Introduction
Renal cell carcinoma (RCC) is the seventh most
common type of cancer worldwide, and its incidence in different
countries, including Canada, Saudi Arabia and Belarus, is on the
increase (1). The 5-year overall
survival (OS) is ~74%, although ~30% of patients with RCC who
present with evidence of distant metastasis upon initial diagnosis
have a 5-year OS of only 8% (2).
Despite the fact that >10 agents have now been approved for the
treatment of metastatic RCC, the survival rates have not improved
drastically. This may be attributed to the fact that these agents
are often used without guided biomarkers that may be used to ensure
that the most effective treatments are administered to selected
patients. Thus, there is a need to further elucidate the genomic
landscape of metastatic RCC and to identify biomarkers for guided
therapy.
Due to the valuable contributions of The Cancer
Genome Atlas (TCGA) and other researchers (3,4), there
is currently a clear picture of the genomic landscape of several
RCCs; however, nearly all the samples from these studies were
derived from primary nephrectomies and from earlier stages of
disease. It is likely that the genomic landscape of metastatic RCCs
differs from than that of primary RCCs, as the cancers may evolve
in more advanced stages and through selective pressures from
treatment. While repeat biopsies of metastatic lesions are
challenging due to financial and medical reasons, circulating tumor
DNA (ctDNA) offers the opportunity to assess genomic alterations in
metastatic cancers. This approach has been used in a number of
different malignancies to identify changes in tumors over time
(5). Indeed, a recent investigation
of ctDNA from metastatic RCC samples demonstrated clear differences
from the genomic landscape of primary RCC and an evolution of
genomic alterations (GAs) under treatment with targeted agents
(6). In this cohort of 220
patients, high frequencies of GAs were identified in TP53, VHL1,
EGFR, NF1, as well as several other genes. A number of these genes
had more frequent GAs in samples from patients with multiple lines
of therapy vs. those receiving only one line of therapy. However,
the genomics of metastatic ctDNA have yet to be fully elucidated.
Furthermore, with the plethora of treatment options and the
presence of rapidly evolving tumors, there is a need to identify
biomarkers to individualize treatment in order to ensure maximum
efficacy. Therefore, the aim of the present study was to determine
whether the patterns of GAs in our cohort were consistent with
previous findings and to further elucidate the genomics of
metastatic RCC in patients with varied treatment patterns using a
more comprehensive ctDNA sequencing panel.
Materials and methods
Sample collection and processing
As part of the IRB-approved study 17089, patients
included in the present series had a diagnosis of metastatic RCC
and consented to blood and urine collection for ctDNA assessment as
a part of routine clinical care at City of Hope Medical Center
(Duarte, USA). Urine was collected in a standard collection cup and
frozen within 2 h. Blood was collected in Cell-Free DNA BCT
collection tubes (Streck). To separate plasma, whole blood was
centrifuged at 300 x g for 20 min at room temperature. The upper
plasma layer was removed and transferred to a new conical tube
which was then centrifuged at 5,000 x g for 10 min at 20˚C.
Cell-free DNA (cfDNA) was isolated using the QIAamp Circulating
Nucleic Acid Kit (Qiagen, Inc.); the median collection amount was
359.7 ng (range, 222.4-1,329.7 ng) for plasma and 2,111.4 ng
(range, 456.9-3,101.8 ng) for urine. cfDNA was shipped on dry ice
to Beijing Scisoon Biotechnology Ltd., Co. for analysis. The
concentration of cfDNA was determined using Qubit fluorometer 3.0
(Thermo Fisher Scientific, Inc.), KAPA Hyper Prep Kit (KK8504,
Roche Diagnostics) was used for library construction. Library
quality control was measured using 2100 Bioanalyzer (Agilent
Technologies, Inc.) and then 150 base pair-end sequencing was
performed on Illumina Novaseq 6000 (Illumina, Inc.). Reads were
aligned to human reference genome GRCh37/hg19 using BWA (v0.7.17;
https://github.com/lh3/bwa) and
single-nucleotide variants were determined by VarScan (v2.4.1;
https://github.com/dkoboldt/varscan).
The sequenced genes are shown in Table
SI.
Statistical analysis
The mean depth of sequencing coverage by sample had
a mean ± SD of 2,979.514x±1,420.744 (range, 1,008.54-6,353.28x) for
plasma and 2,756.0±3,338.112 (range, 361.4-14,180.0) for urine. The
frequency of GAs was assessed in the overall cohort and compared
across line of therapy and histology (when available).
Multivariable regression was used to evaluate the associations
among clinical variables and GAs. The frequencies of individual GAs
in subgroup analyses were compared using χ2 tests.
P<0.05 was considered to indicate statistically significant
differences.
Results
Patient characteristics and
association with distribution of GAs
ctDNA was collected from the urine and plasma from a
total of 50 patients with mRCC between June 2018 and December 2018
(Table I). The median age of the
cohort was 65 years (range, 36-89 years), and the patients included
40 men and 10 women. Samples were collected from patients with
varying therapies, lines of treatment and histological subtypes
(Tables I and SII). Using an investigational ctDNA panel
that covers part or the entirety of 120 genes (Table SI), it was observed that all 50
plasma samples contained GAs compared to the consensus sequences at
these loci (median GA no. 2; range, 1-6). The number of GAs was
significantly associated with the number of lines of therapy, both
on univariate regression analysis (P=0.038) and on multivariable
regression analysis (P=0.047), which included age, sex, T stage,
Fuhrman grade, and histological subtype; no other variables were
associated with the number of GAs (Tables SIII and SIV).
 | Table IPatient characteristics. |
Table I
Patient characteristics.
| Clinical
variables | Patient no. |
|---|
| Sex | |
|
Male | 40 |
|
Female | 10 |
| Lines of therapy | |
|
0 | 1 |
|
1 | 28 |
|
2 | 9 |
|
3 | 6 |
|
4+ | 6 |
| T stage | |
|
1 | 9 |
|
2 | 12 |
|
3 | 22 |
|
4 | 2 |
|
Unknown | 5 |
| Fuhrman grade | |
|
1 | 0 |
|
2 | 7 |
|
3 | 19 |
|
4 | 18 |
|
Unknown | 6 |
| Histology | |
|
Clear
cell | 40 |
|
Papillary | 7 |
|
Chromophobe | 3 |
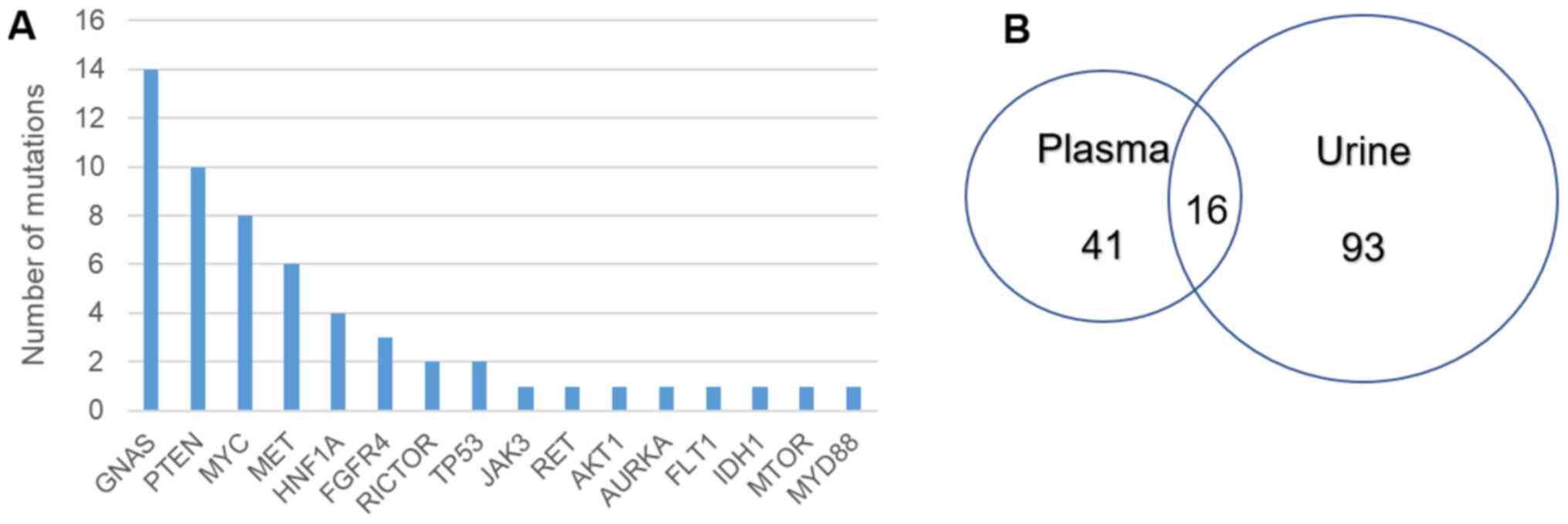
The genes with the most frequent GAs were GNAS,
PTEN, MYC, MET and HNF1A (Fig. 1A). Full lists of GAs found in plasma
samples are in provided in Table
SV. Our findings bear some similarities to the largest
published analysis of ctDNA from mRCC patients to date (6), despite using different sequencing
panels (Table II). A similar high
frequency of GAs was observed in MET and MYC, and a
similar low frequency of GAs in RET, JAK3, IDH1 and
AKT1. Pal et al (6)
reported a higher frequency of GAs in TP53 compared with
that of the present study, while we identified a higher frequency
of GAs in GNAS and PTEN. Additional GAs were
identified in RICTOR, FGFR4, AURKA, MYD88 and
FLT1, none of which were sequenced by Pal et al.
| Table IIComparison of mutation frequency
across datasets. |
Table II
Comparison of mutation frequency
across datasets.
| | Samples with
genomic alterations (%) |
|---|
| Gene | Present study | Pal et
al | TCGA clear
cell | TCGA papillary |
|---|
| MET | 12 | 9.5 | 0.7 | 13 |
| MYC | 16 | 7.7 | 1.2 | 0 |
| PTEN | 20 | 3.6 | 5 | 2.9 |
| TP53 | 4 | 35 | 2.1 | 2.5 |
| GNAS | 28 | 3.2 | 0.7 | 0 |
| HNF1A | 8 | 0 | 0 | 1.5 |
| FGFR4 | 6 | N/S | 0.7 | 0.7 |
| RICTOR | 4 | N/S | 1 | 1.1 |
| MTOR | 2 | 0 | 5.4 | 1.4 |
| AKT1 | 2 | 0.5 | 0.5 | 0 |
| RET | 2 | 0.9 | 0.2 | 0 |
| FLT1 | 2 | N/S | 1.4 | 1.4 |
| JAK3 | 2 | 0.9 | 1 | 0.4 |
| AURKA | 2 | N/S | 0 | 0 |
| IDH1 | 2 | 1.4 | 1.2 | 0.7 |
| MYD88 | 2 | N/S | 3 | 0 |
Pal et al (6)
also observed significant increases in the number of patients with
GAs in TP53, VHL, EGFR, NF1 and PIK3CA among patients
who had received multiple lines of therapy vs. those receiving only
first-line therapy. We searched for increased GAs in patients who
had multiple lines of therapy (n=22) vs. those with only first-line
therapy (n=28); however, the only significant difference identified
by χ2 analysis was an increased frequency of GAs in
GNAS in patients with only first-line therapy (P=0.025).
Site of metastasis data were available on 43/50 patients in our
database. A total of 7 patients had multiple metastatic sites, and
the overall involvement included 21 lymph node, 31 lung, 2 brain, 4
liver, 7 bone and 9 ‘other soft tissue’ lesions. Subgroup analysis
by metastatic site for enrichment in GAs in PTEN, MYC, GNAS
or MET did not identify any significant differences by
χ2 analyses (Table
SVI).
Matched urine samples were also obtained from the
patients, 45 of which yielded ctDNA sequencing results that passed
quality control filters. All 45 samples had at least one GA
(median, 3; range, 1-8). Regression analysis demonstrated a
significant association between the number of GAs detected in
matched plasma and urine samples (P=0.047). While several GAs were
unique to urine samples (Fig. 1B),
28.1% (16/57) of the GAs detected in plasma samples were also
detected in matched urine samples.
Discussion
In the largest assessment of ctDNA from metastatic
RCC patients to date, Pal et al (6) reported high rates of GAs in several
genes, including TP53, VHL, EGFR, PIK3CA and NF1, and
that the rate of GAs in these genes was higher in patients
receiving multiple lines of therapy vs. first-line therapy. The
present study, although from a smaller cohort, identified several
similarities to this previous study, with high frequencies of
mutations in MET and MYC. There were also several
differences, including higher GA frequencies in the GNAS and
PTEN genes, and several GAs we identified in genes that were
not sequenced in the previous study, including RICTOR, FGFR4,
AURKA, MYD88 and FLT1.
GNAS was the most frequently mutated gene in
our cohort, with the majority of mutations occurring at two
residues. The A448D mutation has been previously identified in
prostate and pancreatic adenocarcinomas (7), while, to the best of our knowledge,
the mutation at aa 371 has not been previously identified. The
GNAS gene is located on chromosome 20q13.3 and encodes the α
subunit of the heterotrimeric G protein complex (Gsα), which
couples seven-transmembrane receptors to the cAMP-generating enzyme
adenylyl cyclase (8). Activating or
inactivating mutations and epigenetic changes at the GNAS
locus have been described in a number of cancers (9), but little is known regarding
GNAS mutations in RCC. The findings of the present study
must be replicated in a larger cohort and the mutations confirmed
by additional methods. However, our data warrant further
investigation into the role of GNAS mutation in RCC.
A recurrent mutation at Y55 was identified in the
PTEN gene. Mutation of this residue has been previously
identified in chromophobe RCC, uterine endometrioid carcinoma,
glioblastoma multiforme, breast invasive ductal carcinoma (IDC) and
cervical squamous cell carcinoma (SCC), and is predicted to be
oncogenic (7). It is well
established that PTEN is a frequently mutated gene in RCC,
particularly clear cell RCC (10).
Moreover, several studies have demonstrated that mutations in
PTEN are associated with worse overall survival in patients
with RCC (10,11). Therefore, it was not surprising to
find a relatively high frequency of PTEN mutation in our
cohort.
The RICTOR A3V mutation has been identified
in breast IDC, and while the R8H mutation has not been previously
identified, the R8C mutation has been identified in adenoid cystic
carcinoma and hepatocellular carcinoma. RICTOR is the mTOR partner
in the mTORC2 complex, which has been shown to serve an important
function in the development and progression of RCC (12); thus, the identification of mutations
in this gene are expected in RCC. None of the FGFR4
mutations have been previously identified in cancer, but the FGFR
pathway is involved in driving VEGF-independent tumor angiogenesis
as a mechanism to escape VEGF-targeted treatment (13). As several patients in our cohort had
received such treatment, it is possible that these mutations arose
in response to this treatment. The same may be the case for the
mutation identified in FLT1, the gene that encodes VEGFR1.
The AURKA mutation also has not been previously identified
in cancer, but the role of Aurora kinase A has been described in
advanced RCC (14), which may also
be of relevance.
A subgroup analysis of patients who had been exposed
to any MET inhibitor (n=29) vs. those who had not (n=21) was
performed, and revealed that only patients who had been exposed to
these agents had GAs in MET (n=6) and RET (n=1);
these GAs were not identified in any patients who had not received
MET inhibitor treatment. The two tyrosine kinase-encoding genes
RET and MET are targets of cabozantinib and other MET
inhibitors (15), and RET
mutations have been shown experimentally to cause resistance to
cabozantinib in model systems (16), as have mutations in MET
(17). Of the mutations in our
cohort, the RET E366X mutation has been found in head and
neck SCC and lung SCC. Mutation at aa63 has been found in uterine
endometrioid carcinoma and mutation at aa375 has been found in
prostate cancer, while the V1220I mutation has not been previously
identified in cancer. It is possible that these mutations arose in
response to treatment with the MET inhibitors; however, we did not
identify significant differences in response or time-to-progression
in MET inhibitor-treated patients with or without mutations in
MET and RET, which may be attributed to the small
sample size.
The present study was unable to confirm the findings
of Pal et al (6), which
demonstrated that GAs in several genes were more common among
patients treated with multiple lines of treatment vs. those
receiving only one line of treatment, but this was likely due to
the smaller cohort and smaller number of mutations identified in
the present study. Of note, GAs in GNAS were not found to be
more frequent in patients receiving only one line of therapy, but
the relevance of this finding is unclear.
Although the majority of studies extract ctDNA from
plasma for sequencing, other body fluids, including the urine, also
contain ctDNA. Urine has several advantages compared to blood, as
it is a non-invasive sample source, which is ideal for patients who
require repeated sampling to monitor cancer progression or
therapeutic efficacy, and the collection of urine does not require
specialized facilities or expensive equipment (18). Several studies have demonstrated
that there is a high degree of correlation between the detection of
specific GAs in the urine and plasma ctDNA (19,20).
We herein observed that, while there was a significant correlation
between the number of GAs detected in matched urine and plasma
samples, only 28.1% of GAs detected in plasma samples were also
detected in matched urine samples. This may be due to technical
complications from isolating ctDNA from urine (18), or it may reflect different subsets
or clones of cancers that preferentially shed DNA into these
fluids. Further research is needed to determine the association
between ctDNA isolated from urine and plasma before urine ctDNA can
reliably be used to monitor metastatic RCC.
Certain differences from the study of Pal et
al (6) were to be expected, as
a different sequencing platform was used and the patient population
was also different. The present study included fewer patients, and
they were all from a single institution. Furthermore, the treatment
differed significantly between the two studies, as the patients in
the Pal et al study received primarily VEGF-targeted
therapies, while the treatments received by patients in the present
study varied widely. The ctDNA sequencing platform used in the
present study detected only mutations, not amplifications, which
may partially explain why fewer GAs we identified in certain genes
compared with the Pal et al study.
There were certain limitations to the present study.
First, including only 50 patients from a single institution
precludes wide generalizations of the findings, and it also limits
subgroup analysis between different treatments and histological
subtypes. The present study did not have primary tumor tissue to
compare ctDNA samples, and serial samples were not collected, so it
was not feasible to directly assess tumor evolution in individual
patients. Furthermore, DNA from normal cells was not available for
comparing ctDNA results; therefore, GAs were determined by
comparison to a reference database. In addition, the
investigational ctDNA sequencing panel used is marketed in China,
but has not undergone testing as rigorous as that of its
US-approved counterparts, such as those from Guardant or Foundation
Medicine. Several of the GAs identified in the present study are
variants of unknown significance and must be further investigated
to determine whether they are truly oncogenic.
In conclusion, despite its limitations, the present
study largely supports the findings of Pal et al and has
identified potential new targets in metastatic RCC.
Supplementary Material
Scisoon ctDNA sequencing panel.
Therapy on blood collection date.
Univariate regression analysis.
Multivariate regression analysis.
Full list of genomic alterations in
patient samples.
Site of mutation subgroup
analysis.
Acknowledgements
The authors would like to thank the clinicians at
City of Hope Medical Center for their help with enrolling patients
in the present study.
Funding
The present study was funded by Beijing USCI Medical
Laboratory.
Availability of data and materials
De-identified data are available from the
corresponding author upon reasonable request.
Authors' contributions
JZ, YL, BX, FL, YW, ML, RD and YZ performed sample
preparation, sequencing, data analysis and edited the manuscript.
MS and LY collected and processed biological samples. JOJ assisted
with sample processing, data analysis and manuscript preparation.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the City of Hope
IRB and informed consent was obtained from all individual
participants.
Patient consent for publication
Not applicable.
Competing interests
JZ, YL, BX, FL, YW, ML, RD and YZ are employees of
Scisoon, a subsidiary of Beijing USCI Medical Laboratory. JOJ is a
compensated advisor for Scisoon.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386.
2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gupta K, Miller JD, Li JZ, Russell MW and
Charbonneau C: Epidemiologic and socioeconomic burden of metastatic
renal cell carcinoma (mRCC): A literature review. Cancer Treat Rev.
34:193–205. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Cancer Genome Atlas Research Network.
Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis
C, Wheeler DA, Murray BA, Schmidt L, et al: Comprehensive molecular
characterization of papillary renal cell carcinoma. N Engl J Med.
374:135–145. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Cancer Genome Atlas Research Network.
Comprehensive molecular characterization of clear cell renal cell
carcinoma. Nature. 499:43–49. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gainor JF, Dardaei L, Yoda S, Friboulet L,
Leshchiner I, Katayama R, Dagogo-Jack I, Gadgeel S, Schultz K,
Singh M, et al: Molecular mechanisms of resistance to first- and
second-generation ALK inhibitors in ALK-rearranged lung cancer.
Cancer Discov. 6:1118–1133. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Pal SK, Sonpavde G, Agarwal N, Vogelzang
NJ, Srinivas S, Haas NB, Signoretti S, McGregor BA, Jones J, Lanman
RB, et al: Evolution of circulating tumor DNA profile from
first-line to subsequent therapy in metastatic renal cell
carcinoma. Eur Urol. 72:557–564. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Weinstein LS, Liu J, Sakamoto A, Xie T and
Chen M: Minireview: GNAS: Normal and abnormal functions.
Endocrinology. 145:5459–5464. 2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Furukawa T, Kuboki Y, Tanji E, Yoshida S,
Hatori T, Yamamoto M, Shibata N, Shimizu K, Kamatani N and
Shiratori K: Whole-exome sequencing uncovers frequent GNAS
mutations in intraductal papillary mucinous neoplasms of the
pancreas. Sci Rep. 1(161)2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fan C, Zhao C, Wang F, Li S and Wang J:
Significance of PTEN mutation in cellular process, prognosis, and
drug selection in clear cell renal cell carcinoma. Front Oncol.
9(357)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Que WC, Qiu HQ, Cheng Y, Liu MB and Wu CY:
PTEN in kidney cancer: A review and meta-analysis. Clin Chim Acta.
480:92–98. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sun B, Chen L, Fu H, Guo L, Guo H and
Zhang N: Upregulation of RICTOR gene transcription by the
proinflammatory cytokines through NF-κB pathway contributes to the
metastasis of renal cell carcinoma. Tumor Biol. 37:4457–4466.
2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Massari F, Ciccarese C, Santoni M,
Lopez-Beltran A, Scarpelli M, Montironi R and Cheng L: Targeting
fibroblast growth factor receptor (FGFR) pathway in renal cell
carcinoma. Expert Rev Anticancer Ther. 15:1367–1369.
2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Pal SK, He M, Tong T, Wu H, Liu X, Lau C,
Wang JH, Warden C, Wu X, Signoretti S, et al: RNA-seq reveals
aurora kinase-driven mTOR pathway activation in patients with
sarcomatoid metastatic renal cell carcinoma. Mol Cancer Res.
13:130–137. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Osanto S and van der Hulle T: Cabozantinib
in the treatment of advanced renal cell carcinoma in adults
following prior vascular endothelial growth factor targeted
therapy: Clinical trial evidence and experience. Ther Adv Urol.
10:109–123. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liu X, Shen T, Mooers BHM, Hilberg F and
Wu J: Drug resistance profiles of mutations in the RET kinase
domain. Br J Pharmacol. 175:3504–3515. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bahcall M, Sim T, Paweletz CP, Patel JD,
Alden RS, Kuang Y, Sacher AG, Kim ND, Lydon CA, Awad MM, et al:
Acquired METD1228V mutation and resistance to MET inhibition in
lung cancer. Cancer Discov. 6:1334–1341. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lu T and Li J: Clinical applications of
urinary cell-free DNA in cancer: Current insights and promising
future. Am J Cancer Res. 7:2318–2332. 2017.PubMed/NCBI
|
|
19
|
Reckamp KL, Melnikova VO, Karlovich C,
Sequist LV, Camidge DR, Wakelee H, Perol M, Oxnard GR, Kosco K,
Croucher P, et al: A highly sensitive and quantitative test
platform for detection of NSCLC EGFR mutations in urine and plasma.
J Thorac Oncol. 11:1690–1700. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fujii T, Barzi A, Sartore-Bianchi A,
Cassingena A, Siravegna G, Karp DD, Piha-Paul SA, Subbiah V,
Tsimberidou AM, Huang HJ, et al: Mutation-enrichment
next-generation sequencing for quantitative detection of KRAS
mutations in urine cell-free DNA from patients with advanced
cancers. Clin Cancer Res. 23:3657–3666. 2017.PubMed/NCBI View Article : Google Scholar
|