Introduction
Tumor-cells apoptosis factor (TCApF) is a novel
human 84-amino acid hormone peptide naturally expressed in the
thymus, colon and frontal lobe of the brain (1). TCApF was identified through screening
of the human genome with bioinformatics tools, and was found to
have anticancer activity in acute myeloid leukemia (AML),
glioblastoma, breast, lung and prostate cancer cell lines (1). Studies investigating the mechanism
through which TCApF exerts its anticancer activity in AML
demonstrated that it binds to the T1/ST2 receptor, which is a
member of the Toll/Interleukin-1 receptor superfamily expressed in
macrophages, dendritic cells and mast cells, and was implicated in
various diseases, including cancer, inflammatory and cardiovascular
diseases (2,3). The T1/ST2 receptor is a stable marker
of T-helper 2 (Th2) cells, but not of polarized thymocytes, and is
important in Th2-associated immune response. Differential mRNA
processing within the ST2 gene generates three ST2 isoforms, namely
a full transmembrane receptor (ST2L), a truncated form associated
with the membrane (ST2V), and a secreted soluble form (sST2)
(4). The activation of the T1/ST2
receptor has been shown to lead to apoptosis in proliferating cells
(but not in non-proliferating cells) through activation of caspases
8 and B-cell lymphoma (Bcl)-2-mediated apoptotic pathways (1).
dTCApFs (Nerofe™, Immune System Key Ltd., Jerusalem,
Israel) is a short, 14-amino acid derivative of TCApF that retains
its anticancer activity. The objective of the present study was to
investigate the mechanism of action (MOA) of dTCApFs, referring
particularly to its potential activity in solid tumors, in order to
provide the necessary framework for clinical studies evaluating its
utility as a potential treatment for advanced/metastatic solid
tumors.
Materials and methods
dTCApFs
dTCApFs was synthesized by Novetide Ltd. (Haifa,
Israel) at Asia Chemical Industries Ltd. (Ramat Hovav, Beer Sheva,
Israel). The polypeptide was assembled on a solid support using
protected amino acids; the peptide was then cleaved from the resin
and purified using preparative high-performance liquid
chromatography, followed by desalting and counter-ion exchange to
generate dTCApFs acetate.
Cell lines
Human cell lines including PANC-1 (pancreatic
cancer), OV-90 (ovarian cancer) and MDA-MB-231 (breast
adenocarcinoma) were obtained from American Type Culture Collection
and used within 6 months. Cell lines were maintained in DMEM
(Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine
serum (FBS; Biological Industries, Beit Haemek, Israel), 50 µg/ml
gentamycin sulfate (Biological Industries) and 250 µg/ml
amphotericin B (Biological Industries).
Cell culture and dTCApFs
treatment
For immunofluorescence (IF) assays, 10,000
PANC-1/MDA-MB-231/OV-90 cells were seeded into 8-well chamber
slides (Nunc®, Roskilde, Denmark) in 0.5 ml 10% FBS in
DMEM. Following incubation for 24 h, the medium was changed to 2.5%
FBS in DMEM and 5% mannitol, and the cells were treated with 25
µg/ml dTCApFs for 48 h, after which time another dose of dTCApFs
(25 µg/ml) was added to the medium. After an additional 24 h, cells
were collected for IF analysis.
IF
For IF studies, cells were grown on glass chamber
slides, fixed with 3% formaldehyde in phosphate-buffered saline
(PBS), permeabilized with 0.25% Triton X-100 in PBS, blocked with
1% bovine serum albumin in PBS plus Tween-20, incubated overnight
at 4°C with the primary antibody in blocking media, and stained for
1 h with secondary antibody. The primary antibodies used included
anti-dTCApFs (1:1,000 rabbit, polyclonal, custom made by Genemed
Synthesis Inc., San Antonio, TX, USA) and anti-soluble T1/ST2
(1:500 rabbit, polyclonal, custom made by Genemed Synthesis Inc.).
Additional primary antibodies used included anti-58k Golgi protein
antibody (ab27043, 1:500 mouse monoclonal, Abcam Cambridge, UK),
anti-γ-adaptin (ab11328, 1:500 mouse monoclonal, Abcam),
anti-coatomer subunit β (β-COP) antibody (ab6323, 1:250 mouse
monoclonal, Abcam), anti-inositol-requiring enzyme (IRE)1 antibody
(9F2, ab96481, 1:500 mouse monoclonal, Abcam), antibody against
Golgi anti-apoptotic protein (GAAP) (ab169461, 1:100 mouse
polyclonal, Abcam), and anti-C/EBP homologous protein (CHOP)
antibody (no. 2895), anti-binding immunoglobulin protein (BiP)
(C50B12) rabbit mAB (no. 3177, 1:400 rabbit monoclonal, Cell
Signaling Technology, Danvers, MA, USA), and
anti-phospho-eukaryotic translation initiation factor (eIF) 2α
(Ser51) (D9G8) XP rabbit mAB (no. 3398, pEIF2a rabbit monoclonal,
Cell Signaling Technology). Secondary antibodies included
anti-rabbit IgG (H+L), F(ab')2 Fragment (no. 4413, 1:1,000 goat,
Cell Signaling Technology), and anti-mouse IgG (H+L), F(ab')2
fragment (no. 4408, 1:1,000 goat, Cell Signaling Technology), goat
anti-rabbit IgG H+L (ab150077) and goat anti-mouse (ab150118) (both
diluted 1:1,000, Abcam). Slides were mounted with fluoroshield
mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; ab104139,
Abcam). The microscopes used included BX41 and FV-1200 (Olympus,
Tokyo, Japan).
Immunohistochemistry (IHC)
IHC examination of paraffin-embedded tumor sections
was performed by deparaffinizing the sections with washes of
xylene, and rehydrating them with serial washes of absolute
ethanol, 95% ethanol, and then with distilled water. Antigen
retrieval was performed by boiling the sections in Tris-EDTA (pH 9)
for ST2 staining and in acetate (pH 6) for other antibodies.
Endogenous peroxidase was blocked with 3% oxygen peroxidase.
Non-specific binding was blocked with Background Buster (Innovex
Biosciences, Richmond, CA). The primary antibody was incubated on
slide overnight at 4°C in SignalStain antibody diluent (Cell
Signaling Technology). Secondary antibody binding and
3,3′-diaminobenzidine (DAB) development were performed the
following day. The nuclei were stained with hematoxylin. The slides
were dehydrated, mounted and visualized under an Olympus BX41
microscope (Olympus) with a CCD camera. Primary antibodies included
those used in the IF experiments, as well as an antibody against
the full-length ST2 (prepared for the present study by Genemed
Synthesis Inc.). Secondary antibodies included anti-rabbit
horseradish peroxidase (HRP; Cell Signaling Technology) diluted in
SignalStain® Boost IHC Detection Reagent (Cell Signaling
Technology) with DAB as the substrate, and Eukitt® as
the mounting media (both from Sigma-Aldrich, St. Louis, MO,
USA).
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis, ELISA, dihydroethidium (DHE) assay,
western blotting and cell viability studies
RT-PCR analysis was performed as previously
described (1). For the ELISA
studies, 100,000 cells were incubated in 6-well plates in 2 ml of
10% FBS in DMEM medium and, 24 h later, the medium was changed to 1
ml of 2.5% FBS and 5% mannitol in DMEM. Cells were treated with
dTCApFs as in the IF experiments, and the supernatant was
collected. Cell debris was removed by centrifugation. Soluble
T1/ST2 peptide (for calibration) or cell supernatant was added to
96-well microplate (MaxiSorp; Nunc) and incubated in PBS at 4°C
overnight. The primary antibody was anti-soluble T1/ST2 (as for the
IF experiments), and the secondary antibody was anti-rabbit IgG
HRP-linked (Cell Signaling Technology). For DHE experiments, cells
were seeded at concentrations of 20,000/well into black 96-well
plates in 100 µl of growing media (DMEM with 4,500 mg/l glucose, 4
mM L-glutamine, 1 mM sodium pyruvate, 10% FBS, 250 ng/ml
amphotericin B, 100 U/ml penicillin, and 100 µg/ml streptomycin)
overnight. The medium was then changed to the growing media with or
without dTCApFs (50 µg/ml), and with or without 4-phenylbutyrate
(PBA; 5 µM). Following overnight incubation, the cells were
incubated with growing media containing 10 µM DHE, incubated for 20
min and washed 3 times with PBS. The plates were read using a
fluorescent spectrophotometer (BIO-TEK Synergy HT, BioTek
Instruments, Inc., Winooski, VT, USA) with an excitation/emission
wavelength of 590/530 nm. The experiment was performed in
triplicate. For western blot analysis, the cells were grown as
described above. After treatment [dCTApFs, PBA, control (5%
mannitol)], the cells were washed twice with PBS and collected (10
min, 300 × g, 4°C). Lysis buffer containing RIPA buffer and
phosphatase inhibitors (both from Sigma-Aldrich), and protease
inhibitor (Sigma-Aldrich), were added for 10 min and the cells were
incubated for 10 min on ice. The cells were then broken using a 21
G needle. Supernatant was collected after centrifuging the cells
(15,000 × g) for 10 min, LDS sample buffer and reducing agent (both
from Invitrogen, Carlsbad, CA, USA) were added to each sample. The
samples were boiled for 5 min and loaded to 4–20% Tris-Glycine gel
(Nusep, New South Wales, Australia). Western blot analysis was
performed according to standard procedures. The primary antibodies
used included anti-nuclear factor (NF)-κB and anti-pNF-κB (SC-3033
and SC-8242, respectively, Santa Cruz Biotechnology, Dallas, TX,
USA). The secondary antibody was anti-rabbit IgG HRP-linked (Cell
Signaling Technology).
For viability assays, cells
(PANC-1/MDA-MB-231/OV-90) were seeded at concentrations of 80,000
cells/flask into 75 cm2 in 25 ml 10% FBS in DMEM, and 24
h later the medium was changed to 25 ml 2.5% FBS in DMEM with
dTCApFs (0, 25, 50 or 75 µg/ml) in 5% mannitol. After 48 h, another
dose of dTCApFs (at the same concentration as the initial dose) was
added to each well and the cells were incubated with dTCApFs for
another 24 h. Viability was measured using the Annexin V FITC assay
(Medical &Biological Laboratories Co. Ltd., Nagoya, Japan
(according to the manufacturer's instructions, and was read using
flow cytometry.
Murine xenograft models of human
pancreatic cancer
A total of 8 Hsd:Athymic Nude-Foxn1nu female mice
(age, 5 weeks; weight, 20 gr), acquired from Harlan Laboratories
Israel Ltd. (Jerusalem, Israel), were injected subcutaneously with
8×106 PANC-1 cells. dTCApFs treatment commenced after
the tumor size had reached 0.5 cm at the largest dimension. The
mice received intraperitoneal injections of dTCApFs 300 µg per 20
gr 3 times a week. During treatment, tumor size was documented. At
the end of treatment, the mice were euthanized by isofluran
exposure (Primal Critical Care, Bethlehem, PA, USA) followed by
cervical dislocation. Serum was collected for ELISA and the tumors
were placed in 4% formaldehyde and sent to Pathovet Ltd. (Rehovot,
Israel), where slides were prepared for IHC analysis. To determine
the effect of dTCApFs on tumor size, the mice were injected
subcutaneously with 1×106 PANC-1 cells. After the tumor
volume had exceeded 64 mm3, the mice were randomly
divided into dTCApFs treatment (n=4, 15 mg/kg dTCApFs administered
intraperitoneally) and control (n=4, 5% mannitol) groups, and the
tumor size was measured every 4 days.
All animal studies were approved by and performed in
accordance with the guidelines of the Institutional Animal Care and
Use Committee.
Tumor tissue from a dTCApFs-treated
patient
Samples before and after dTCApFs treatment were
obtained from a patient who participated in a phase I study
investigating dTCApFs in patients with pathologically confirmed
locally advanced and/or metastatic solid tumors, who failed/could
not tolerate standard therapy (personal communication). For this
patient, a pre-treatment tumor sample was obtained from a biopsy of
the primary tumor in the spinal cord and a post-treatment tumor
sample was obtained following a partial sacrectomy. The phase I
study in which this patient participated (clinicaltrials.gov identifier: NCT01690741) was
approved by the Institutional Review Board of Rabin Medical Center
and the Ministry of Health of Israel, it was conducted in
accordance with the Declaration of Helsinki, and all the patients
had signed an informed consent prior to enrollment.
Results
Cell culture studies
In order to elucidate the MOA of the anticancer
activity of dTCApFs, we first investigated (by IF staining) its
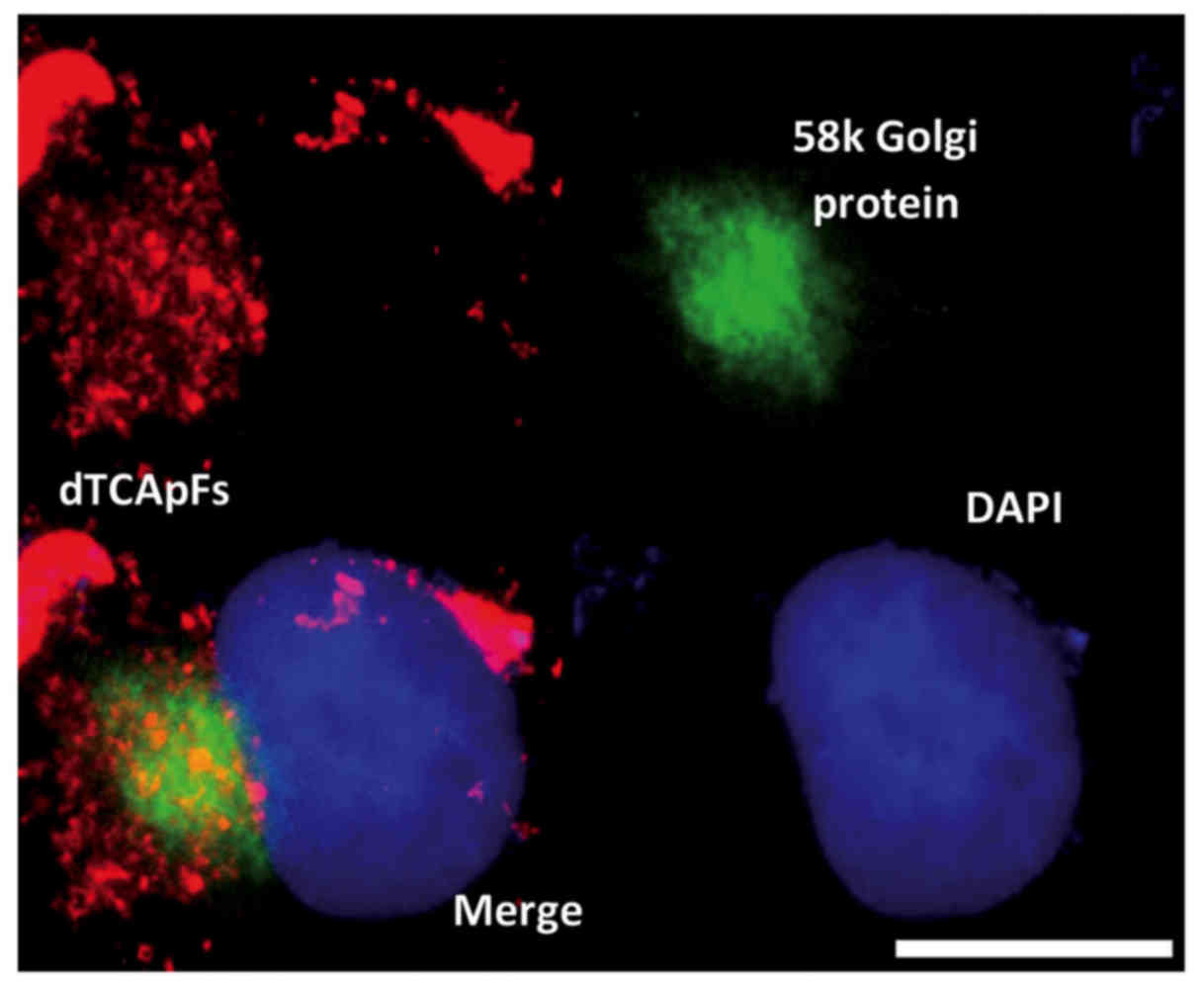
localization in PANC-1 cells. dTCApFs was found to be located in
the Golgi apparatus of treated cells (Fig. 1).
IF staining was then used to investigate the
subcellular changes induced by dTCApFs; the location of its soluble
receptor (soluble T1/ST2) and the structure of the Golgi apparatus
in treated vs. untreated cells were examined. Prior to treatment,
soluble T1/ST2 was found in the nucleus, the ER, and the Golgi
apparatus (Fig. 2A and B). Following
dTCApFs treatment, soluble T1/ST2 was found only in the nucleus
(Fig. 2C). Confocal microscopy
studies using the 58-kDa Golgi protein and DAPI for DNA staining,
demonstrated that, in MDA-MB-23 cells, the Golgi apparatus was
intact without dTCApFs treatment and diffused following dTCApFs
treatment (Fig. 2D). Similar results
were observed in the OV-90 and PANC-1 cell lines (data not shown).
Using DAPI, the Golgi marker γ-adaptin and the ER marker β-COP in
PANC-1 cells, it was demonstrated that the Golgi apparatus
structure became separated from the ER following dTCApFs treatment
(Fig. 2E).
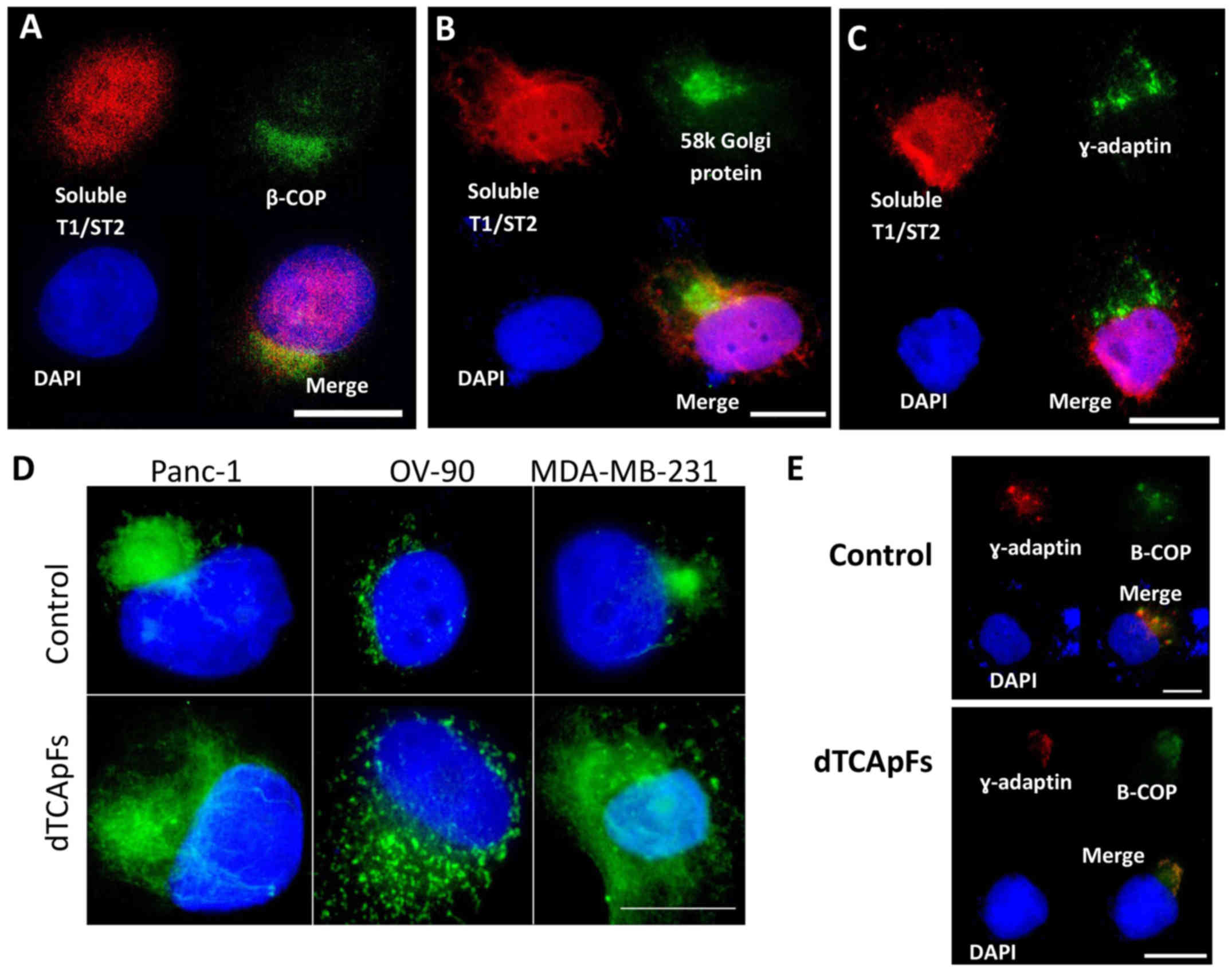 | Figure 2.dTCApFs causes a structural change in
the Golgi apparatus, in untreated cells, soluble T1/ST2 is in the
ER/Golgi, whereas in dTCApFs-treated cells it is in the nucleus,
and the Golgi apparatus is diffused and separated from the ER. (A)
Soluble T1/ST2 was detected in the ER of untreated PANC-1 cells (IF
staining). Soluble T1/ST2, red; DAPI (DNA), blue, β-COP (ER
marker), green. (B) Soluble T1/ST2 was detected in the Golgi
apparatus of untreated PANC-1 cells (IF staining). Soluble T1/ST2,
red; DAPI (DNA), blue; 58 kDa Golgi protein, green. (C) Soluble
T1/ST2 was detected (IF staining) only in the nucleus of PANC-1
cells treated with dTCApFs (25 µg/ml for 48 h followed by an
additional dose of 25 µg/ml for a further 24 h). Soluble T1/ST2,
red; DAPI (DNA), blue; γ-adaptin (a Golgi marker), green. (D) In
MDA-MB-231 cells treated with dTCApFs (25 or 50 µg/ml for 24 h
followed by an additional dose of 25 or 50 µg/ml for a further 72
h), the Golgi apparatus structure was diffused, compared with
controls (no dTCApFs treatment). DAPI (DNA), blue; 58 kDa Golgi
protein, green. (E) In PANC-1 cells treated with dTCApFs (25 µg/ml
for 48 h followed by an additional dose of 25 µg/ml for a further
24 h), the Golgi apparatus structure was separated from the ER.
DAPI (DNA), blue; γ-adaptin (a Golgi marker), red; β-COP (ER
marker), green. Scale bars in all the panels, 25 μm. dTCApFs,
14-amino acid derivative of tumor-cells apoptosis factor; ER,
endoplasmic reticulum; DAPI, 4′,6-diamidino-2-phenylindole; β-COP,
coatomer subunit β; IF, immunofluorescence. |
Subsequently, it was investigated whether the
structural changes observed in the Golgi apparatus were associated
with loss of Golgi function and ER stress. To this end, IF staining
was used to determine the levels of soluble T1/ST2 in untreated and
dTCApFs-treated PANC-1cells, and ELISA experiments to determine the
levels of soluble T1/ST2 in the supernatant of untreated and
treated cells (PANC-1, OV-90 and MDA-MB-23) (Fig. 3A and B). With dTCApFs treatment, the
levels of soluble T1/ST2 inside the cells remained the same
(Fig. 3A), whereas its secretion
from the cells into the supernatant decreased (Fig. 3B), suggesting loss of Golgi function.
To further investigate the possibility of ER stress-related MOA,
IF/IHC was used to evaluate the known molecular effects of such
stress, including increased levels of CHOP [a transcription factor
in the ER stress response (5)] and
BiP [an abundant ER chaperone (6)]
in the cells, expression of pIRE1 [an ER stress sensor protein
(7)] in the cytoplasm and increased
phosphorylation of eIF2a [which represses protein synthesis during
ER stress response (8)]. As shown in
Fig. 3C-F, dTCApFs treatment was
associated with all these effects. Using IF staining in OV-90 cells
revealed that the structural changes in the Golgi apparatus
observed in dTCApFs-treated cells (but not in untreated controls),
were not observed when the cells were incubated with dTCApFs and
the ER stress inhibitor PBA (Fig.
3G). Similar results were observed with the OV-90 cell line
(data not shown). The generation of reactive oxygen species (ROS),
which has been associated with ER stress, was also evaluated in
PANC-1, OV-90, and MDA-MB-23 cells treated with dCTApFs. An
increase was found in ROS levels (by 1.2–2.1-fold) with dCTApFs
treatment, which was attenuated with PBA (Fig. 3H), further implicating ER stress in
the dCTApFs MOA. In addition, western blotting was used to evaluate
the effect of dCTApFs treatment on the levels of NF-κB and pNF-κB,
both of which are implicated in ER stress. The experiment was
performed on the OV-90 and MDA-MB-231 cell lines, and both cell
lines were found to exhibit increased NF-κB and pNF-κB levels with
dCTApFs treatment compared with untreated controls (Fig. 3I), consistent with a mechanism
involving ER stress.
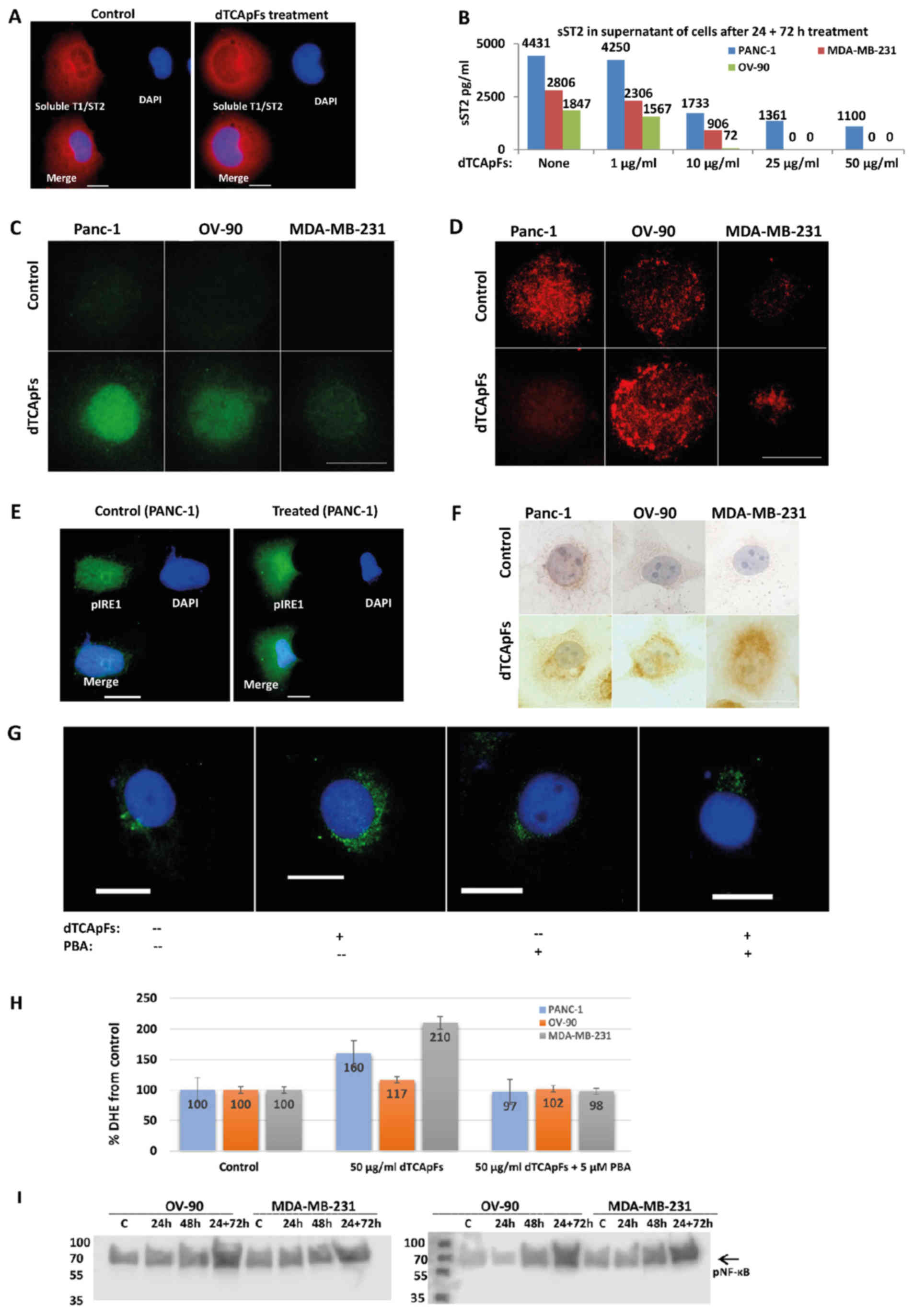 | Figure 3.The dTCApFs-induced structural change
in the Golgi apparatus results in loss of Golgi function and ER
stress, In treated cells, the cellular levels of proteins (soluble
T1/ST2) remain the same, whereas their secretion from the cells
decreases, and molecular effects associated with ER stress are
observed. (A) The soluble T1/ST2 levels were similar between
untreated and treated (dTCApFs 50 µg/ml for 24 h) PANC-1 cells (IF
staining). Soluble T1/ST2; red; DAPI (DNA), blue. Scale bar, 25 µm.
(B) Soluble T1/ST2 levels in the supernatant of cells treated with
dTCApFs for 24 h followed by another dose for 72 h, decreased with
increasing concentrations of dTCApFs (ELISA). (C-E) IF/IHC staining
of untreated and treated cells. For PANC-1 cells, treatment
involved incubation with dTCApFs 50 µg/ml for 48 h, followed by an
additional dose of 50 µg/ml for 24 h. For the OV-90 and MDA-MB-231
cell lines, the dTCApFs concentration was 25 µg/ml. The cells
exhibited increased levels of (C) CHOP, (D) BiP and (E) pIRE1 in
the cytoplasm and (F) e1F2a phosphorylation with dTCApFs treatment.
(G) The Golgi apparatus became diffused in OV-90 cells treated with
dTCApFs (50 µg/ml for 24 h followed by an additional dose of 50
µg/ml for 72 h) compared with controls. Treatment with PBA (5 µM
for 48 h) did not affect the Golgi apparatus, and treatment with
both dTCApFs and PBA (in the concentrations described above)
attenuated the effect of dTCApFs. Scale bars, 25 µm. (H) PANC-1,
OV-90 and MDA-MB-231 cells were treated with dTCApFs (50 µg/ml)
overnight, after which ROS levels were determined using the DHE
assay and compared to untreated cells (controls). Treatment with
dTCApFs (50 µg/ml) plus 5 µM PBA attenuated the observed ROS
increase. Experiments were performed in triplicate. Error bars
represent standard deviation (SD). (I) NF-κB and pNF-κB levels
increased with dTCApFs treatment. OV-90, and the MDA-MB-231 cells
were treated with dTCApFs (50 µg/ml) for the times indicated (for
control, cells were incubated with 5% mannitol for 24 h) and cell
lysates were subjected to western blot analysis. dTCApFs, 14-amino
acid derivative of tumor-cells apoptosis factor; CHOP, C/EBP
homologous protein; BiP, binding immunoglobulin protein; pIRE1,
phosphorylated inositol-requiring enzyme 1; eIF2α, eukaryotic
translation initiation factor 2α; PBA, 4-phenylbutyrate; DHE,
dihydroethidium; ER, endoplasmic reticulum; DAPI,
4′,6-diamidino-2-phenylindole; β-COP, coatomer subunit β; IF,
immunofluorescence; IHC, immunohistochemistry; ROS, reactive oxygen
species; NF-κB, nuclear factor-κB. |
Next, the mechanism by which dTCApFs-induced ER
stress may be associated with apoptosis was investigated by
examining the ER stress repair mechanism. RT-PCR was used to
determine the levels of spliced X-box-binding protein 1 (sXBP1) [a
key player in the ER repair mechanism that upregulates ER-stress
repair genes (9)] with dTCApFs
treatment. It was observed that long-term exposure to dTCApFs led
to downregulation of sXBP1 (Fig.
4A). The anti-apoptotic factor GAAP (10) was also found (by IF staining) to be
downregulated with dTCApFs treatment (Fig. 4B), and cell viability (measured by
the Annexin V FITC assay) was found to be reduced (Fig. 4C). In the cell viability experiments,
dTCApFs was found to cause apoptosis in 50% of PANC-1 cells.
Similar results were observed in OV-90 and MDA-MB-231 cell
lines.
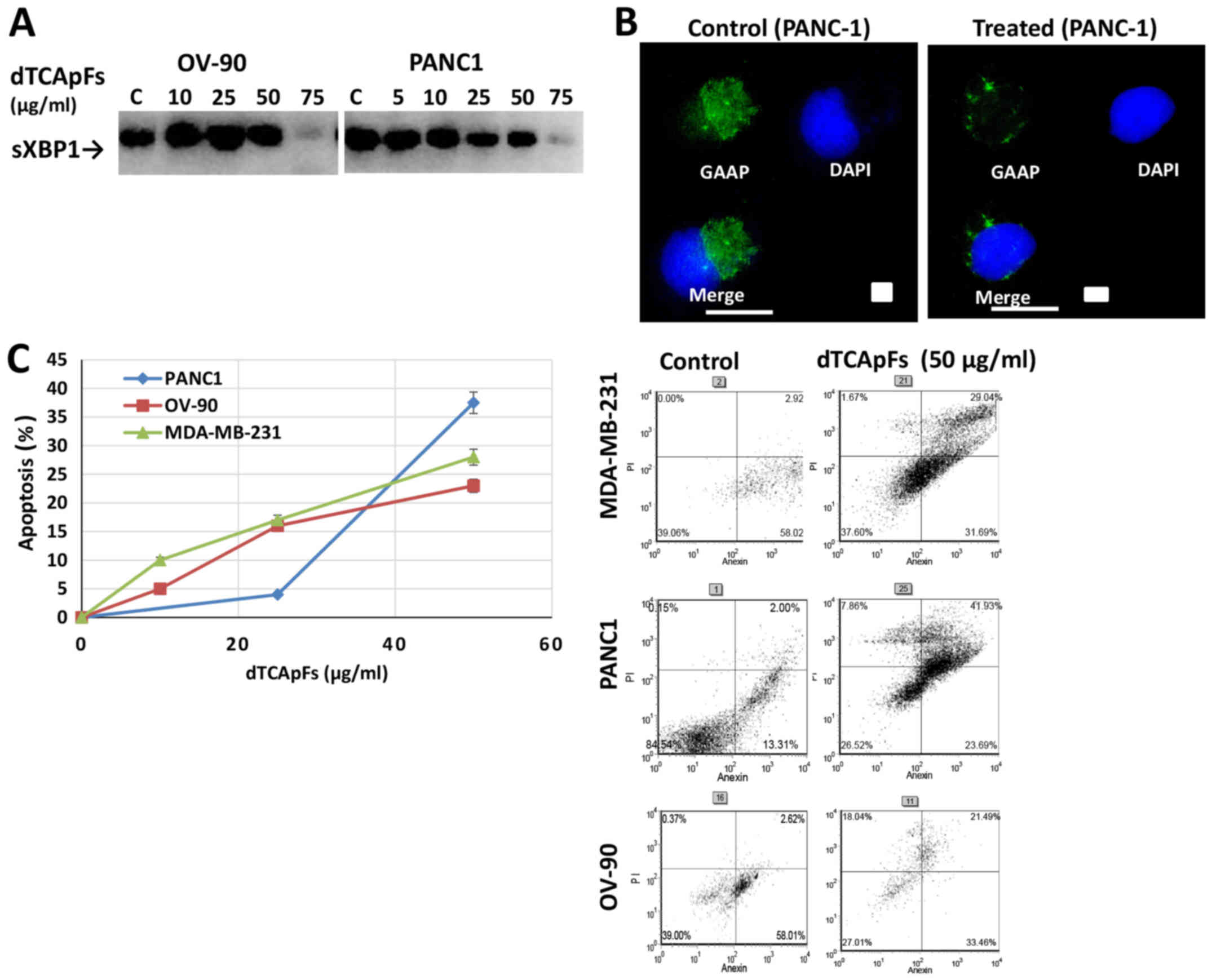 | Figure 4.Long-term exposure to dTCApFs
downregulates the ER stress repair mechanism and as a result
apoptosis is induced. In treated cells, the sXBP1 and GAAP levels
decreased. Cell viability also decreased. (A) RT-PCR-detected sXBP1
levels in OV-90, PANC-1 and MDA-MB-23 cells treated with dTCApFs
for 72 h decreased with increasing dTCApFs concentrations (equal
amounts of RNA/cDNA were used in the analysis). (B) The GAAP levels
were higher in dTCApFs-treated PANC-1 cells (25 µg/ml for 48 h
followed by an additional dose of 25 µg/ml for a further 24 h)
compared with untreated cells (IF staining). Scale bar, 25 µm. (C)
Apoptosis was observed in PANC-1, OV-90 and MDA-MB-231 cells
treated with increasing concentrations of dTCApFs for 24 h followed
by another dTCApFs dose for 72 h. Viability was measured with the
Annexin V FITC assay. Error bars, standard deviation. dTCApFs,
14-amino acid derivative of tumor-cells apoptosis factor; ER,
endoplasmic reticulum; RT-PCR, reverse transcription-polymerase
chain reaction; sXBP1, spliced X-box-binding protein 1; GAAP, Golgi
anti-apoptotic protein; IF, immunofluorescence. |
In vivo studies: Murine xenograft
models of human pancreatic cancer
In vivo studies were next performed to verify
the findings of the cell culture. First, the Golgi apparatus
structure was checked in tumor sections from the PANC-1 murine
xenograft model, comparing dTCApFs-treated mice and controls.
Similar to the cell culture findings, structural changes were
observed in the ER/Golgi (Fig. 5A).
Two molecular markers for ER stress [pIRE1 and BiP (6,7)] were
also investigated; again, the in vivo results were
consistent with the cell culture findings, as an increase in the
number of cells expressing the markers (as detected by IHC) was
observed with dTCApFs treatment (Fig.
5B). Of note, the effect of dTCApFs treatment on tumor size in
the PANC-1 murine xenograft model was also examined, and it was
observed that the increase in tumor size observed in control mice
(no dTCApFs treatment) was attenuated in dTCApFs-treated mice
(Fig. 5C).
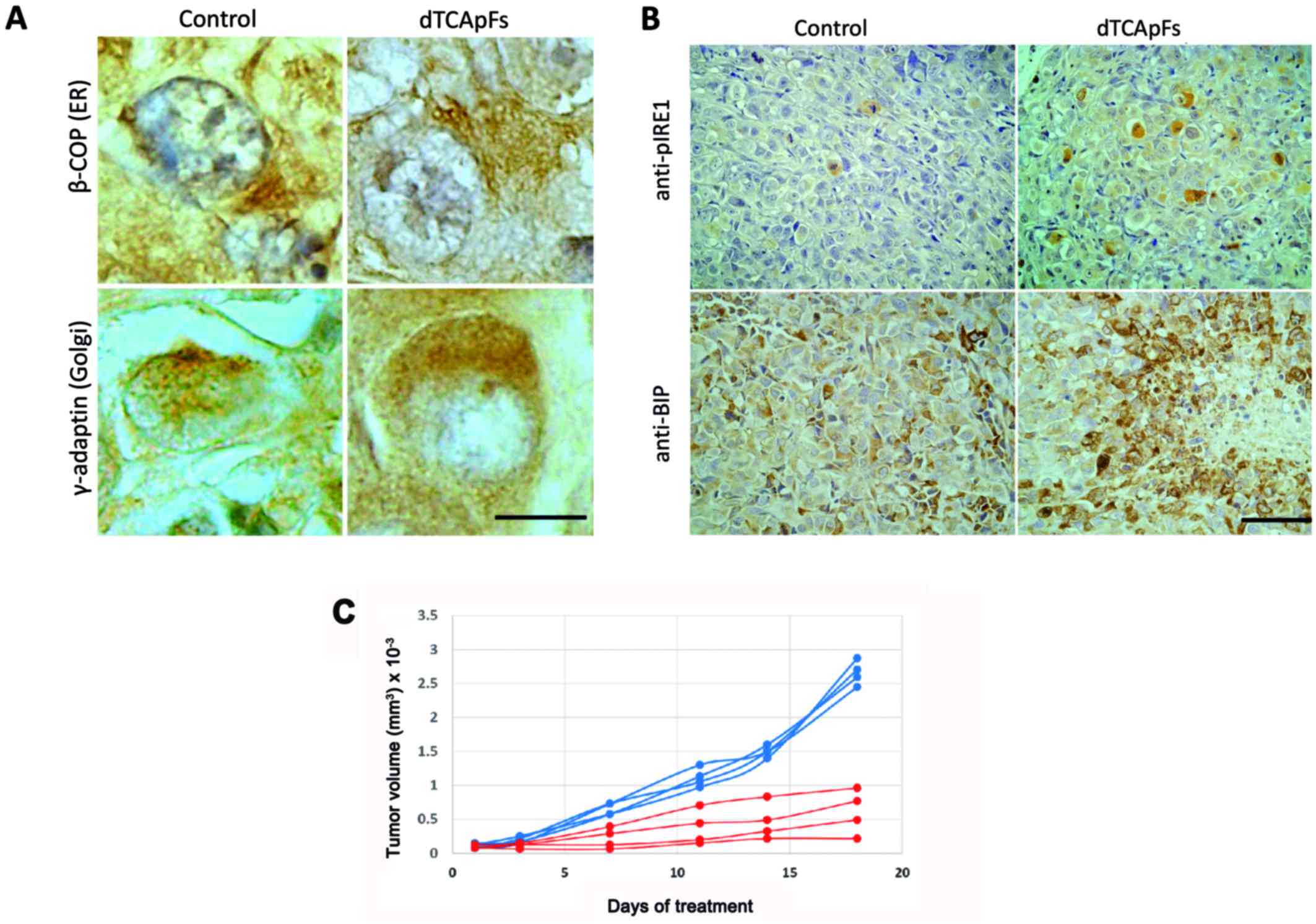 | Figure 5.PANC-1 xenograft murine model studies
showing that dTCApFs causes a structural change in the ER/Golgi, ER
stress, and decreased tumor size. (A) IHC staining using anti-β COP
antibody (ER marker) or anti-γ-adaptin (Golgi marker) of tumor
sections from untreated and dTCApFs-treated PANC-1 xenograft mice
model demonstrating diffused ER/Golgi with dTCApFs treatment. Scale
bar, 12 µm. (B) IHC staining with anti-pIER1 or anti-BiP of tumor
sections from untreated and dTCApFs-treated PANC-1 murine xenograft
model demonstrating increase in the number of cells expressing
these ER stress markers. Scale bar, 50 µm. (C) Tumor size over time
in PANC-1 mouse xenograft model that were dTCApFs-treated (15 mg/kg
intraperitoneally twice a week) vs. controls (treatment with 5%
mannitol). Eight nude mice were inoculated subcutaneously with 1
million PANC-1 cells. When the tumor size exceeded 64
mm3, the animals were randomly assigned to treatment vs.
control and tumor size was measured over time. Each line represents
one animal in the experiment. dTCApFs, 14-amino acid derivative of
tumor-cells apoptosis factor; ER, endoplasmic reticulum; IHC,
immunohistochemistry; β-COP, coatomer subunit β; BiP, binding
immunoglobulin protein; pIRE1, phosphorylated inositol-requiring
enzyme 1. |
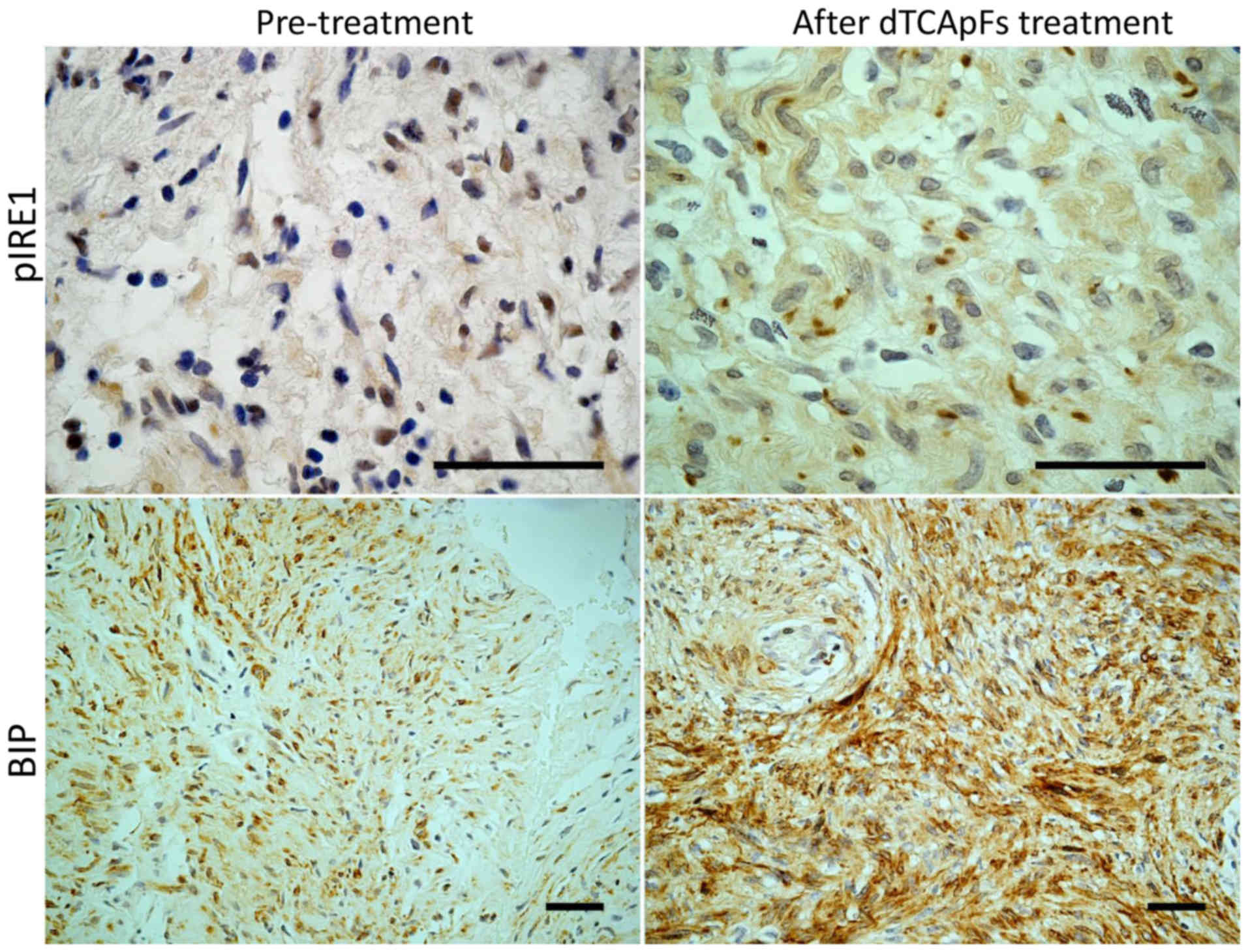
Preliminary evidence on dTCApFs MOA
from human studies
In the final set of experiments, the cell culture
and in vivo findings were confirmed by analyzing human
tissue samples obtained from a patient who received dTCApFs 3 times
a week at increasing concentrations of 12, 24, and 48
mg/m2 for 11 months, as part of a recently completed
phase I study. IHC analysis of tumor sections pre- and
post-treatment revealed an increase in the number of cells
expressing pIRE1 and BiP (Fig. 6),
supporting a MOA involving ER stress.
Discussion
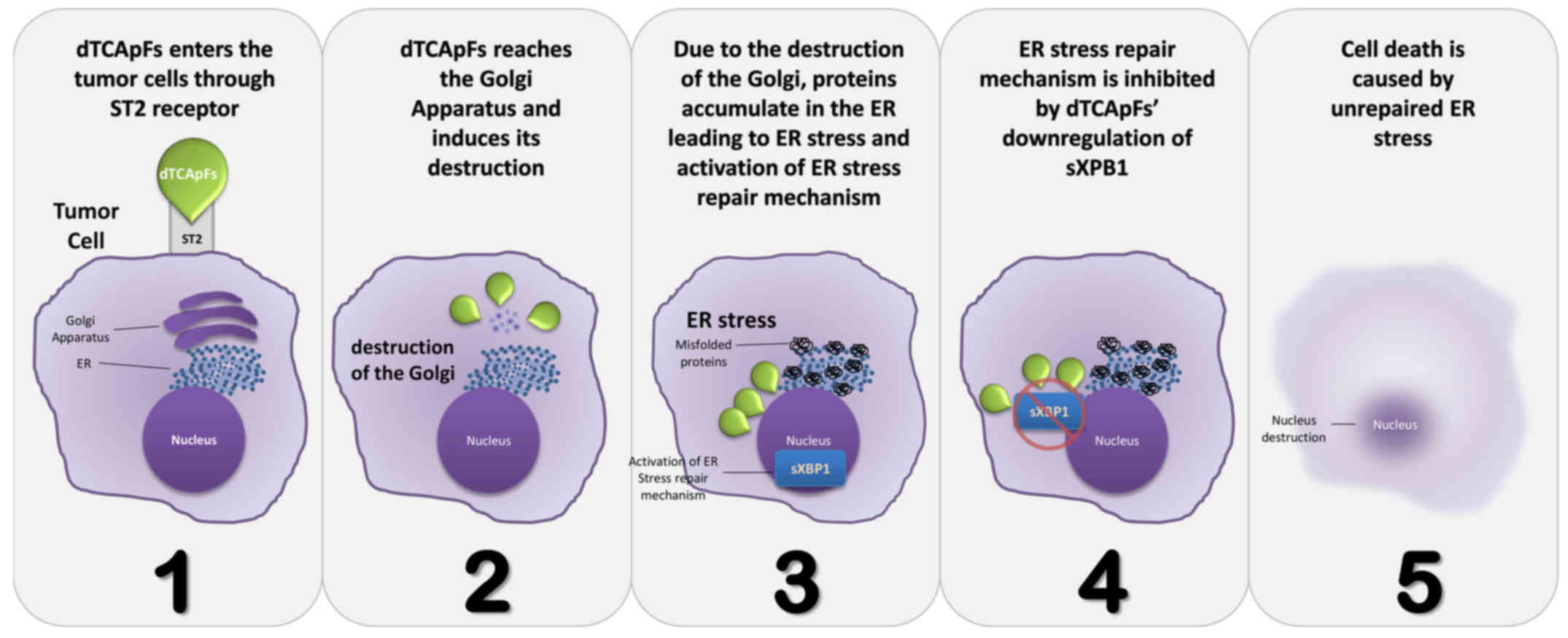
The present study demonstrated that dTCApFs exerts a
direct effect on tumor cells by inducing apoptosis through a novel
mechanism involving the Golgi apparatus. The entry of the dTCApFs
molecules into the cells is mediated through the membrane receptor
ST2. Inside the cells, dTCApFs molecules bind the soluble T1/ST2
receptor, which is located in the Golgi apparatus, and induce
structural changes leading to destruction of the Golgi apparatus
and loss of Golgi function. Consequently, proteins accumulate in
the ER, and ER stress is generated. dTCApFs also downregulates
sXBP1, thus inhibiting the ER stress repair mechanism. These
opposing effects of dTCApFs trigger apoptosis (an illustration of
the suggested MOA is presented in Fig.
7). To the best of our knowledge, this is the first time that
this unique MOA is reported for an anticancer agent. The mechanism
described herein is consistent with our previous study in AML,
where induction of apoptotic pathways was observed, including those
mediated by caspase 8 and Bcl-2, with TCApF treatment (1).
The entry of dTCApFs into the cells through the
T1/ST2 receptor has also been demonstrated in ST2-gene knockout
ovarian cancer cells (OV-90), where TCApFs treatment exerted no
effect on the Golgi apparatus (unpublished data), whereas the Golgi
was completely diffused with dTCApFs treatment in OV-90 control
cells. Notably, the entry through the ST2 receptor also explains
the selectivity of dTCApFs, as overexpression of the T1/ST2
receptor occurs in several tumor types, whereas only a weak
expression of this receptor is documented in normal cells (11–13).
From a clinical perspective, the ability of dTCApFs
to strongly induce ER stress supports its potential use in
combination therapy, where its role would be to enhance the effect
of the concurrently administered chemotherapy agents (e.g.,
platinum-based therapy). This approach may be useful for tumor
types that are difficult to treat, such as triple-negative breast
cancer. Indeed, such an approach was investigated in a phase I
clinical trial evaluating the ER stress inducer nelfinavir
[Viracept®, Agouron Pharmaceuticals, Inc., La Jolla, CA,
USA; approved by the Food and Drug Administration for the treatment
of human immunodeficiency virus infection (14)], in combination with concurrent
chemotherapy (cisplatinum and etoposide) plus radiation in patients
with stage IIIA/IIIB non-small-cell lung cancer (15). This phase I trial demonstrated
acceptable toxicity and promising activity for this regimen
(15).
Notably, dTCApFs also exerts its anticancer activity
through other MOAs. Preliminary unpublished data from ongoing
research suggest that these mechanisms involve indirect effects
through specific activation of the immune system against cancer
cells. This mechanism is compatible with the known role of ST2 in
the immune response.
Molecules that destroy the Golgi apparatus and
activate ER-stress mediated apoptosis were previously identified.
These include the antibiotics monensin (GolgiStop™) and brefeldin A
(BFA; GolgiPlug™) (both from BD Biosciences, San Jose, CA, USA)
(16). Monensin has been shown to
destroy the Golgi apparatus by interacting with the Golgi
sodium/proton exchanger, whereas BFA has been shown to cause
disassembly of the Golgi complex and redistribution of secretory
proteins from the cis/medial Golgi complex to the ER, thereby
lePleaseading to ER stress. Both these antibiotics are very toxic
and, hence, their clinical development was not pursued (17). In contrast to monensin and BFA,
TCApFs was found to be safe in preclinical toxicity studies
(personal communication) and was approved for human studies.
Indeed, the recently completed phase I clinical trial that
evaluated dTCApFs treatment in 17 patients with advanced/metastatic
solid tumors demonstrated tolerability and suggested efficacy for
dTCApFs in this setting (personal communication).
In conclusion, dTCApFs induces apoptosis of cancer
cells through a novel MOA involving two opposing effects: Inducing
ER stress, while downregulating the ER stress repair mechanism.
These findings provide the framework for the ongoing clinical
evaluation of dTCApFs as a novel anticancer therapy.
Acknowledgements
The present study was supported by ISK and Israel
Chief Scientist grants (grant no. 54811). Dr J. Ohana, Dr U.
Sandler, Dr G. Kass and Dr Y. Devary are employed by ISK Ltd.
References
|
1
|
Sandler U, Devary O, Braitbard O, Ohana J,
Kass G, Rubinstein AM, Friedman ZY and Devary Y: NEROFE-a novel
human hormone-peptide with anti-cancer activity. J Exp Ther Oncol.
8:327–339. 2010.PubMed/NCBI
|
|
2
|
Yanagisawa K, Naito Y, Kuroiwa K, Arai T,
Furukawa Y, Tomizuka H, Miura Y, Kasahara T, Tetsuka T and Tominaga
S: The expression of ST2 gene in helper T cells and the binding of
ST2 protein to myeloma-derived RPMI8226 cells. J Biochem.
121:95–103. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kumar S, Tzimas MN, Griswold DE and Young
PR: Expression of ST2, an interleukin-1 receptor homologue, is
induced by proinflammatory stimuli. Biochem Biophys Res Commun.
235:474–478. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garlanda C, Dinarello CA and Mantovani A:
The interleukin-1 family: Back to the future. Immunity.
39:1003–1018. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nishitoh H: CHOP is a multifunctional
transcription factor in the ER stress response. J Biochem.
151:217–219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gething MJ: Role and regulation of the ER
chaperone BiP. Semin Cell Dev Biol. 10:465–472. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Novoa I, Zhang Y, Zeng H, Jungreis R,
Harding HP and Ron D: Stress-induced gene expression requires
programmed recovery from translational repression. EMBO J.
22:1180–1188. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hirota M, Kitagaki M, Itagaki H and Aiba
S: Quantitative measurement of spliced XBP1 mRNA as an indicator of
endoplasmic reticulum stress. J Toxicol Sci. 31:149–156. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Carrara G, Saraiva N, Gubser C, Johnson BF
and Smith GL: Six-transmembrane topology for Golgi anti-apoptotic
protein (GAAP) and Bax inhibitor 1 (BI-1) provides model for the
transmembrane Bax inhibitor-containing motif (TMBIM) family. J Biol
Chem. 287:15896–15905. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kakkar R and Lee RT: The IL-33/ST2
pathway: Therapeutic target and novel biomarker. Nat Rev Drug
Discov. 7:827–840. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jovanovic IP, Pejnovic NN, Radosavljevic
GD, Arsenijevic NN and Lukic ML: IL-33/ST2 axis in innate and
acquired immunity to tumors. Oncoimmunology. 1:229–231. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bergis D, Kassis V, Ranglack A, Koeberle
V, Piiper A, Kronenberger B, Zeuzem S, Waidmann O and Radeke HH:
High serum levels of the interleukin-33 receptor soluble ST2 as a
negative prognostic factor in hepatocellular carcinoma. Transl
Oncol. 6:311–318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Viracept (package isert). Agouron
Pharmaceuticals, Inc.; La Jolla, CA: 2011
|
|
15
|
Rengan R, Mick R, Pryma D, Rosen MA, Lin
LL, Maity AM, Evans TL, Stevenson JP, Langer CJ, Kucharczuk J, et
al: A phase I trial of the HIV protease inhibitor nelfinavir with
concurrent chemoradiotherapy for unresectable stage IIIA/IIIB
non-small cell lung cancer: A report of toxicities and clinical
response. J Thorac Oncol. 7:709–715. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rosa P, Mantovani S, Rosboch R and Huttner
WB: Monensin and brefeldin A differentially affect the
phosphorylation and sulfation of secretory proteins. J Biol Chem.
267:12227–12232. 1992.PubMed/NCBI
|
|
17
|
Torii S, Banno T, Watanabe T, Ikehara Y,
Murakami K and Nakayama K: Cytotoxicity of brefeldin A correlates
with its inhibitory effect on membrane binding of COP coat
proteins. J Biol Chem. 270:11574–11580. 1995. View Article : Google Scholar : PubMed/NCBI
|