Introduction
Malignant gastrointestinal neuroectodermal tumor
(GNET) is a term designated by Stockman et al for rare,
clear-cell sarcoma-like tumors of the gastrointestinal tract
(CCSLGT) (1). They reappraised
CCSLGT cases and clarified the following features. This tumor is
histologically characterized by a sheet-like or nested population
of epithelioid or oval-to-spindle-shaped cells with small nucleoli
and scattered mitoses. These tumor cells are positive for S-100
protein (S-100), SRY-related HMG-box 10 (SOX10), and vimentin and
sometimes positive for CD56, synaptophysin, neuroblastoma 84,
neuron-specific enolase (NSE), and neurofilament proteins. They
usually contain EWSR1 gene rearrangements, such as EWSR1-ATF1 or
EWSR1-CREB1. In general, GNET lacks melanocyte-specific markers,
making it clearly different from clear-cell sarcoma (CCS) of the
tendons and aponeuroses.
Because GNETs are extremely rare and reports on them
are only available in the form of case reports or small size
reviews, their morbidity is unclear. They tend to occur mainly in
young to middle-aged adults. The most common site of tumor origin
is the small intestine (57.9%), followed by the stomach, colon, and
other sites of the gastrointestinal tract (2). Surgery is often the choice for
resectable lesions, but there are currently no standard
chemotherapeutic or targeted therapeutic options for this disease
in the metastatic setting. The prognosis is generally poor, and the
median survival was reported to be 9.5 months (3). Here, we present a unique case of a
GNET that has a history of desmoplastic malignant melanoma
exhibiting a BRAF mutation, which later transformed into a GNET of
the small intestine with both a BRAF mutation and two subtypes of
the EWSR1-ATF1 fusion gene.
Case report
In April 2018, a 66-year-old woman with multiple
metastatic tumors was referred to our hospital for further
diagnosis and treatment. She had suffered from lower abdominal pain
in February 2018 and consulted a nearby hospital. Imaging
examinations revealed a tumor in the small intestine, a soft-tissue
mass in her left forearm, a bilateral pleural mass, and a left
breast mass. A small intestinal tumor of 8 cm in diameter was
resected in early March 2018, and the histological diagnosis was
undifferentiated carcinoma. She was diagnosed with carcinoma of
unknown primary and transferred to our hospital.
The patient also suffered from idiopathic
thrombocytopenic purpura and had a history of cutaneous malignant
melanoma (MM). Her MM history was as follows: She underwent
extended resection of MM at her left thigh in April 2010 at another
hospital; she also received adjuvant combination chemotherapy
containing dacarbazine, nimustine hydrochloride, vincristine and
interferon-β. However, the patient experienced a local recurrence
of MM in May 2011 and underwent additional extended resection,
though no adjuvant chemotherapy was administered after the second
operation.
The patient's pleural mass was rapidly growing;
hence, she was admitted to our hospital in April 2018. On
admission, she was slightly obese (the body mass index was 28.06).
Physical examination demonstrated a soft, painful mass of 5 cm in
diameter in the flexor muscle side of her left forearm. In
addition, a hard mass of 2 cm in diameter was palpable in the left
upper portion of her left breast. Computed tomography revealed a
large necrotic tumor of 15 cm in diameter in the left upper portion
of her chest, with bilateral multiple lung metastatic masses and
left pleural effusion. Furthermore, a 47-mm tumor without contrast
effect in the left forearm and a 17-mm nodule in the left breast
were detected, but no lymphadenopathy was found.
Laboratory studies yielded the following results:
White blood cells=7.96x109/l, hemoglobin=12.8 g/dl,
platelet count=79x109/l (normal range [NR]:
158-348x109/l), lactate dehydrogenase=438 IU/l (NR:
124-222 IU/l), C-reactive protein=6.03 mg/dl (NR ≤0.14 mg/dl),
CA-125=38 U/ml (NR ≤35 U/ml), NSE=19.4 ng/ml (NR ≤16.3 ng/ml), and
platelet-associated IgG=59 ng/107 cells (NR ≤46
ng/107 cells).
Reexamination of the resected small intestinal tumor
by our hospital pathologists resulted in the new diagnosis of a
GNET (details of the findings are described in the next section),
as reverse-transcription polymerase chain reaction (RT-PCR) of the
specimen revealed an EWSR1 exon8/ATF1 exon4 fusion transcript,
which is disease-specific for GNET or CCS. Additional biopsies of
her left forearm mass and left breast mass were performed at our
hospital. The histopathological diagnosis of the left forearm mass
was the same as that of the tumor of the small intestine. On the
other hand, the breast mass was diagnosed as invasive ductal
carcinoma (papillotubular carcinoma).
At this point, we questioned whether the previous
diagnosis of MM was valid because it showed phenotypic and
immunohistochemical overlap with GNET and CCS. Therefore, we asked
the hospital which had treated her MM to provide samples. Because
of the rapid progression of her pleural mass, chemotherapy with
doxorubicin was started even before the samples were examined. Two
courses of doxorubicin were administered, stabilizing the case.
However, the patient further suffered from pneumocystis pneumonia,
and it was important to discontinue chemotherapy. She was further
treated using a drug combination of sulfamethoxazole and
trimethoprim and recovered from pneumonia.
In June 2018, the histological diagnosis of the
previous samples at the time of recurrence of MM was finally
established as desmoplastic MM with a BRAF mutation. Therefore, the
BRAF gene status of the small intestinal and left forearm tumors
were checked, and both tissues were found to exhibit a BRAF
mutation. Her GNET was diagnosed as double-positive for BRAF
mutation and EWSR1-ATF1 fusion transcript. Dabrafenib mesylate and
trametinib dimethyl sulfoxide were started, a combination therapy
that proved effective and achieved a partial response three months
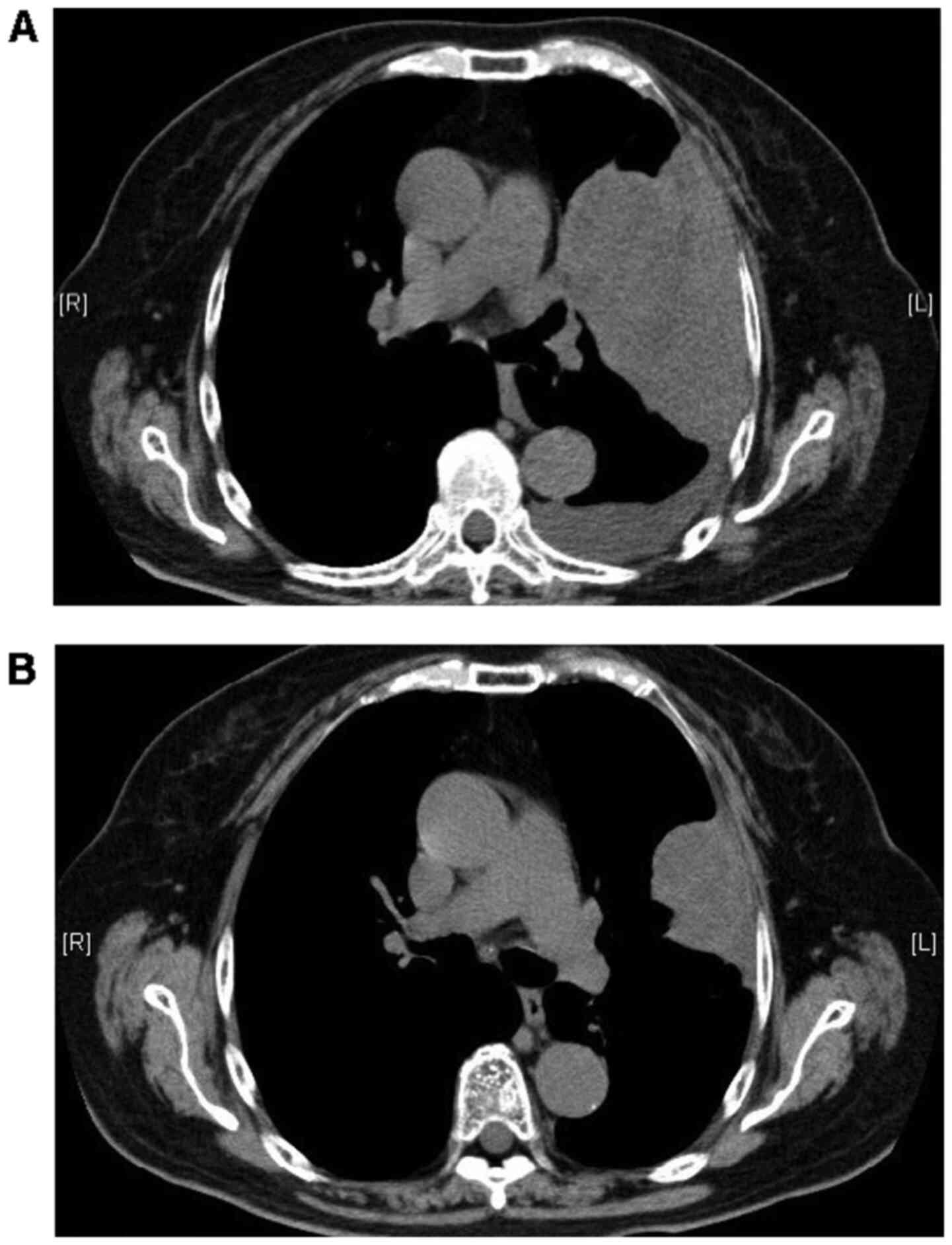
later (Fig. 1). The size of the
pleural mass increased in January 2019, and these drugs were
replaced by immunotherapy with nivolumab and ipilimumab. This
immunotherapy was performed only once because of the rapid
progression of her pleural mass, the occurrence of multiple brain
metastases, and the deterioration of her health. She was
transferred to a hospice facility in February 2019 and died in
January 2020 because of the deterioration of her brain
metastasis.
Pathological findings
In total, four tissue samples were reviewed
(Table I): A primary cutaneous
lesion of the left thigh resected in April 2010 (Sample 1), a
recurrent tumor embedded in the former operation scar excised in
July 2011 (Sample 2), a small intestinal mass resected in March
2018 (Sample 3) and a core-needle biopsy of a soft-tissue mass that
arose in the left forearm in April 2018 (Sample 4).
 | Table ISummary of immunohistochemical and
genetic findings. |
Table I
Summary of immunohistochemical and
genetic findings.
| Variables | Sample 1 (Fig. 2) | Sample 2 | Sample 3 (Fig. 3) | Sample 4 (Fig. 4) |
|---|
| Resection date
Site | April 2010 Left thigh
(primary lesion) | July 2011 Operation
scar in the left thigh (recurrent site) | March 2018 Small
intestine | April 2018
Soft-tissue mass in the left forearm |
| Pathological
diagnosis | Desmoplastic MM | Recurrence of
desmoplastic MM | GNET | Metastatic lesion of
GNET |
| IHC stain:
Positive | S-100 | S-100, SOX10, Melan A
(+, focal) | S-100 (-/+, a few),
SOX10 (+, scattered), CK AE1/3 (+, focal), CK CAM5.2 (+, focal),
CD56 (+, focal) | CD56 (+, focal) |
| IHC stain:
Negative | HMB45 | HMB45, cKIT | CK7, CK20, melan A,
HMB45, desmin, α-SMA, HHF35, caldesmon, cKIT, DOG1, CD34, TTF-1,
GCDFP-15, EMA, MyoD1, myogenin, synaptophysin, chromogranin A,
PAX8, ER | S-100, SOX10, HMB45,
Melan A |
| Ki-67 labeling
index | No sample | 5% | 50% | Not performed |
| Genetic findings of
BRAF | No sample | BRAF mutation
(V600E) | BRAF mutation
(V600E) | BRAF mutation
(V600E) |
| Genetic findings of
EWSR1/ATF1 | No sample | - | EWSR1 exon8/ATF1
exon4 | EWSR1 exon10/ATF1
exon5 |
i) Primary cutaneous lesion of the
left thigh (Sample 1)
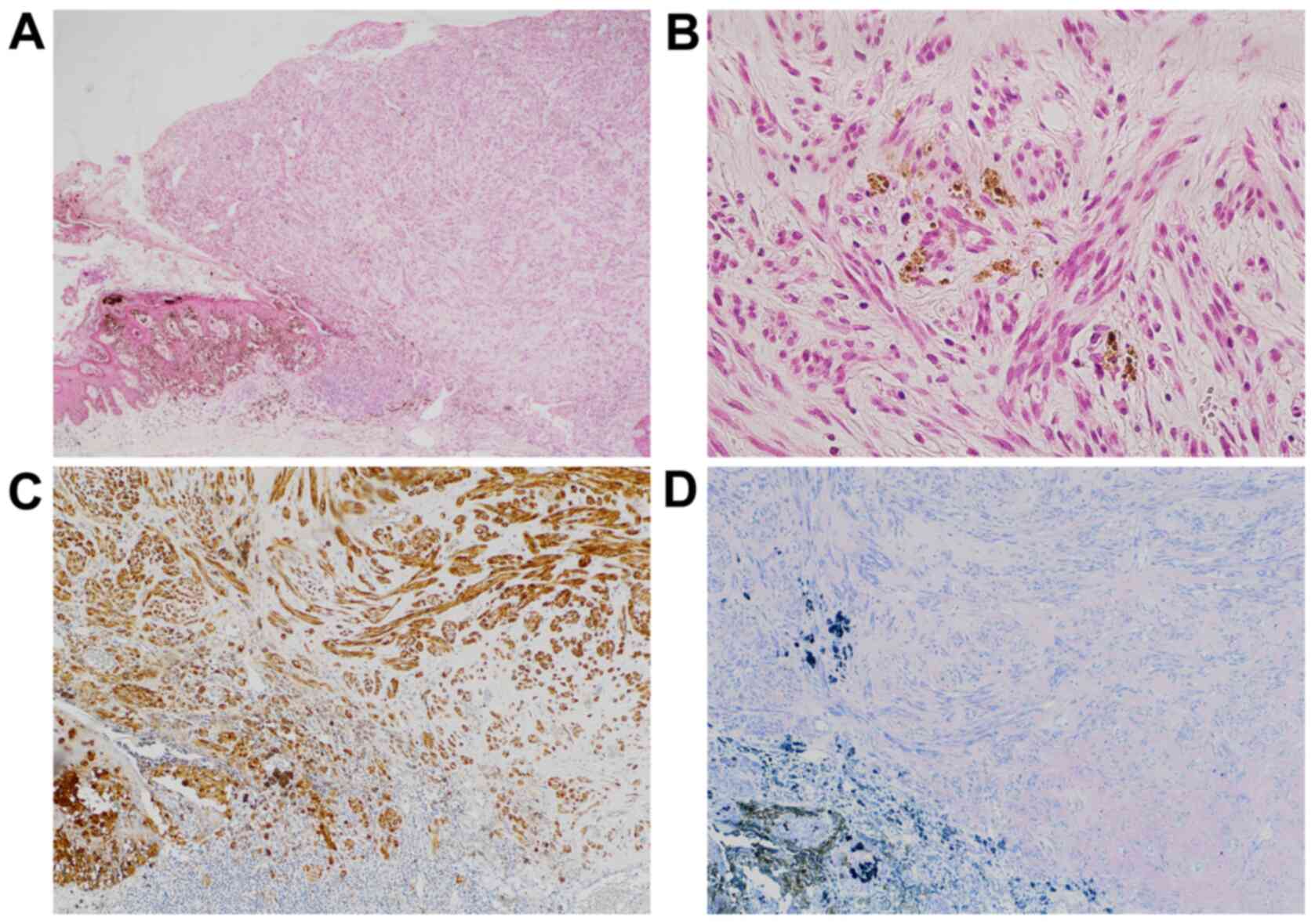
Sections stained with hematoxylin and eosin
(H&E) showed a subpedunculated polypoid lesion with a maximum
diameter of 6 mm associated with intraepidermal neoplasm harboring
brown pigments at the tumor border (Fig. 2A). The protrusion was mainly
composed of spindle cells with relatively low atypia proliferating
in fascicles (Fig. 2B). No
lymphovascular invasion was identified. Immunohistochemically, the
tumor cells were positive for S-100 but negative for human melanin
black-45 (HMB45) (Fig. 2C and
D). The histological diagnosis was
desmoplastic MM with an unclear surgical margin. No tissue blocks
were available for additional studies.
ii) Recurrent tumor beneath operation
scar in the left thigh (Sample 2)
Sections stained with H&E showed the
proliferation of atypical spindle cells in the subcutaneous tissue.
The morphological features resembled that of the invasive component
of the previous tumor (Sample 1). Infiltrating cells were
immunoreactive to S-100 and SOX10. Melan A was focally positive,
although HMB45 was consistently negative. The Ki-67 labeling index
was 5%. These features are compatible with the local recurrence of
desmoplastic MM.
iii) Small intestinal tumor (Sample
3)
Sections stained with H&E demonstrated a
well-demarcated tumor localized in the intestinal subserosa, which
was composed of small round cells with a high nuclear/cytoplasm
(N/C) ratio and frequent mitoses accompanied by central necrosis,
focal hemorrhage, and myxoid changes (Fig. 3A). Neoplastic cells showed an
immunophenotype with limited expressions of S-100 and SOX10 and
negativity for HMB45 and Melan A (Fig.
3B-E). Other immunohistochemical findings were as follows: CK
AE1/3(+, focal), CK CAM5.2(+, focal), CD56(+, focal; Fig. 3F), CK7(-), CK20(-), Desmin(-),
α-SMA(-), HHF35(-), Caldesmon(-), cKIT(-), DOG1(-), CD34(-),
TTF-1(-), GCDFP-15(-), EMA(-), MyoD1(-), Myogenin(-),
Synaptophysin(-), Chromogranin A(-), PAX8(-) and ER(-). The Ki-67
labeling index was 50%. These features were suggestive of GNET,
although the diagnosis was inconclusive.
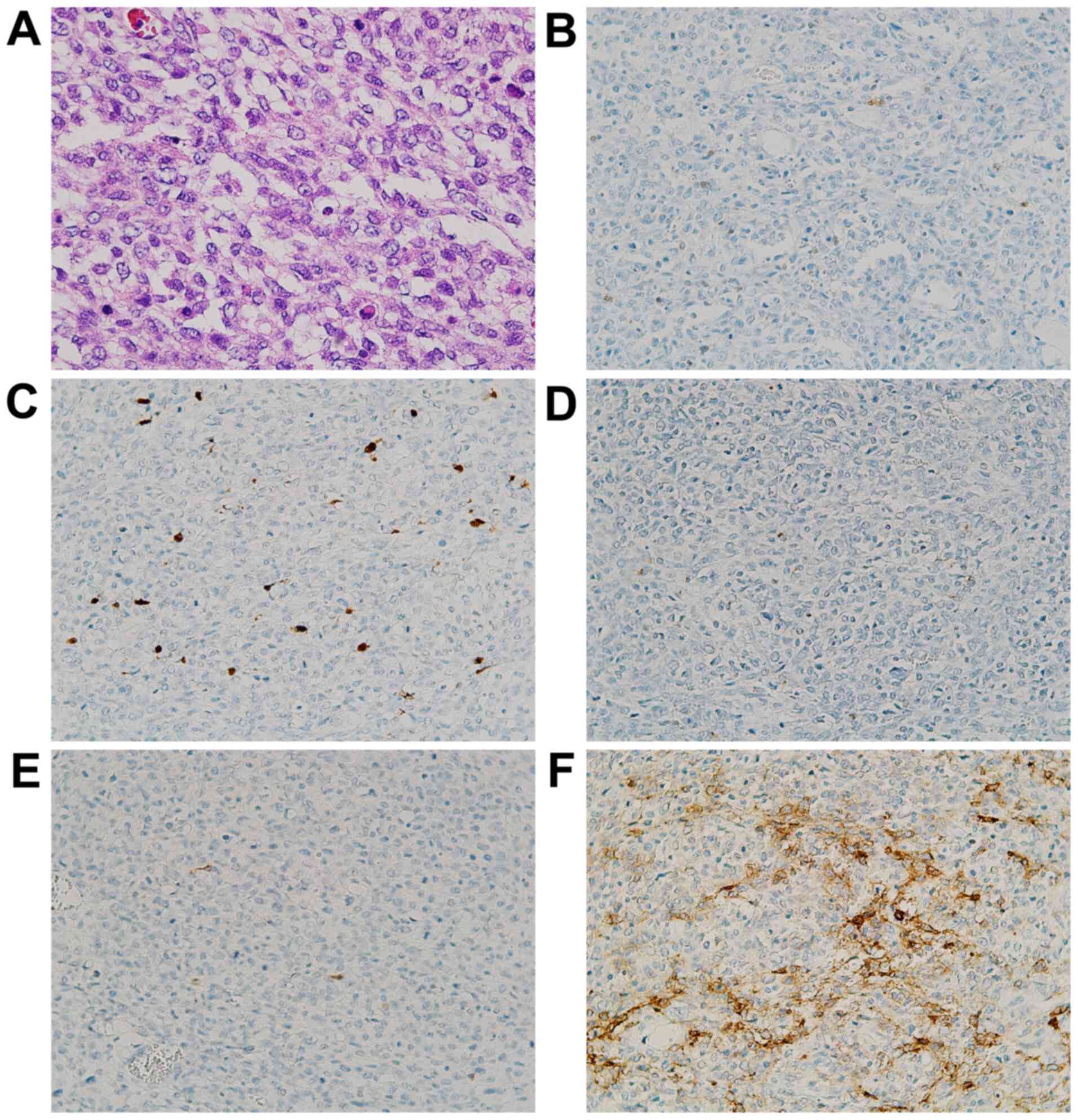 | Figure 3Photomicrographs of the small
intestinal tumor. (A) A patternless proliferation of atypical cells
characterized by scant pale cytoplasm with microvesicular
degeneration (H&E stain; magnification, x200). The tumor was
immunohistochemically partially positive for (B) S-100 (IHC stain;
magnification, x100) and (C) SOX10 (IHC stain; magnification,
x100), but negative for (D) HMB45 (IHC stain; magnification, x100)
and (E) Melan A (IHC stain; magnification, x100). Additionally, the
tumor was positive for (F) CD56 (IHC stain; magnification, x100).
IHC, immunohistochemical; SOX10, SRY-related HMG-box 10; HMB45,
human melanin black-45. |
iv) Soft-tissue mass of the left
forearm (Sample 4)
Sections stained with H&E revealed morphological
features resembling those of Sample 3, composed of compact
dysplastic cells with a high N/C ratio, which were
immunohistochemically negative for S-100, SOX10, HMB45 and Melan A
but focally positive for CD56 (Fig.
4A-F). A metastatic tumor was considered based on molecular
rearrangement similar to that seen in the small-intestinal tumor
(Sample 3), as described below.
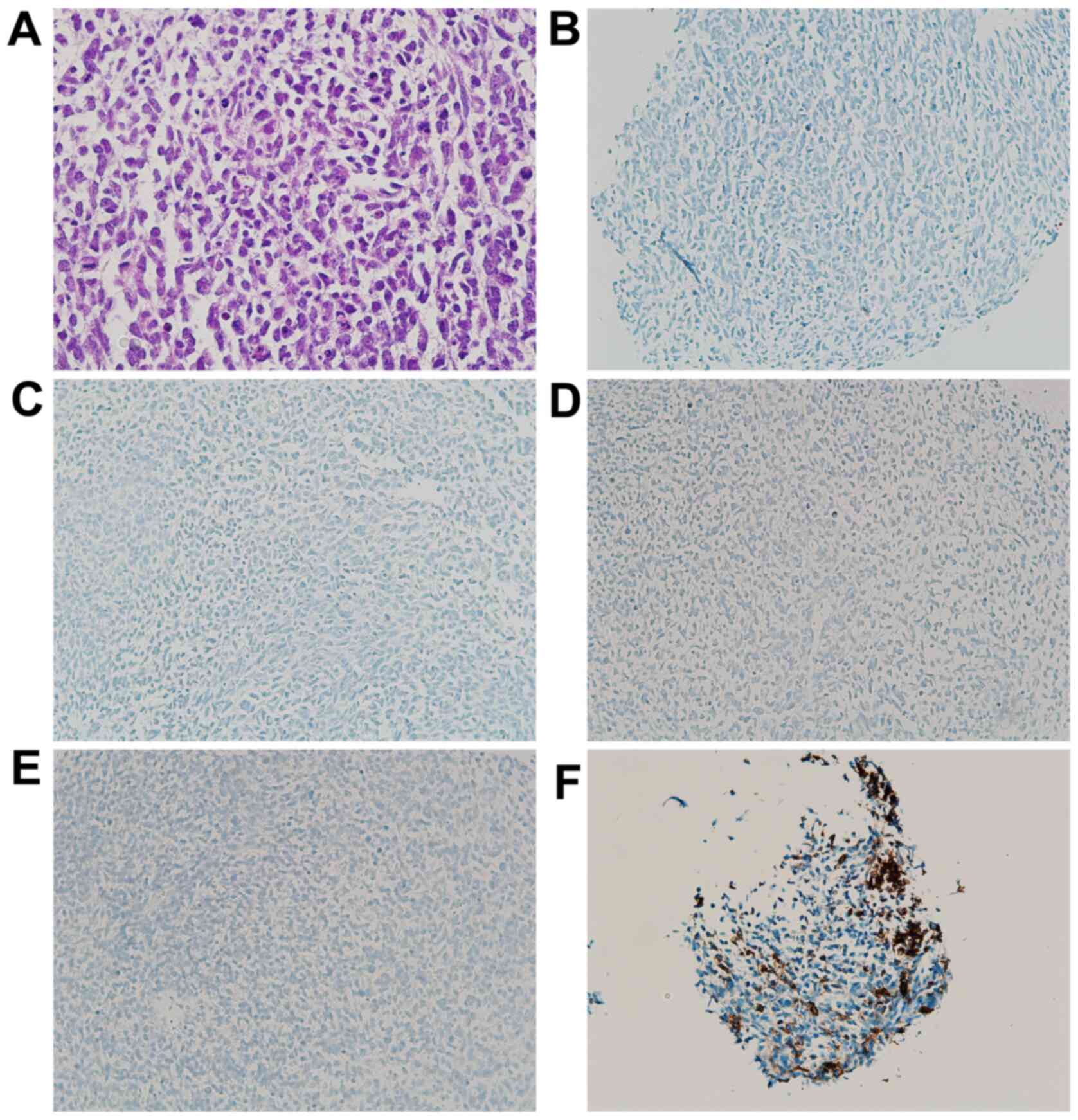 | Figure 4Photomicrographs of the left forearm
tumor. (A) Diffuse proliferation of small round or polygonal cells
with a high nuclear/cytoplasm ratio (H&E stain; magnification,
x200). The tumor was immunohistochemically negative for (B) S-100
(IHC stain; magnification, x100), (C) SOX10 (IHC stain;
magnification, x100), (D) HMB45 (IHC stain; magnification, x100)
and (E) Melan A (IHC stain; magnification, x100). The sample was
also partially positive for (F) CD56 (IHC stain; magnification,
x100). IHC, immunohistochemical; SOX10, SRY-related HMG-box 10;
HMB45, human melanin black-45. |
Genetic findings
All BRAF mutation analyses were consigned to LSI
Medience Corporation (Tokyo) and were performed using real-time PCR
techniques. BRAF mutations (V600E) were detected in Samples 2, 3
and 4. To detect the type of EWSR1-ATF1 fusion transcript, RT-PCR
was performed at our laboratory, as previously reported (4). In Sample 2, no fusion transcript was
detected. However, in Sample 3, an EWSR1 exon8/ATF1 exon4 fusion
transcript was identified. In addition, RT-PCR of Sample 4 revealed
an EWSR1 exon10/ATF1 exon5 fusion transcript.
Discussion
Clear-cell sarcoma is a rare soft-tissue sarcoma
that was first described by Enzinger (5). It typically involves the deep soft
tissues of the extremities in close proximity with tendons and
aponeurotic structures. Its distinctive features include a nested
growth pattern and consistent melanocytic differentiation (6). From a pathological point of view, CCS
and MM share many histological and immunohistochemical features.
However, CCS and MM are currently considered to be two distinct
disease entities because, in most cases, CCS involves specific
fusion genes such as EWSR1-ATF1 or EWSR-CREB1. On the other hand,
CCSLGT is an extremely rare condition that was originally described
by Zambrano et al in 2003 as an osteoclast-rich tumor of the
gastrointestinal tract with features resembling those of CCS of
soft parts (7). Stockman et
al (1) described the
clinicopathological, immunohistochemical, ultrastructural and
molecular analysis of 16 cases with CCSLGT. They reported that
these tumors are positive for S-100 and SOX10 but lack
melanocyte-specific markers. Genetically, these tumors were
characterized by EWSR1 gene rearrangements, including EWSR1-ATF1 or
EWSR1-CREB1 fusion, similar to CCS of the tendons and aponeuroses.
At the ultrastructural level, they lacked evidence of melanocytic
differentiation and showed features of neural differentiation. In
their study, Stockman et al suggested designating these
tumors as ‘malignant gastrointestinal neuroectodermal tumors’
(GNETs).
Our case exhibited the following unique
characteristics: i) the patient had a history of BRAF-mutated
desmoplastic MM; ii) her tumor cells were weakly immunoreactive or
negative for S-100 and SOX10; iii) there was no evidence of
melanocytic differentiation; iv) focal positivity for CD56 was
present; and v) her tumor exhibited both a BRAF mutation and two
subtypes of EWSR1-ATF1. Thway and Fisher (8) reviewed the clinicopathological and
molecular features of five diverse neoplasms that frequently
exhibit EWSR1-CREB1 or EWSR1-ATF1 genetic fusion: Angiomatoid
fibrous histiocytoma, conventional CCS (of tendons and
aponeuroses), CCSLGT, hyalinizing CCS of the salivary gland and
primary pulmonary myxoid sarcoma. Among these five neoplasms, S-100
negativity was a finding consistent with angiomatoid fibrous
histiocytoma and hyalinizing CCS of the salivary gland, but their
pathological and clinical features were clearly different from
those of our case. Clinicopathologically, our case was more likely
to be diagnosed with CCS or GNET, but both neoplasms were reported
to be positive for S-100 without exception. Therefore, we could not
reach a definite diagnosis for our case. However, considering the
tumor site (small intestine), the absence of melanocytic
differentiation, and the weak positivity for CD56, we determined
that her tumor was very similar to a GNET.
There were some limitations in our diagnosis. First,
the patient already had three large tumors when she visited the
previous hospital for the first time: A pleural mass, a small
intestinal mass, and a soft-tissue mass in her left forearm. No
biopsy of her pleural mass was performed. The EWSR1-ATF1 fusion
gene subtypes were different between the small intestinal mass and
the forearm mass. As for CCS, several cases with two or three
different types of EWS-ATF1 fusion have previously been reported
(9,10). To the best of our knowledge, no case
report of a GNET with more than two subtypes of EWSR1-ATF1 fusion
exists so far. Therefore, it cannot be verified whether her tumor
is really a single tumor or a collection of similar tumors. In
addition, this tumor exhibited both a BRAF mutation and EWSR1-ATF1
fusion. Such cases have rarely been reported with CCS (11,12);
however, to the best of our knowledge, no case report of a GNET
with both a BRAF mutation and EWSR1-ATF1 fusion gene exists so far.
This tumor is similar to GNET, but it may be a different type of
neoplasm.
The unique highlight in our case is the patient's
history of desmoplastic MM. It should be noted that the patient's
past MM and GNET are morphologically and immunohistochemically
different neoplasms, but both tumors exhibited BRAF mutations.
Therefore, our case suggests one hypothesis for the pathogenetic
process of her GNET: Her desmoplastic MM exhibiting a BRAF mutation
acquired an additional EWSR1-ATF1 fusion gene and changed its
morphology to a GNET-like one. However, this theory has a
limitation. The recurrent MM sample (Sample 2), which had been
resected nearly seven years earlier, may have been too old for the
detection of the EWSR1-ATF1 fusion gene. Fusion gene detection may
have been falsely negative due to a sampling error.
Therefore, the combination of BRAF mutation analysis
and EWSR1 fusion gene detection has attracted a significant amount
of attention as a means of differentiating CCS from MM (13). Our patient's GNET exhibited both a
BRAF mutation and EWSR1-ATF1 fusion genes, and combination therapy
with dabrafenib mesylate and trametinib dimethyl sulfoxide proved
to be temporarily effective. This combination of tests for GNET may
contribute to the diagnosis as well as the choice of treatment
method.
In summary, we reported the case of a woman with a
history of desmoplastic MM exhibiting a BRAF mutation, which later
transformed into a GNET exhibiting both a BRAF mutation and two
subtypes of EWSR1-ATF1 fusion genes. Combination therapy with
dabrafenib mesylate and trametinib dimethyl sulfoxide proved to be
temporarily effective for this type of tumor. Further accumulation
of similar cases will be necessary to elucidate the pathological
significance of this type of tumor.
Acknowledgements
The authors would like to thank Dr Norifumi Naka
(NachiKatsuura Town Onsen Hospital, Japan) for the supportive
reading of this manuscript and useful suggestions.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TYag conceived the present study. SN pathologically
diagnosed the patient and revised the manuscript. TYam performed
the gene analysis. TW, YI, HT, HS, KY and ST collected the clinical
data. All authors read and approved the final manuscript prior to
submission.
Ethics approval and consent to
participate
The soft-tissue mass sample was collected after
obtaining written informed consent from the patient according to a
protocol approved by the Osaka International Cancer Institute
(Osaka, Japan).
Patient consent for publication
Written informed consent was obtained from the
patient's husband for the publication of her data and associated
images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stockman DL, Miettinen M, Suster S,
Spagnolo D, Dominguez-Malagon H, Hornick JL, Adsay V, Chou PM,
Amanuel B, Vantuinen P and Zambrano EV: Malignant gastrointestinal
neuroectodermal tumor: Clinicopathologic, immunohistochemical,
ultrastructural, and molecular analysis of 16 cases with a
reappraisal of clear cell sarcoma-like tumors of the
gastrointestinal tract. Am J Surg Pathol. 36:857–868.
2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chang B, Yu L, Guo WW, Sheng WQ, Wang L,
Lao I, Huang D, Bai QM and Wang J: Malignant gastrointestinal
neuroectodermal tumor: Clinicopathologic, immunohistochemical, and
molecular analysis of 19 cases. Am J Surg Pathol. 44:456–466.
2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Green C, Spagnolo DV, Robbins PD, Fermoyle
S and Wong DD: Clear cell sarcoma of the gastrointestinal tract and
malignant gastrointestinal neuroectodermal tumor: Distinct or
related entities? A review. Pathology. 50:490–498. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Antonescu CR, Tschernyavsky SJ, Woodruff
JM, Jungbluth AA, Brennan MF and Ladanyi M: Molecular diagnosis of
clear cell sarcoma: Detection of EWS-ATF1 and MITF-M transcripts
and histopathological and ultrastructural analysis of 12 cases. J
Mol Diagn. 4:44–52. 2002.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Enzinger FM: Clear-cell sarcoma of tendons
and aponeuroses An analysis of 21 cases. Cancer. 18:1163–1174.
1965.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Antonescu CR: Clear cell sarcoma of soft
tissue. In: WHO Classification of Tumours of Soft tissue and Bone.
4th edition. Fletcher CDM, Bridge JA, Hogendoorn PCW and Mertens F
(eds). International Agency for Research on Cancer (IARC), Lyon,
pp221-222, 2013.
|
|
7
|
Zambrano E, Reyes-Mugica M, Franchi A and
Rosai J: An osteoclast-rich tumor of the gastrointestinal tract
with features resembling clear cell sarcoma of soft parts: Reports
of 6 cases of a GIST simulator. Int J Surg Pathol. 11:75–81.
2003.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Thway K and Fisher C: Tumors with
EWSR1-CREB1 and EWSR1-ATF1 fusions: The current status. Am J Surg
Pathol. 36:e1–e11. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Panagopoulos I, Mertens F, Dêbiec-Rychter
M, Isaksson M, Limon J, Kardas I, Domanski H, Sciot R, Perek D,
Crnalic S, et al: Molecular genetic characterization of the
EWS/ATF1 fusion gene in clear cell sarcoma of tendons and
aponeuroses. Int J Cancer. 99:560–567. 2002.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang WL, Mayordomo E, Zhang W, Hernandez
VS, Tuvin D, Garcia L, Lev DC, Lazar AJF and López-Terrada D:
Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1
chimeric transcripts in clear cell sarcoma (melanoma of soft
parts). Mod Pathol. 22:1201–1209. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hocar O, Le Cesne A, Berissi S, Terrier P,
Bonvalot S, Vanel D, Auperin A, Le Pechoux C, Bui B, Coindre JM and
Robert C: Clear cell sarcoma (malignant melanoma) of soft parts: A
clinicopathologic study of 52 cases. Dermatol Res Pract.
2012(984096)2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Park BM, Jin SA, Choi YD, Shin SH, Jung
ST, Lee JB, Lee SC and Yun SJ: Two cases of clear cell sarcoma with
different clinical and genetic features: Cutaneous type with BRAF
mutation and subcutaneous type with KIT mutation. Br J Dermatol.
169:1346–1352. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yang L, Chen Y, Cui T, Knösel T, Zhang Q,
Geier C, Katenkam D and Petersen I: Identification of biomarkers to
distinguish clear cell sarcoma from malignant melanoma. Hum Pathol.
43:1463–1470. 2012.PubMed/NCBI View Article : Google Scholar
|