Introduction
Colorectal cancer (CRC) is the second leading cause
of cancer death worldwide (1). The
estimated number of deaths due to CRC in Europe was 243,000 in
2018(2). Approximately 25% of
patients present metastases at the time of diagnosis (synchronous
disease) and about half of the remaining patients will develop
metachronous metastases, contributing to the high mortality rates
reported for CRC (3).
The treatment of metastatic CRC (mCRC) is based on
chemotherapy when metastases cannot be removed by surgery.
Treatment is guided by molecular information. Microsatellite
unstable tumors are treated with immunotherapy (4), while microsatellite stable tumors,
which account for 95% of mCRC, are treated by cytotoxic agents and
targeted therapies. The principal cytotoxic agents currently used
are fluoropyrimidines, irinotecan and oxaliplatin. The chemotherapy
regimen often consists of fluorouracil and folinic acid combined
with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) or both
(FOLFOXIRI). These chemotherapies are associated with two different
classes of target therapies: Anti-angiogenesis drugs, and
anti-epidermal growth factor receptor (EGFR). While antiangiogenics
can be used broadly (5,6), anti EGFR therapy is only effective in
patients with RAS wild type status (3,7).
It is currently established that mCRC is a
heterogeneous disease. Tumor sidedness carries strong prognostic
value (8) while synchronous mCRC is
associated with a particularly poor prognosis. These data suggest
that the biological characteristics of these tumors are different.
Several papers have introduced CRC molecular subtyping systems,
which are currently summarized in consensus molecular subtypes
(9). This transcriptomic typology
classifies patients into 4 consensual subtypes with different
prognostic behavior. Using large panel sequencing data, a recent
analysis detailed the molecular landscape of mCRC (10). This study observed different gene
variations between left and right-sided tumors. However, the
genomic structural pattern was not assessed in this study.
Here, using exome analysis of 77 mCRC patients, we
aimed to determine genomic structural patterns and gene or pathway
variations associated with overall survival, sidedness, RAS
mutation status and synchronous disease.
Materials and methods
Study population
Seventy-seven patients with mCRC in whom WES (Whole
Exome Sequencing) analysis was performed in 2018 as part of routine
care, and interpreted according to the Molecular Tumor Board of the
Georges François Leclerc Cancer Center, were included in this
single-centre, retrospective study. WES analyses were performed
during first or second line therapy. WES analysis is performed as
part of routine care in our center in order to find potential
targetable mutations for second line therapy. Before patients
consented to WES of their tumoral tissue, they were informed by
their oncologist. Germline testing was performed after counseling
by a clinical geneticist.
Only patients from whom informed consent was
obtained and recorded in the medical chart were included in this
retrospective study. The study was approved by the CNIL (French
national commission for data privacy) and the local ethics
committee, and was performed in accordance with the Helsinki
Declaration and European legislation.
Sample selection
Physicians selected an archival tumor sample
(primary or metastasis) for genomic analysis. At the discretion of
the physician, a new tumor biopsy could be proposed to the patient.
Tumor cellularity was assessed by a senior pathologist on a
hematoxylin and eosin slide from the same biopsy core used for
nucleic acid extraction and molecular analysis.
DNA isolation
DNA was isolated from archival tumor tissue using
the Maxwell 16 FFPE Plus LEV DNA purification kit (Promega Corp.).
DNA from whole blood (germline DNA) was isolated using the Maxwell
16 Blood DNA Purification kit (Promega Corp.) following the
manufacturer's instructions. The quantity of extracted genomic DNA
was assessed by a fluorometric method with a Qubit device.
Whole exome capture and
sequencing
Two hundred ng of genomic DNA were used for library
preparation, using the Agilent SureSelectXT reagent kit (Catalog
number G9642B, Agilent Technologies, Inc.)and the All Exon v5
probeset (5190-8863, Agilent Technologies, Inc.). Following
hybridization, the libraries were purified according to the
manufacturer's recommendations and amplified by polymerase chain
reaction (12 cycles). DNA integrity was verified using TapeStation
(SCREENTAPE D1000 tapestation 5067-5582, reagents D1000 tapestation
5067-5583). Concentrations were measured using Qubit®
dsDNA BR Assay Q32853. Loading concentrations were 22 nM for
fragmentation and 6 pM for NextSeq injection. Normalized libraries
were pooled, and DNA was sequenced on an Illumina NextSeq500 device
using 2x111-bp paired-end reads and multiplexed. Names, catalog
numbers and suppliers of the Illumina sequencing kit were
following: NextSeq 500 High Output Kit FC404-2004/2140817 and
NextSeq 500 Mid Output Kit FC404-2003/2140816. More than 90% of the
target sequence was covered with a read depth of at least 10X for
somatic DNA.
Exome analysis pipeline
Reads in FASTQ format were aligned to the reference
human genome GRCh37 using the Burrows-Wheeler aligner (BWA
v.0.7.15). Local realignment was performed using the Genome
Analysis Toolkit (GATK v.3.6). Duplicate reads were removed using
Picard v.2.5. To identify somatic single-nucleotide variants
(SNVs), a validated pipeline was used that integrates mutation
calls from three different mutation callers. Single Nucleotide
Variants (SNVs) were called with VarScan (v2.4.3) (11) and Mutect (v1.1.7) (12) insertion/deletions (indels) were
called with VarScan and Strelka (v2.9.2) (13). Tumor Mutational Burden (TMB) was
calculated using the number of significant SNV (UTRs, synonyms,
introns and intergenic SNVs filtered out) divided by the number of
megabases covered at a defined level. TMB was calculated with and
without splicing sites mutations. Splicing site mutations were
excluded because it has been demonstrated that variants present in
splice regions have predominantly no impact (14). To identify tumor-specific mutant
peptides, pVAC-Seq v4.0.3(15)
(personalized Variant Antigens by
Cancer Sequencing) was used; pVAC-Seq is based on HLA
typing obtained by HLAminer (16).
TITAN (17) was used to infer the
number of copy number alterations (CNA) subclones, the number of
large deletions, as well as loss of heterogeneity (LOH)>15 Mb
from whole-exome sequencing data. It was also used to estimate
tumor ploidy. SNV signatures were generated using DeconstructSigs
(v1.8.0) (18) and COSMIC
signatures identified by Alexandrov et al (19). CNV signatures were inferred
according to the methodology of Macintyre et al (20). MSI score was computed using
MSIsensor (21) HRD score was
obtained through scarHRD (22)
pipeline.
Statistical analysis
Patient and disease characteristics were compared
across the different groups of interest using the Chi-2 or Fisher's
exact test for qualitative variables and the Wilcoxon test for
continuous variables, as appropriate. Enrichr analysis using KEGG
database was performed on genes differentially mutated given
sidedness, metastases and KRAS mutation status (23). Genes with a P-value <0.1 were
selected for this analysis. Enrichr is a web-based tool for
analysing gene sets; it returns any enrichment of common annotated
biological features, here KEGG database.
Survival analysis was performed using the survival R
library. Continuous variables were dichotomised using Lausen et
al (24) methodology through
the maxstat library (25). The
prognostic value of the different variables was tested using
univariate Cox regression for overall (OS) survival. OS was defined
as the time from diagnosis to death from mCRC. Survivors were
censored at the end of study. Survival probabilities were estimated
using the Kaplan-Meier method and survival curves were compared
using the log-rank test.
Statistical analyses were performed using the R
software (http://www.R-project.org/) and graphs
were drawn using GraphPad Prism version 7.03 (GraphPad Software,
LLC).
Results
Patients' clinical
characteristics
We included 27 (35%) patients with right primary
colon cancer and 50 (65%) with rectal or left primary colon cancer.
Twenty-seven (35%) patients had metachronous metastasis and 50
(65%) were synchronous. Forty-six (60%) patients were RAS mutated.
The most frequent metastasis site was the liver (54 patients, 70%).
Liver metastasis were more frequently synchronous (44 patients,
57%), than metachronous (10 patients, 12%) (P-value=0.01). The most
common sites of first metastasis were the liver (74%) and the lung
(27%), two metastatic sites that are potentially curable by
resection. The other identified sites of metastases were the lymph
nodes, peritoneal, adrenal and bone metastasis.
The presence of lung or liver metastases at time of
diagnosis of metastatic disease did not vary significantly by
primary tumour site (19% of right-sided tumors versus 32% of
left-sided mCRC for lung metastasis, and 18% of right-sided tumors
versus 36% of left-sided mCRC for liver metastasis). In contrast,
peritoneal and omental metastases were more frequent among
right-sided primary tumors (P-value=0.001). The main differences
between clinical variables according to tumor sidedness are
presented in Table SI, according
to synchronous or metachronous status in Table SII, and between RAS/WT mutations in
Table SIII.
All patients received a doublet or triplet of
chemotherapy as first-line therapy; fluorouracil and folinic acid
were used consistently. Patients with synchronous metastasis
significantly more frequently received oxaliplatin in their
chemotherapy regimens (P-value=0.01). Thirty patients were treated
with anti-epidermal growth factor receptor (EGFR), without any
significant difference concerning the sidedness or the time to
metastasis (P-values 0.13 and 0.61 respectively). Sixty-four
patients (83%) were treated with anti-angiogenesis drugs
(bevacizumab or aflibercept). Fifty-four patients (70%) received a
therapeutic proposition from the molecular tumor board. Only 18
patients (23%) received a treatment based on the molecular tumour
board recommendations, most frequently using an oral MEK-inhibitor
called Trametinib (Table I). Three
patients yielded significant clinical benefit from these
therapeutic strategies, with more than 6 months of progression-free
survival. Two of these three patients had unstable microsatellite
status and were treated with immunotherapy.
 | Table IDescription of treatment based on
molecular tumour board recommendation. |
Table I
Description of treatment based on
molecular tumour board recommendation.
| Sample | Somatic
mutation | Nucleotide
variant | Protein
variant | Impact | Treatment |
|---|
| 1 | BARD1, BRIP1 | c.266C>T,
c.2665C>A | p.Pro89Leu,
p.Gln889Lys | Unknown | OLAPARIB |
| 2 | NF1 | c.1007G>A | p.Trp336Ter | Unknown | TRAMETINIB |
| 3 | BRAF | c.1799T>A | p.Val600Glu | Activating
function | VEMURAFENIB |
| 4 | RAD51C | c.859A>G | p.Thr287Ala | Loss of
function | OLAPARIB |
| 5 | MSH6 | c.2017C>A | p.Pro673Thr | Unknown | NIVOLUMAB |
| 6 | TOP1 | c.852G>A | p.Lys284Lys | Unknown | IRINOTECAN |
| 7 | TP53 | c.450_451delAC |
p.Pro152AlafsTer28 | Loss of
function | AFLIBERCEPT |
| 8 | KRAS | c.35G>C | p.Gly12Ala | Activating
function | TRAMETINIB |
| 9 | MSH6 | .3254delC |
p.Phe1088SerfsTer2 | Loss of
function | DURVAUMAB |
| 10 | TP53 | c.638G>T | p.Arg213Leu | Loss of
function | BEVACIZUMAB |
| 11 | MSH6 | c.2731C>T | p.Arg911Ter | Unknown | NIVOLUMAB |
| 12 | High number of
variant | - | - | - | NIVOLUMAB |
| 13 | KRAS | c.35G>T | p.Gly12Val | Activating
function | TRAMETINIB |
| 14 | MTOR | c.6352C>T | p.Leu2118Phe | Unknown | EVEROLIMUS |
| 15 | TP53 | c.637C>T | p.Arg213X | Loss of
function | REGORAFENIB |
| 16 | KRAS | c.38G>A | p.Gly13Asp | Activating
function | TRAMETINIB |
Patients' genomic characteristics
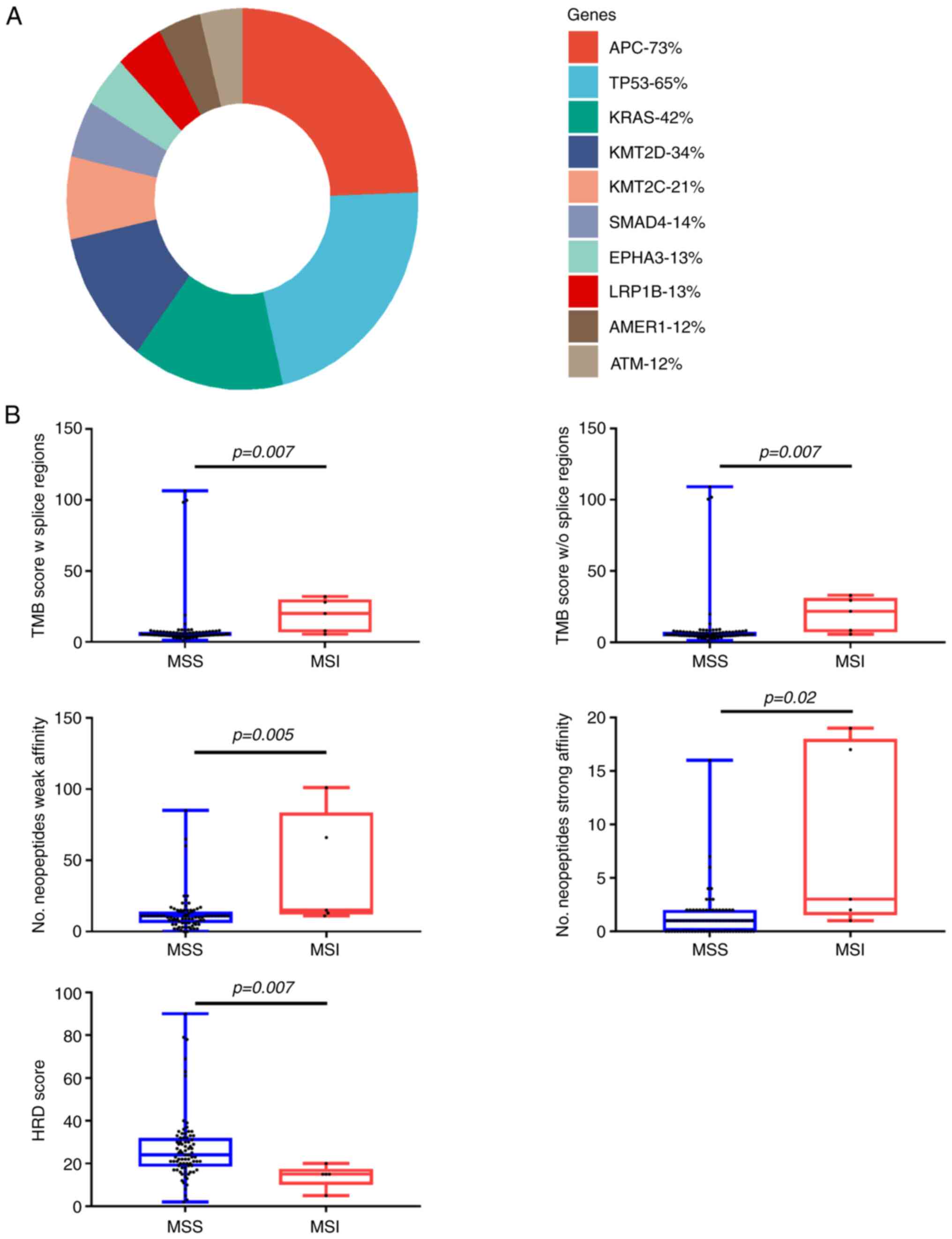
The most frequent mutation in the whole cohort was
APC followed by TP53 and RAS (Fig.
1A). A summary of the genomic characteristics is presented in
Table II. Thirty one patients
presented WT mutational status, 44 patients presented KRAS and 2
patients NRAS mutational status. We identified 8 patients (10%)
with BRAF mutation. The most frequent RAS mutation was KRAS
(60%).
| Table IISummary of genomic characteristics by
primary side, metachronous/synchronous and RAS/WT status. |
Table II
Summary of genomic characteristics by
primary side, metachronous/synchronous and RAS/WT status.
| | Primary side | Moment of
metastasis diagnosis | RAS status |
|---|
| Variable | Whole population
(n=77) | Right side
(n=27) | Left side
(n=50) | P-value | Metachronous group
(n=27) | Synchronous group
(n=50) | P-value | RAS mutated
(n=46) | RAS WT (n=31) | P-value |
|---|
| MSI categories, n
(%) | | | | >0.99 | | | 0.46 | | | >0.99 |
|
MSS (score
≤5) | 72 (93.5) | 25(92) | 47(94) | | 24(89) | 48(98) | | 44(96) | 29(94) | |
|
MSI low and
high | 5 (6.5) | 2(8) | 3(6) | | 3(11) | 2(2) | | 2(4) | 2(6) | |
| TMB without
splicing categories, n (%) | | | | 0.41 | | | 0.66 | | | >0.99 |
|
Low (score
≤40) | 71(92) | 26(96) | 45(90) | | 24(89) | 47(94) | | 42(91) | 29(94) | |
|
High | 6(8) | 1(4) | 5(10) | | 3(11) | 3(6) | | 4(9) | 2 6) | |
| RAS mutation, n
(%) | 46(60) | 18(69) | 28(56) | 0.65 | 17(63) | 29(58) | 0.46 | 46(100) | - | - |
| BRAF mutation, n
(%) | 8(10) | 5(19) | 3(6) | 0.18 | 5(19) | 3(6) | 0.12 | - | 8(10) | >0.99 |
| MSI score, median
(IQR) | 0.08 (0.25) | 0.03 (0.27) | 0.09 (0.22) | 0.35 | 0.23 (0.43) | 0.09 (0.22) | 0.35 | 0.06 (0.21) | 0.10 (0.24) | 0.40 |
| TMB without
splicing regions score, median (IQR) | 5.5 (3.1) | 5.7 (2.8) | 5.4 (2.9) | 0.55 | 5.7 (2.8) | 5.4 (2.9) | 0.5 | 5.6 (3.1) | 5.4 (2.7) | 0.7 |
| HRD score, median
(IQR) | 23(13) | 23(14) | 23. (11.7) | 0.87 | 23(14) | 23(12) | 0.87 | 21(13) | 28(14) | 0.07 |
| Ploidy, median
(IQR) | 2.1 (0.5) | 2.1 (0.5) | 2.1 (0.5) | 0.73 | 2.1 (0.4) | 2.1 (0.5) | 0.73 | 2.1 (0.2) | 2.1 (0.6) | 0.3 |
| Clonality, n
(%) | | | | 0.06 | | | 0.06 | 2 (1.75) | 1 (1.75) | 0.3 |
|
1 | 39(51) | 9(33) | 30(60) | | 9(33) | 30(60) | | - | - | |
|
>1 | 37(49) | 17(63) | 20(40) | | 17(63) | 20(40) | | - | - | |
| Deletions with LOH,
median (IQR) | 3(8) | 1(7) | 4(9) | 0.21 | 1(7) | 4(9) | 0.21 | 5(9) | 1(5) | 0.1 |
| Microdeletions,
median (IQR) | 3(4) | 3 (3.) | 3(5) | 0.64 | 3(3) | 3(5) | 0.64 | 3(5) | 2(4) | 0.1 |
| Neoantigens, median
(IQR) | 11(8) | 11(8) | 11(7) | 0.7 | 11(8) | 11(7) | 0.88 | 11(6) | 11(10) | 0.9 |
| Strong neoantigens,
median (IQR) | 1(2) | 1(1) | 1(2) | 0.07 | 1(2) | 1(2) | 0.63 | 1(2) | 1(2) | 0.9 |
Mean tumor mutational burden (TMB) without splicing
regions in the whole cohort was 8.87 (median=5.50, IQR=3.07). The
mean number of neoantigens was 13 (median=11, IQR=7.75). Five
patients had a high HRD score, using the classical cut off value
42(26). The two most frequent SNV
signatures were 1 and 25. The two most frequent CNV signatures were
3 and 5.
Seventy-two (94%) patients had proficient mismatch
repair status (pMMR) and 5 (6%) had deficient mismatch repair
status (dMMR) on immunohistological analysis (Table SIV). Median MSI score and TMB score
were significantly higher in the dMMR group (respectively
P-value=0.02 and P-value=0.01) (Fig.
1B). There was no significant enrichment of signature 6 in
patients with dMMR status (P-value=0.7).
Association of genomic variables with
sidedness
To evaluate whether we could isolate a genetic basis
for the difference in survival between disease that originates in
the right versus left side of the colon, we analysed genomic
structural and gene alterations for the primary tumor site. The
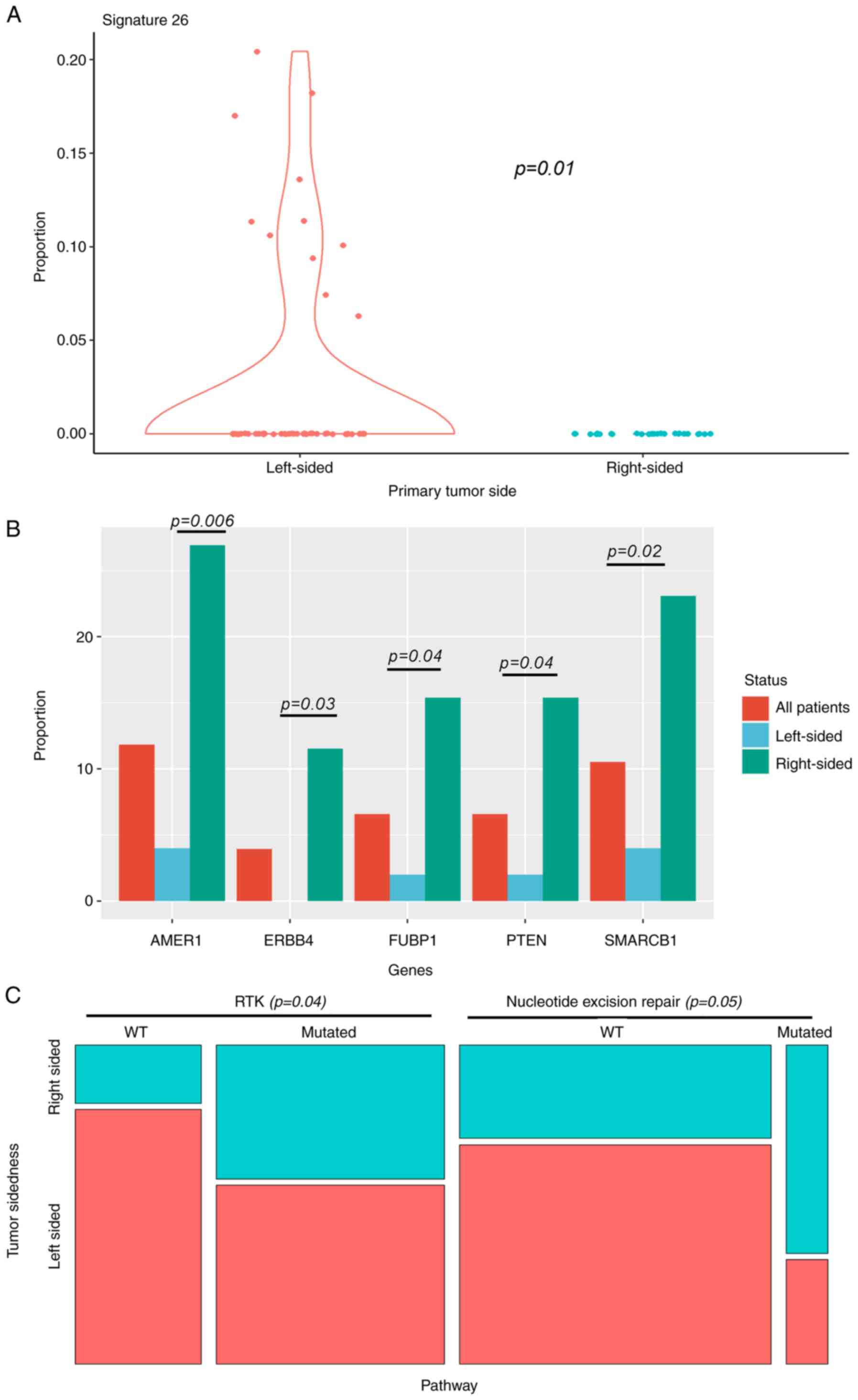
only difference in structural variants was significant enrichment
of signature 26 in left-sided primary tumors (Fig. 2A). Signature 26 is associated with
homologous repair deficiency (defective DNA mismatch repair).
For gene base analysis (Fig. 2B), there was significant enrichment
of oncogenic alterations in AMER1, SMARCB1, ERBB4, FUBP1 and PTEN
in right-sided primary tumors. Nevertheless, we did not identify
any significant differences according to sidedness or frequencies
of KRAS/BRAF mutations. Mutations observed in these genes are
further described in Table SV.
Results of Enrichr analysis using KEGG database are described in
Table SVI.
Beyond the gene-level associations, analysis at the
level of oncogenic pathways showed that mutations related to
certain pathways differed according to primary tumor site. These
pathways consisted in significant enrichment of Nucleotide excision
repair for right-sided tumors, and RTK for left-sided ones
(Fig. 2C). No significant
association was found between sidedness and TP53/ATM, WNT/CTNNB1,
TGF-beta or IGF2/PI-3-kinase pathways, which are frequently mutated
in CRC.
Association of genomic variables with
synchronous versus metachronous presentation
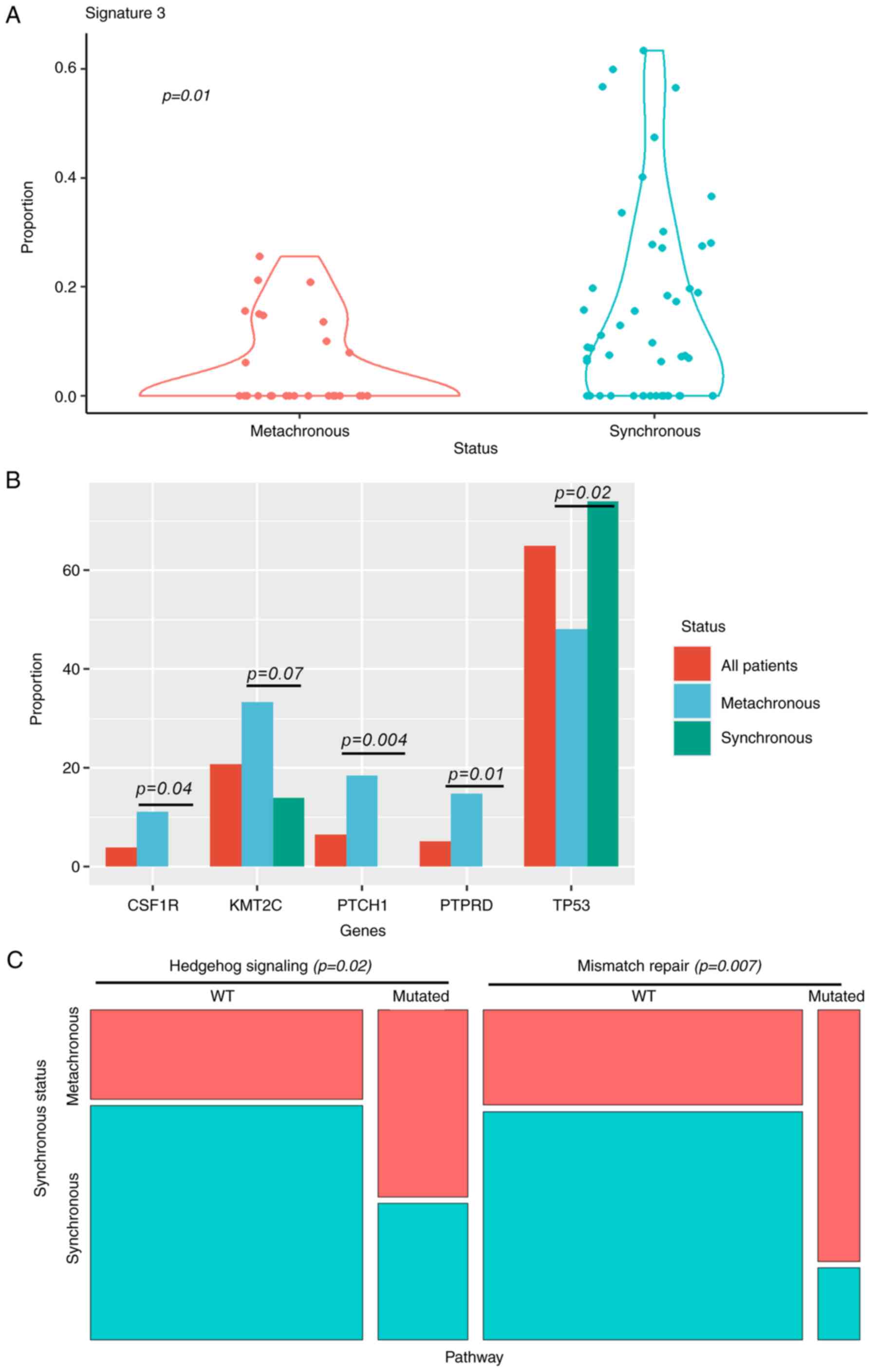
Using structural genome analysis, SNV signature 3,
which is associated with failure of DNA double-strand break repair
by homologous recombination, was significantly associated with
synchronous presentation (Fig. 3A).
We did not find additional differences at this genomic level.
P53 loss of function mutations were significantly
more frequent in synchronous metastasis. In contrast, metachronous
metastasis was associated with PTCH, PTPRD and CSF1R mutations
(Fig. 3B). Mutations observed in
these genes are further described in Table SV. Results of Enrichr analysis
using KEGG database are described in Table SVI.
When pooling mutations in gene pathways, we observed
that Mismatch repair and Hedgehog signaling were significantly more
frequently affected in metachronous tumors (Fig. 3C).
Association of genomic variables with
RAS status
In our series, the prevalence of RAS mutations was
not significantly different according to tumor location. On gene
variation, KMT2B and RET were significantly associated with RAS
wild type tumors. In contrast, KMT2C and GNAS were significantly
more frequent with RAS mutated tumors (Fig. S1A). Mutations observed in these
genes are further described in Table
SV. Results of Enrichr analysis using KEGG database are
described in Table SVI.
An analysis at the level of oncogenic pathways
demonstrated that ERK signaling and chromatin organisation pathways
were significantly associated with RAS mutation, while
Phosphatidylinositol signaling was associated with WT tumors
(Fig. S1B).
Association of genomic variables with
survival
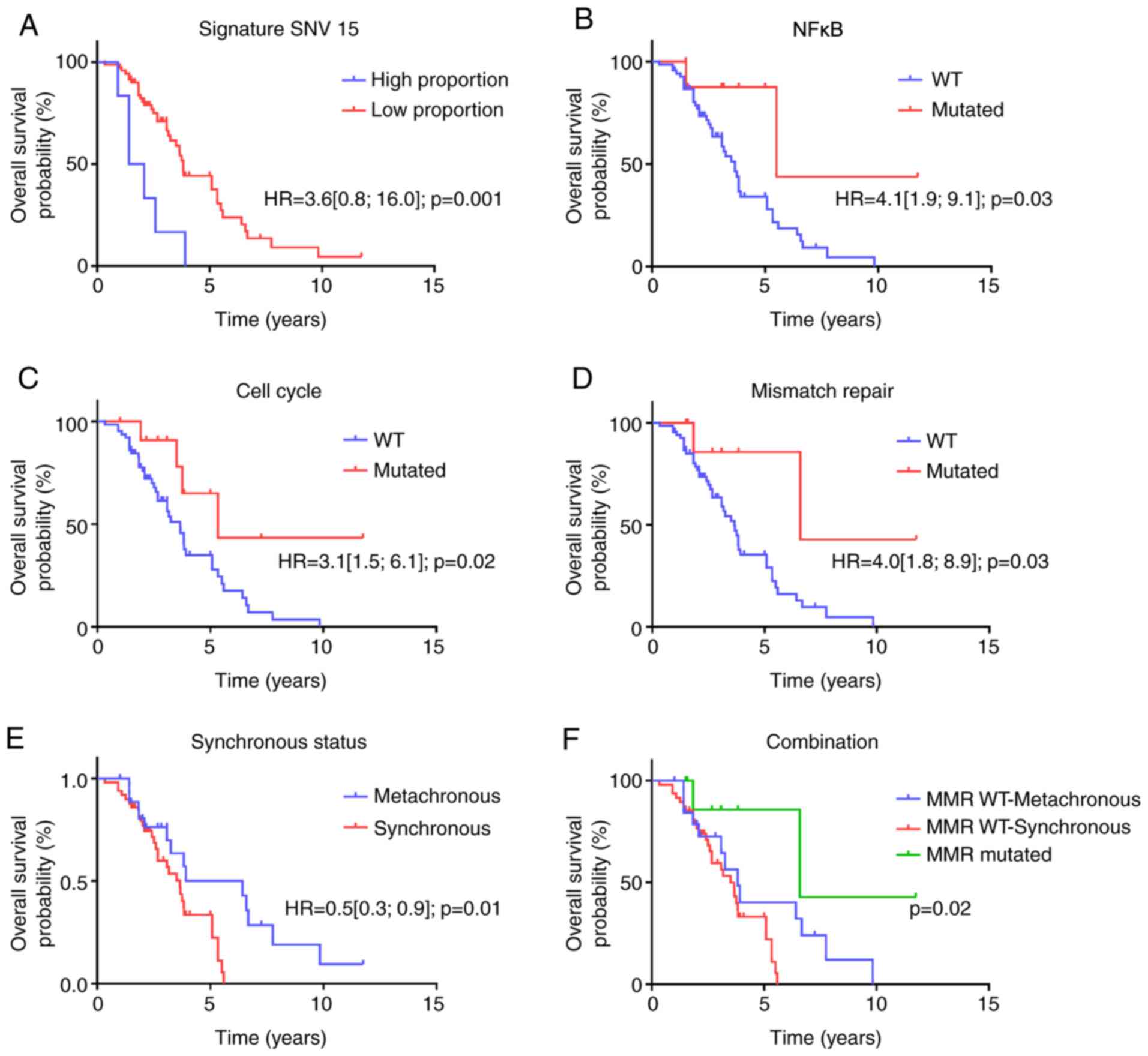
Median OS was 3.75 years. Regarding genomic
structural variants, low SNV signature 15 was associated with poor
overall survival (Fig. 4A). In gene
variation, PDGFRA, SMAD4, PTEN, MYCN mutations were significantly
associated with poor prognosis (Fig.
S2A-D). We also estimated a multivariate model involving the 4
genes; all genes remain significant. At the level of oncogenic
pathways, cell-cycle, NFKappaB signaling and mismatch repair
pathways were significantly associated with better survival
(Fig. 4B-D). We did not observe a
significant association between any WNT-signaling pathway and CRC
survival.
Overall survival was significantly better in
patients with metachronous metastatic disease, but this difference
was no longer significant after removing MMR deficient tumors, thus
suggesting a link between the prognosis of metachronous disease and
enrichment in MMR deficient tumors (Fig. 4E and F).
Discussion
CRC is caused by multiple risk factors, including
environmental, lifestyle and genetic risks. All these elements
cause mutations and epigenetic alterations, conferring on cells the
capacity to transform and grow, with aberrant DNA editing and
defective DNA maintenance.
Next-generation sequencing (NGS) has identified a
diversity of driver mutations in genes and altered signaling
pathways in CRC (10,27). In addition, genomic structural
patterns within gene pathway mutations could be used to identify
prognostic features. Recent studies have revealed a mutational
landscape of colorectal cancer and defined different subtypes that
could guide therapeutic decisions (28). Increasing access to WES and the
increasing rapidity of analysis offers new opportunities to
implement such tests in the care of mCRC. WES analysis is performed
in routine care in our center in order to find potentially
targetable mutations for second line therapy.
Like in previous studies of precision medicine, we
found that only very few mCRC patients yielded a benefit from
precision medicine. In our study, we used WES analysis to identify
genomic mutational profiles linked to tumor sidedness and
metastatic occurrences, as well as genomic features related to
prognosis.
In a systemic review and metanalysis (8), left-sided primary tumors were
associated with improved prognosis, for both localized and
metastatic tumors, in comparison to right-sided disease. Peritoneal
and omental metastases, which are metastatic sites known to be
associated with poor survival (29), were more frequent among right-sided
primary tumors. Moreover, the survival differences seen between
patients with right-sided vs. left-sided primary tumor sites in
mCRC are supported by differences in transcriptomic patterns
(8,30).
Like other authors (30,31),
we showed that metachronous metastatic status is associated with
better outcome. However, the molecular mechanism remains obscure,
and the prognostic role seems to be less impactful than that of MMR
status.
Our findings indicate that differences in survival
could also be explained by genomic differences. Our analysis relied
on structural information and on mutated genes grouped in pathways.
We showed in particular that SNV signature 26, associated with MMR
deficiency 29 (https://cancer.sanger.ac.uk/cosmic/signatures_v2.tt),
was surprisingly associated with left-sided mCRC, while SNV
signature 3 was associated with synchronous metastasis. Signature 3
is associated with better overall survival in ovarian (32) and breast cancer (33), by the better response to platinum
therapy. At present, no data link signature 3 to oxaliplatin
efficacy in mCRC. This question may be relevant for further
clinical trials.
Moreover, somatic mutations in TP53 genes, which are
present in the majority of cancers, are associated with poorer
clinical outcomes, in several cancer types, including CRC (28). P53 loss of function mutations were
significantly more frequent in synchronous metastasis.
In contrast, when we looked at more classical
genomic features such as tumor mutational burden, there was no
significant difference between tumors according to sidedness or
time to metastasis occurrence. Conversely, we observed that MMR
deficiency was associated with better outcome. MMR deficiency is
more frequent in metachronous tumors and this genetic event may
explain the better prognosis of these patients.
Oncogenic alterations-KRAS, NRAS and BRAF, which
exhibit resistance to EGFR therapy with panitumumab and cetuximab,
did not differ in our cohort, as previously reported (34), according to sidedness or metastatic
occurrence, thus suggesting that these parameters are probably weak
prognostic factors. The RAS pathway is a cell signaling pathway
that plays a key role in regulation of cellular proliferation,
apoptosis, cellular differentiation and migration and angiogenesis.
This pathway is often dysregulated in mCRC. The association between
RAS mutations and activation of the ERK signaling pathway is
already well known.
When we looked at classical somatic mutations in
mCRC, we observed that APC was frequently mutated in our series.
However, APC was not associated with survival. The WNT signaling
pathway is also frequently mutated in CRC and we did not observe a
significant association between any WNT-signaling pathway and CRC
survival. SMAD4 loss was found to be associated with poor outcome
in our series. Similarly, a recent report also showed an
association between SMAD4 loss and poor CRC survival, resistance to
chemotherapy and decreased tumor immune infiltration (35).
Both PDGF and PDGFR families play an important role
in colorectal carcinogenesis, and PDGFR is frequently overexpressed
in CRC. Activation of this pathway is frequently related to
angiogenesis, invasion, metastasis and poor survival (36). In our study, mutations in PDGFRA
were associated with poor prognosis.
The main limitation of our work is the low number of
patients included, which may impact the statistical significance,
and precluded multivariate analysis. Moreover, WES was made during
first or second line of treatment, so mutation profiles were
generated under therapeutic pressure, which changed the cancer
characterizations. Unfortunately, we do not dispose of samples
before treatments as a baseline. In addition, considering the
sequencing result of PTEN, PDGFRA, MYCN or SMAD4, the lack of PCR
verification step is a limitation of this study and should be done
in future works. Thus, our results should be considered as
descriptive and exploratory, and warrant confirmation in further
studies including larger sample sizes.
In conclusion, with the development of targeted
therapies, it seems necessary to be able to rapidly identify the
molecular status of mCRC tumors. Large panel or exome sequencing is
slightly effective in improving patient care with precision
medicine; however, such analyses could reveal structural and
pathway genomic features that are associated with sidedness,
synchronous status, and overall survival. Although the cost of NGS
is steadily declining, WES nonetheless remains expensive, and
currently, this is one of the major limitations on the routine use
of exome sequencing. Moreover, the number of tumour mutations that
could be used to treat a given patient is limited in conventional
clinical practice or clinical trials, and it is difficult to
determine which patients will clinically benefit from exome
sequencing. In all likelihood, before being used in routine
practice, exome sequencing will be reserved for patients with
advanced mCRC, after one or more lines of treatment, or for
patients with very poor outcomes, who fail to respond to classical
targeted therapy and chemotherapy. Indeed, the knowledge of the
molecular status could lead to inclusion in therapeutic clinical
trials with a direct benefit for our patients. More generally,
patients with right-sided mCRC, synchronous metastasis or
peritoneal and omental metastasis may benefit the most from such
analysis. This important information could be used to improve
patient stratification for clinical trials, and will lead to a new
molecular classification of patients that could be helpful to
finetune the future of mCRC therapy.
Supplementary Material
Genetic characteristics according to
RAS status. (A) Proportion of patients presenting at least one
mutation in the most frequently mutated genes, by RAS status. (B)
Distribution of patients presenting an alteration in significant
signaling pathways according to RAS status. Only pathways for which
the Fisher tests were considered significant (P<0.05) are
presented. WT, wild-type.
Gene mutation and overall survival.
Kaplan Meier curves for genes with significantly different overall
survival according to their mutation status, namely (A) PDGFRA, (B)
SMAD4, (C) PTEN and (D) MYCN. HR, hazard ratio; WT, wild-type.
Summary of clinical characteristics
according to primary tumour side (right/left).
Summary of clinical characteristics
according to metachronous/synchronous status.
Summary of clinical characteristics
according to RAS mutational status.
Description of genomic characteristics
according to MSI/MSS groups.
Description of mutations observed in
genes differentially mutated in function of sidedness, synchronous
or metachronous status, and KRAS mutated status.
Results of Enrichr analysis based on
Kyoto Encyclopedia of Genes and Genomes pathways and genes
differentially mutated in function of sidedness, synchronous or
metachronous status, and KRAS mutated status.
Acknowledgements
The authors would like to thank Ms. Sandy Chevrier
(Department of Medical Oncology, Georges François Leclerc Cancer
Center-UNICANCER, Dijon, France), Dr Romain Boidot (Department of
Medical Oncology, Georges François Leclerc Cancer Center-UNICANCER,
Dijon, France) for providing whole exome sequencing data, Mr. Hugo
Mananet (Department of Medical Oncology, Georges François Leclerc
Cancer Center-UNICANCER, Dijon, France) for pre-processing data,
and Dr Juliette Albuisson (Department of Medical Oncology, Georges
François Leclerc Cancer Center-UNICANCER, Dijon, France) for her
valuable comments. The authors would also like to thank Dr Fiona
Ecarnot (EA3920, University of Franche-Comté, Besançon, France) for
English correction and helpful comments.
Funding
No funding was received.
Availability of data and materials
The datasets generated and analyzed during the
current study are available in the Sequence Read Archive
repository, (https://www.ncbi.nlm.nih.gov/sra/?term=SRP318854).
Authors' contributions
MDGDA, LG, ZT, CT and FG contributed to the design
and implementation of the research, to the analysis of the results
and to the writing of the manuscript. CT and FG confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the CNIL (French
national commission for data privacy) and the Georges François
Leclerc Cancer Center (Dijon, France) local ethics committee
(13.085), and was performed in accordance with the Helsinki
Declaration and European legislation. Written informed consent was
obtained from all subjects involved in the study. Only patients
from whom informed consent was obtained and recorded in the medical
chart were included in this retrospective study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ferlay J, Colombet M, Soerjomataram I,
Dyba T, Randi G, Bettio M, Gavin A, Visser O and Bray F: Cancer
incidence and mortality patterns in Europe: Estimates for 40
countries and 25 major cancers in 2018. Eur J Cancer. 103:356–387.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Van Cutsem E, Cervantes A, Adam R, Sobrero
A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson
A, Bodoky G, et al: ESMO consensus guidelines for the management of
patients with metastatic colorectal cancer. Ann Oncol.
27:1386–1422. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Andre T, Shiu KK, Kim TW, Jensen BV,
Jensen LH, Punt CJA, Smith DM, Garcia-Carbonero R, Benavides M,
Gibbs P, et al: Pembrolizumab versus chemotherapy for
microsatellite instability-high/mismatch repair deficient
metastatic colorectal cancer: The phase 3 KEYNOTE-177 study. J Clin
Oncol. 38 (Suppl 18)(LBA4)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Giantonio BJ, Catalano PJ, Meropol NJ,
O'Dwyer PJ, Mitchell EP, Alberts SR, Schwartz MA and Benson AB III:
Eastern Cooperative Oncology Group Study E3200. Bevacizumab in
combination with oxaliplatin, fluorouracil, and leucovorin
(FOLFOX4) for previously treated metastatic colorectal cancer:
Results from the eastern cooperative oncology group study E3200. J
Clin Oncol. 25:1539–1544. 2007.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Van Cutsem E, Rivera F, Berry S,
Kretzschmar A, Michael M, DiBartolomeo M, Mazier MA, Canon JL,
Georgoulias V, Peeters M, et al: Safety and efficacy of first-line
bevacizumab with FOLFOX, XELOX, FOLFIRI and fluoropyrimidines in
metastatic colorectal cancer: The BEAT study. Ann Oncol.
20:1842–1847. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Van Cutsem E, Lenz HJ, Köhne CH, Heinemann
V, Tejpar S, Melezínek I, Beier F, Stroh C, Rougier P, van Krieken
JH and Ciardiello F: Fluorouracil, leucovorin, and irinotecan plus
cetuximab treatment and RAS mutations in colorectal cancer. J Clin
Oncol. 33:692–700. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Petrelli F, Tomasello G, Borgonovo K,
Ghidini M, Turati L, Dallera P, Passalacqua R, Sgroi G and Barni S:
Prognostic survival associated with left-sided vs right-sided colon
cancer: A systematic review and meta-analysis. JAMA Oncol.
3:211–219. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Guinney J, Dienstmann R, Wang X, de
Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda
G, Angelino P, et al: The consensus molecular subtypes of
colorectal cancer. Nat Med. 21:1350–1356. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Yaeger R, Chatila WK, Lipsyc MD, Hechtman
JF, Cercek A, Sanchez-Vega F, Jayakumaran G, Middha S, Zehir A,
Donoghue MTA, et al: Clinical sequencing defines the genomic
landscape of metastatic colorectal cancer. Cancer Cell.
33:125–136.e3. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cibulskis K, Lawrence MS, Carter SL,
Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES
and Getz G: Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat Biotechnol.
31:213–219. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Kim S, Scheffler K, Halpern AL, Bekritsky
MA, Noh E, Källberg M, Chen X, Kim Y, Beyter D, Krusche P and
Saunders CT: Strelka2: Fast and accurate calling of germline and
somatic variants. Nat Methods. 15:591–594. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Nykamp K, Anderson M, Powers M, Garcia J,
Herrera B, Ho YY, Kobayashi Y, Patil N, Thusberg J, Westbrook M, et
al: Sherloc: A comprehensive refinement of the ACMG-AMP variant
classification criteria. Genet Med. 19:1105–1117. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hundal J, Carreno BM, Petti AA, Linette
GP, Griffith OL, Mardis ER and Griffith M: pVAC-Seq: A
genome-guided in silico approach to identifying tumor neoantigens.
Genome Med. 8(11)2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Warren RL, Choe G, Freeman DJ, Castellarin
M, Munro S, Moore R and Holt RA: Derivation of HLA types from
shotgun sequence datasets. Genome Med. 4(95)2012.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Ha G, Roth A, Khattra J, Ho J, Yap D,
Prentice LM, Melnyk N, McPherson A, Bashashati A, Laks E, et al:
TITAN: Inference of copy number architectures in clonal cell
populations from tumor whole-genome sequence data. Genome Res.
24:1881–1893. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Rosenthal R, McGranahan N, Herrero J,
Taylor BS and Swanton C: DeconstructSigs: Delineating mutational
processes in single tumors distinguishes DNA repair deficiencies
and patterns of carcinoma evolution. Genome Biol.
17(31)2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Alexandrov LB, Nik-Zainal S, Wedge DC,
Campbell PJ and Stratton MR: Deciphering signatures of mutational
processes operative in human cancer. Cell Rep. 3:246–259.
2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Macintyre G, Goranova TE, De Silva D,
Ennis D, Piskorz AM, Eldridge M, Sie D, Lewsley LA, Hanif A, Wilson
C, et al: Copy number signatures and mutational processes in
ovarian carcinoma. Nat Genet. 50:1262–1270. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Middha S, Zhang L, Nafa K, Jayakumaran G,
Wong D, Kim HR, Sadowska J, Berger MF, Delair DF, Shia J, et al:
Reliable pan-cancer microsatellite instability assessment by using
targeted next-generation sequencing data. JCO Precis Oncol.
2017(PO.17.00084)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sztupinszki Z, Diossy M, Krzystanek M,
Reiniger L, Csabai I, Favero F, Birkbak NJ, Eklund AC, Syed A and
Szallasi Z: Migrating the SNP array-based homologous recombination
deficiency measures to next generation sequencing data of breast
cancer. NPJ Breast Cancer. 4(16)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z,
Meirelles GV, Clark NR and Ma'ayan A: Enrichr: Interactive and
collaborative HTML5 gene list enrichment analysis tool. BMC
Bioinformatics. 14(128)2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lausen B, Hothorn T, Bretz F and
Schumacher M: Assessment of optimal selected prognostic factors.
Biom J. 46:364–374. 2004.
|
|
25
|
Hothorn T and Lausen B: On the exact
distribution of maximally selected rank statistics. Comput Stat
Data Anal. 43:121–137. 2003.
|
|
26
|
Takaya H, Nakai H, Takamatsu S, Mandai M
and Matsumura N: Homologous recombination deficiency status-based
classification of high-grade serous ovarian carcinoma. Sci Rep.
10(2757)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cancer Genome Atlas Network. Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zaidi SH, Harrison TA, Phipps AI,
Steinfelder R, Trinh QM, Qu C, Banbury BL, Georgeson P, Grasso CS,
Giannakis M, et al: Landscape of somatic single nucleotide variants
and indels in colorectal cancer and impact on survival. Nat Commun.
11(3644)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Franko J, Shi Q, Goldman CD, Pockaj BA,
Nelson GD, Goldberg RM, Pitot HC, Grothey A, Alberts SR and Sargent
DJ: Treatment of colorectal peritoneal carcinomatosis with systemic
chemotherapy: A pooled analysis of north central cancer treatment
group phase III trials N9741 and N9841. J Clin Oncol. 30:263–267.
2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Boeckx N, Koukakis R, Op de Beeck K, Rolfo
C, Van Camp G, Siena S, Tabernero J, Douillard JY, André T and
Peeters M: Primary tumor sidedness has an impact on prognosis and
treatment outcome in metastatic colorectal cancer: Results from two
randomized first-line panitumumab studies. Ann Oncol. 28:1862–1868.
2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ghiringhelli F, Hennequin A, Drouillard A,
Lepage C, Faivre J and Bouvier AM: Epidemiology and prognosis of
synchronous and metachronous colon cancer metastases: A French
population-based study. Dig Liver Dis. 46:854–858. 2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Pennington KP, Walsh T, Harrell MI, Lee
MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord
AS, et al: Germline and somatic mutations in homologous
recombination genes predict platinum response and survival in
ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer
Res. 20:764–775. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhao EY, Shen Y, Pleasance E, Kasaian K,
Leelakumari S, Jones M, Bose P, Ch'ng C, Reisle C, Eirew P, et al:
Homologous recombination deficiency and platinum-based therapy
outcomes in advanced breast cancer. Clin Cancer Res. 23:7521–7530.
2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bylsma LC, Gillezeau C, Garawin TA, Kelsh
MA, Fryzek JP, Sangaré L and Lowe KA: Prevalence of RAS and BRAF
mutations in metastatic colorectal cancer patients by tumor
sidedness: A systematic review and meta-analysis. Cancer Med.
9:1044–1057. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wasserman I, Lee LH, Ogino S, Marco MR, Wu
C, Chen X, Datta J, Sadot E, Szeglin B, Guillem JG, et al: SMAD4
loss in colorectal cancer patients correlates with recurrence, loss
of immune infiltrate, and chemoresistance. Clin Cancer Res.
25:1948–1956. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Manzat Saplacan RM, Balacescu L, Gherman
C, Chira RI, Craiu A, Mircea PA, Lisencu C and Balacescu O: The
Role of PDGFs and PDGFRs in colorectal cancer. Mediators Inflamm.
2017(4708076)2017.PubMed/NCBI View Article : Google Scholar
|