targeted retroviral
vector is efficacious for cancer gene therapy in mice. Hum Gene
Ther. 12:193–204. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lenz HJ, Anderson WF, Hall FL and Gordon
EM: Tumor site specific phase I evaluation of safety and efficacy
of hepatic arterial infusion of a matrix-targeted retroviral vector
bearing a dominant negative Cyclin G1 construct as treatment for
colorectal carcinoma metastatic to liver. Hum Gene Ther.
13:1515–1537. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Le Tourneau C, Lee JJ and Siu LL: Dose
escalation methods in phase I cancer clinical trials. J Natl Cancer
Inst. 101:708–720. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Gordon EM, Cornelio GH, Lorenzo CC III,
Levy JP, Reed RA, Liu L and Hall FL: First clinical experience
using a ‘pathotropic’ injectable retroviral vector (Rexin-G) as
intervention for stage IV pancreatic cancer. Int J Oncol.
24:177–185. 2004.PubMed/NCBI
|
|
91
|
Gordon EM, Lopez FF, Cornelio GH, Lorenzo
CC III, Levy JP, Reed RA, Liu L, Bruckner HW and Hall FL:
Pathotropic nanoparticles for cancer gene therapy Rexin-G IV:
Three-year clinical experience. Int J Oncol. 29:1053–1064.
2006.PubMed/NCBI
|
|
92
|
Galanis E, Carlso SK, Foster NR, Lowe V,
Quevedo F, McWilliams RR, Grothey A, Jatoi A, Alberts SR and Rubin
J: Phase I trial of a pathotropic retroviral vector expressing a
cytocidal cyclin G1 construct (Rexin-G) in patients with advanced
pancreatic cancer. Mol Ther. 16:979–984. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chawla SP, Chua VS, Mohan V, Alzwahereh K,
Kalra A, Quon D, Gordon EM and Hall FL: Phase I/II study of
targeted gene delivery in vivo-intravenous infusions of Rexin-G
demonstrate significant biologic activity by FDG PET-CT without
toxicity in patients with progressive chemo-resistant sarcoma,
breast cancer and pancreatic cancer. J Clin Oncol. 26
15-Suppl:S14509. 2008. View Article : Google Scholar
|
|
94
|
Chawla SP, Chua VS, Fernandez L, Quon D,
Saralou A, Blackwelder WC, Hall FL and Gordon EM: Evaluation of the
safety and efficacy of ‘pathotropic’ nanoparticles bearing a
dominant-negative Cyclin G1 construct (Rexin-G) as monotherapy for
chemo-resistant osteosarcoma and other sarcomas-phase I/II and
phase II studies. J Clin Oncol. 27:10513. 2009.
|
|
95
|
Chawla SP, Chua VS, Fernandez L, Quon D,
Saralou A, Blackwelder WC, Hall FL and Gordon EM: Phase I/II and
phase II studies of targeted gene delivery in vivo: intravenous
Rexin-G for chemotherapy-resistant sarcoma and osteosarcoma. Mol
Ther. 17:1651–1657. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Chawla SP, Chawla NS, Quon D, Chua-Alcala
V, Blackwelder WC, Hall FL and Gordon EM: An advanced phase 1/2
study using an XC-targeted gene therapy vector for chemotherapy
resistant sarcoma. Sarcoma Res Int. 3:1024–1031. 2016.
|
|
97
|
Chawla SP, Chua VS, Fernandez L, Quon D,
Blackwelder WC, Gordon EM and Hall FL: Advanced phase I/II studies
of targeted gene delivery in vivo: Intravenous Rexin-G for
gemcitabine-resistant metastatic pancreatic cancer. Mol Ther.
18:435–441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Gordon EM and Hall FL: Rexin-G, a targeted
genetic medicine for cancer. Expert Opin Biol Ther. 10:819–832.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Gordon EM, Chan MT, Geraldino N, Lopez FF,
Cornelio GH, Lorenzo CC III, Levy JP, Reed RA, Liu L and Hall FL:
Le morte du tumour: Histological features of tumor destruction in
chemo-resistant cancers following intravenous infusions of
pathotropic nanoparticles bearing therapeutic genes. Int J Oncol.
30:1297–1307. 2007.PubMed/NCBI
|
|
100
|
Gordon EM and Hall FL: A primer on
pathotropic medicine. In ‘one hundred years of the FDA and the
future of global health. Brooklands New Media Ltd; Shopshire UK:
pp. 842007
|
|
101
|
Kim S, Federman N, Gordon EM, Hall FL and
Chawla SP: Rexin-G®, a tumor-targeted retrovector for
malignant peripheral nerve sheath tumor: A case report. Mol Clin
Oncol. 6:861–865. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Feng Z, Zhang C, Wu R and Hu W: Tumor
suppressor p53 meets microRNAs. J Mol Cell Biol. 3:44–50. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Huang S and He X: The role of microRNAs in
liver cancer progression. Br J Cancer. 104:235–240. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Gramantieri L, Ferracin M, Fornari F,
Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E,
Grazi LG, et al: Cyclin G1 is a target of miR-122a, a microRNA
frequently down-regulated in human hepatocellular carcinoma. Cancer
Res. 67:6092–6099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Coulouarn C, Factor VM, Andersen JB,
Durkin ME and Thorgeirsson SS: Loss of miR-122 expression in liver
cancer correlates with suppression of the hepatic phenotype and
gain of metastatic properties. Oncogene. 28:3526–3536. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Fornari F, Gramantieri L, Giovannini C,
Veronese A, Ferracin M, Sabbioni S, Calin GA, Grazi GL, Croce CM,
Tavolari S, et al: MiR-122/cyclin G1 interaction modulates p53
activity and affects doxorubicin sensitivity of human
hepatocarcinoma cells. Cancer Res. 69:5761–5767. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wu X, Wu S, Tong L, Luan T, Lin L, Lu S,
Zhao W, Ma Q, Liu H and Zhong Z: miR-122 affects the viability and
apoptosis of hepatocellular carcinoma cells. Scand J
Gastroenternol. 44:1332–1339. 2009. View Article : Google Scholar
|
|
108
|
Ma L, Liu J, Shen J, Liu L, Wu J, Li W,
Luo J, Chen Q and Qian C: Expression of miR-122 mediated by
adenoviral vector induces apoptosis and cell cycle arrest of cancer
cells. Cancer Biol Ther. 9:554–561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hsu SH, Wang B, Kota J, Yu J, Costinean S,
Kutay H, Yu L, Bai S, La Perle K, Chivukula RR, et al: Essential
metabolic, anti-inflammatory, and anti-tumorigenic functions of
miR-122 in liver. J Clin Invest. 122:2871–2883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Bandopadhyay M, Sarkar N, Datta S, Das D,
Pal A, Panigrahi1 R, Banerjee A, Panda CK, Das C, Chakrabarti S and
Chakravarty R: Hepatitis B virus X protein mediated suppression of
miRNA-122 expression enhances hepatoblastoma cell proliferation
through cyclin G1-p53 axis. Infect Agent Cancer. 11:402016.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Reimer CL, Borras AM, Kurdistani SK,
Garreau JR, Chung M, Aaronson SA and Lee SW: Altered regulation of
Cyclin G in human breast cancer and its specific localization at
replication foci in response to DNA damage in p53+/+ cells. J Biol
Chem. 274:11022–11029. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Perez R, Wu N, Klipfel AA and Beart RW Jr:
A better cell cycle target for gene therapy of colorectal cancer:
Cyclin G. J Gastrointest Surg. 7:884–889. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wen W, Ding J, Sun W, Fu J, Chen Y, Wu K,
Ning B, Han T, Huang L, Chen C, et al: Cyclin G1-mediated
epithelial-mesenchymal transition via phosphoinositide 3-kinase/Akt
signaling facilitates liver cancer progression. Hepatology.
55:1787–1798. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Weinstein B and Joe A: Oncogene addiction.
Cancer Res. 68:3077–3080. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Li H, Okamoto K, Peart MJ and Prives C:
Lysine-independent turnover of Cyclin G1 can be stabilized by
B'alpha subunits of protein phosphatase 2A. Mol Cell Biol.
29:919–928. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Piscopo DM and Hinds PW: A role for the
cyclin box in the ubiquitin-mediated degradation of cyclin G1.
Cancer Res. 68:5581–5590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Seo HR, Kim J, Bae S, Soh JW and Lee YS:
Cdk5-mediated phosphorylation of c-Myc on Ser-62 is essential in
transcriptional activation of cyclin B1 by Cyclin G1. J Biol Chem.
283:15601–15610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Menssen A and Hermeking H:
Characterization of the c-MYC-regulated transcriptome by SAGE:
Identification and analysis of c-MYC target genes. Proc Natl Acad
Sci USA. 99:6274–6279. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Morita N, Kiryu S and Kiyama H:
p53-independent Cyclin G expression in a group of mature neurons
and its enhanced expression during nerve regeneration. J Neurosci.
16:5961–5966. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Sultana R and Butterfield DA: Regional
expression of key cell cycle proteins in brain from subjects with
amnestic mild cognitive impairment. Neurochem Res. 32:655–662.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Yang Y, Mufson EJ and Herrup K: Neuronal
cell death is preceded by cell cycle events at all stages of
Alzheimer's disease. J Neurosci. 23:2557–2563. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Lee MS, Kwon YT, Li M, Peng J, Friedlander
RM and Tsai LH: Neurotoxicity induces cleavage of p35 to p25 by
calpain. Nature. 405:360–364. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Ko J, Humbert S, Bronson RT, Takahashi S,
Kulkarni AB, Li E and Tsai L: p35 and p39 are essential for
cyclin-dependent kinase 5 function during neurodevelopment. J
Neurosci. 21:6758–6771. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Cheung ZH, Gong K and Ip NY:
Cyclin-dependent kinase 5 supports neuronal survival through
phosphorylation of Bcl-2. J Neurosci. 28:4872–4877. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cruz JC, Tseng HC, Goldman JA, Shih H and
Tsai LH: Aberrant Cdk5 activation by p25 triggers pathological
events leading to neurodegeneration and neurofibrillary tangles.
Neuron. 40:471–483. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Tansey WP: Mammalian MYC proteins and
cancer. New J Sci. 2014:Article ID 757534. 2014. View Article : Google Scholar
|
|
127
|
Dang CV, Reddy EP, Shokat KM and Soucek L:
Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer.
17:502–508. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zhang W, Xu J, Ji D, Li Z, He W, Yang F,
Lan H, Wang Y, Wu Z, Liu X, et al: Cyclin G1 amplification enhances
aurora kinase inhibitor-induced polyploid resistance and inhibition
of Bcl-2 pathway reverses the resistance. Cell Physiol Biochem.
43:94–107. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Russell P, Hennessy BT, Li J, Carey MS,
Bast RC, Freeman T and Venkitaraman AR: Cyclin G1 regulates the
outcome of taxane-induced mitotic checkpoint arrest. Oncogene.
31:2450–2460. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Shang Y, Feng B, Zhou L, Ren G, Zhang Z,
Fan X, Sun Y, Luo G, Liang J, Wu K, et al: The
miR27b-CCNG1-P53-miR-508-5p axis regulates multidrug resistance of
gastric cancer. Oncotarget. 7:538–549. 2015.
|
|
131
|
Zhang H, Hao Y, Yang J, Zhou Y, Li J, Yin
S, Sun C, Ma M, Huang Y and Xi JJ: Genome-wide functional screening
of miR-23b as a pleiotropic modulator suppressing cancer
metastasis. Nat Commun. 2:5542011. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Yan J, Jiang JY, Meng XN, Xiu YL and Zong
ZH: MiR-23b targets cyclin G1 and suppresses ovarian cancer
tumorigenesis and progression. J Exp Clin Cancer Res. 35:312016.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Uchihashi T, Ota K, Yabuno Y, Ohno S,
Fukushima K, Naito Y, Kogo M, Yabuta N and Nojima H: ELAS1 induces
apoptotic death in adenocarcinoma DU145 and squamous-cell carcinoma
SAS cancer cells, but not in normal KD cells. Oncotarget.
8:85868–85882. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Brown NR, Noble ME, Endicott JA, Garman
EF, Wakatsuki S, Mitchell E, Rasmussen B, Hunt T and Johnson LN:
The crystal structure of Cyclin A. Structure. 3:1235–1247. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ferro ES, Hyslop S and Camargo AC:
Intracellullar peptides as putative natural regulators of protein
interactions. J Neurochem. 91:769–777. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
de Araujo CB, Russo LC, Castro LM, Forti
FL, do Monte ER, Rioli V, Gozzo FC, Colquhoun A and Ferro ES: A
Novel intracellular peptide derived from g1/s cyclin d2 induces
cell death. J Biol Chem. 289:16711–16726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Russo LC, Araujo CB, Iwai LK, Ferro ES and
Forti FL: A cyclin D2-derived peptide acts on specific cell cycle
phases by activating ERK1/2 to cause the death of breast cancer
cells. J Proteomics. 151:24–32. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Gondeau C, Gerbal-Chaloin S, Bello P,
Aldrian-Herrada G, Morris MC and Divita G: Design of a novel class
of peptide inhibitors of cyclin-dependent kinase/cyclin activation.
J Biol Chem. 280:13793–13800. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Ahuja D, Sáenz-Robles MT and Pipas JM:
SV40 large T antigen targets multiple cellular pathways to elicit
cellular transformation. Oncogene. 24:7729–7745. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ohno S, Naito Y, Mukai S, Yabuta N and
Nojima H: ELAS1-mediated inhibition of the cyclin G1-B’γ
interaction promotes cancer cell apoptosis via stabilization and
activation of p53. Oncogene. 34:5983–5996. 2015. View Article : Google Scholar : PubMed/NCBI
|