Introduction
Autism spectrum disorder (ASD) is a group of
neurodevelopmental disorders characterized by disturbances in
interpersonal relationships and behavior, and is characterized by
severe and pervasive impairment in reciprocal socialization,
qualitative impairment in communication, and repetitive or unusual
behavior (1). It is also known that
ASD is male-biased and present genetic heterogeneity (2). However, it is difficult to identify the
common causes in this heterogeneous disorder. Previous genome-wide
association studies have identified numerous mutations or
variations associated with ASD risk on many chromosome loci and
genes (1,3,4).
Furthermore, a promising development in understanding the genetics
of ASD is the identification of variations in the gene copy number
as a risk factor (5).
Copy-number variation is defined as the structural
variation in the genome in which genetic material is either
duplicated or deleted and appears to be strongly associated with
various nervous system disorders and mental illnesses, such as
intellectual impairment and autism (6,7). While
sequencing of sporadic cases has identified de novo risk variants,
the heritable genetic contribution and mechanisms driving the male
bias are complex and less understood (2,8,9). A previous study highlighted the
male-specific effect of the rs6323 marker and its haplotypes in ASD
etiology, revealing the sexual dimorphic effect of monoamine
oxidase A (responsible for degradation of serotonin) in ASD
(1). From a total sample of 1,008
multiplex families, Werling et al (10) performed genome-wide, non-parametric
linkage analysis in a discovery sample of 847 families separated by
subsets of families with only male affected children (male-only;
MO) or with at least one female affected child (female-containing;
FC), and observed sex-differential linkage at 1p31.3 (MO), 8p21.2
(FC), and 8p12 (FC).
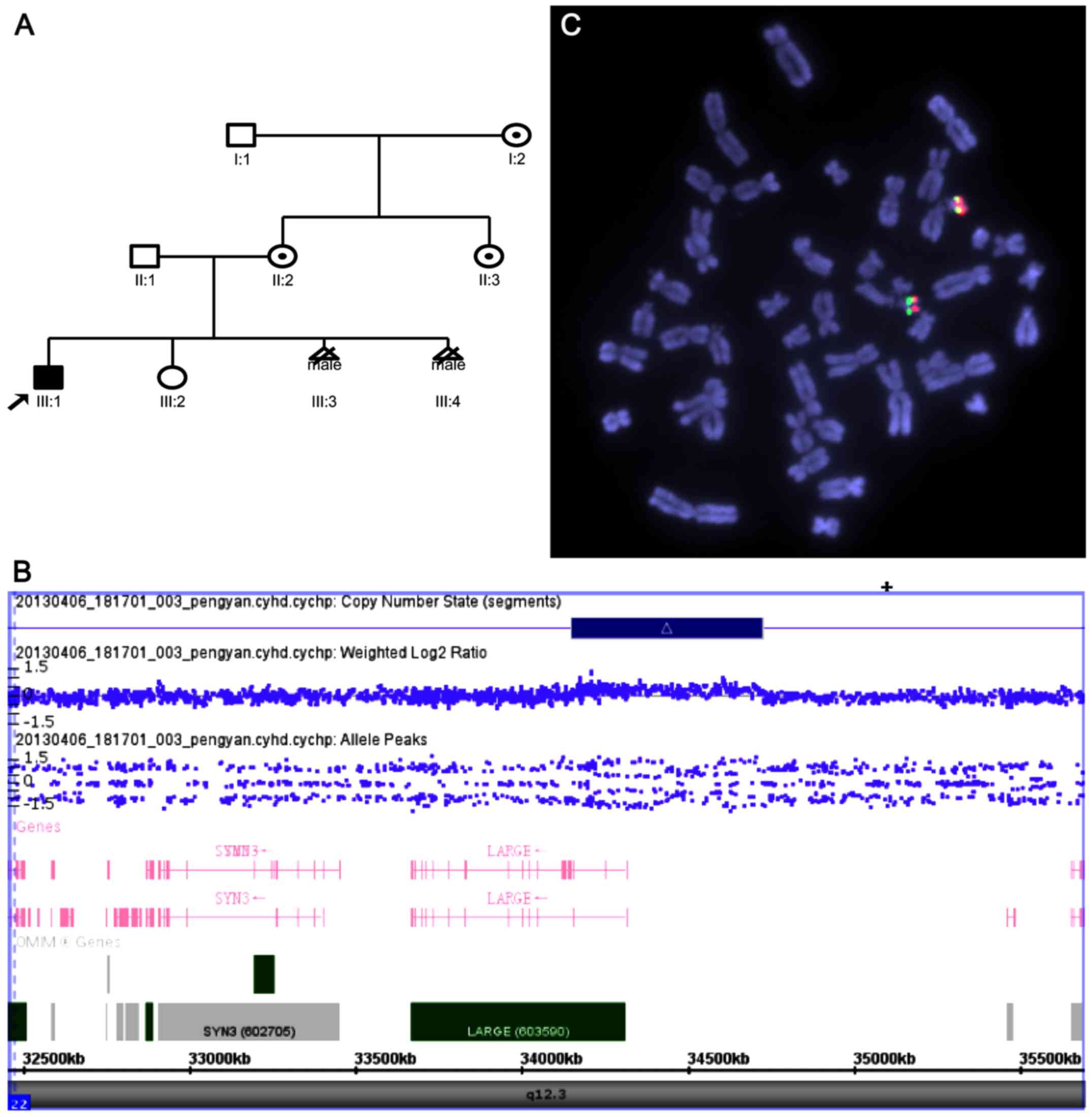
The current study describes a three-generation
Chinese family (Fig. 1A) with one
affected male, who was diagnosed with an ASD disease using
whole-genome single nucleotide polymorphism (SNP)-microarray and
fluorescence in situ hybridization analysis, with special
probes, and determined a 22q12.3 duplication among the six family
members, which included three healthy female carriers, one affected
boy and two male fetuses. In addition, LARGE gene sequencing
indicated that the 22q12.3 duplication overlapped a LARGE
gene. The present study hypothesized that the specific 22q12.3
duplication overlapping the LARGE gene may be a male-only
loci, which is responsible for increasing the nervous system
disorder risk, including ASD.
Materials and methods
Clinical investigation of the patient
and family
A 35-year-old woman (II:2), gravida 4 para 3, was
referred to the First Affiliated Hospital of Sun Yat-sen University
(Guangzhou, China) for genetic consultation at gestational week 28
in March 2013, due to detection of fetal cerebellar vermis by
prenatal ultrasound at gestational week 27. A fetal ultrasound
demonstrated that the cerebellar vermis area was 1.63
cm2 (2.27±0.64 cm2) and polyhydramnios was
observed (amniotic fluid volume, 98 mm). Additionally, fetal
magnetic resonance imaging (MRI) indicated that fetal brain
development was significantly slower than its gestational age, the
hemispheric sulci and gyri were not obvious and lateral cerebral
fissure was shallow. Her husband (II:1) was 42 years old. The
couple was non-consanguineous and had no family history of
congenital malformations on either side.
However, the couple had an abnormal
childbearing history
The first child (III:1) of the couple was a boy who
was diagnosed with ASD. At the time of the study, the proband was 9
years old. He was delivered normally (birth weight, 3.2 kg);
however, the birth head circumference was not available. Mild
hypospadias was noted. At 30 months of age, he showed microcephaly,
social deficits and communication difficulties, stereotyped or
repetitive behaviors and interests, sensory issues, and was
diagnosed as ASD. He was unable to walk until the age of 4 with
marked motor and language delay. He had a long face, periorbital
fullness, a smooth philtrum, and a thin upper lip vermilion. He
also had tapering fingers and camptodactyly. He was characterized
by frequent mood swings and difficulties obeying rules. At the age
of 7, brain MRI indicated a small brain size, focal radial
distribution of defects on the front parts of the two frontal
lobes, the right side of the Brota area and the left side of the
hippocampus.
The second child (III:2) of the couple was a
4-year-old healthy girl. The third sibling was a male fetus.
Ultrasound-guided amniocentesis of the fetuses (III:3 and III:4)
was performed for a cytogenetic analysis and chromosomal microarray
analysis (CMA) was performed at gestational week 18. The CMA result
was as abnormal as the proband. Therefore, subsequent to receiving
genetic counseling, the parents decided to terminate the third
pregnancy.
Umbilical cordocentesis was then performed at
gestation week 28. Umbilical cord blood was collected for
cytogenetic analysis and CMA. Due to the malformation of the fetal
brain and abnormal CMA result, the parents opted to have an
abortion. The pregnancy was subsequently terminated, and a male
fetus was delivered. However, the brain malformation could not be
determined, as the parents did not consent to fetal autopsy.
Approval was obtained for the current study from the
Ethics Committee of the First Affiliated Hospital of Sun Yat-sen
University (Guangzhou, China). Principles outlined in the
Declaration of Helsinki were followed and informed consent was
obtained from the participants.
Cytogenetics and SNP array
analyses
Cytogenetic analysis was performed on G-banded
metaphases at a resolution of 450–550 bands. Chromosome
preparations were established from cultured lymphocytes of the
proband and the couple.
Peripheral venous blood (2 ml) was collected from
the couples, their two children, the sister of the mother and the
mother's mother, and cord blood was collected from the present
fetus (the fourth sibling). Genomic DNA was extracted from the
uncultured blood samples using a QIAamp DNA Blood Mini kit (cat.
no. 51104; Qiagen, Inc., Valencia, CA, USA). DNA (250 ng) was
amplified (CytoScan PCR program), labeled and hybridized to the
CytoScan HD array platform (Affymetrix; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) according to the manufacturer's protocol.
The array is designed specifically for cytogenetic research, which
offers >2,700,000 markers across the whole genome, including
750,000 SNP probes and 1,950,000 probes for detecting the copy
number variations (Cyto-arrays). CEL files obtained by scanning
CytoScan arrays were analyzed with Chromosome Analysis Suite
software (ChAS 2.1, Affymetrix), using annotations of genome
version GRCH37 (hg19). Only those qualified measures were included
in the present analysis. Gains and losses that affected a minimum
of 50 markers in a 100-kb length were initially considered.
Fluorescence in situ hybridization
(FISH)
FISH was performed according to standard protocols.
Single copy DNA probes (Agilent Technologies, Inc., Santa Clara,
CA, USA), 22q12.3 (561 k) located within the duplication region on
22q12.3, and the commercially available probe LSI 22 (Abbott GmbH
& Co. KG, Wiesbaden, Germany) served as a chromosome 22 control
probe.
Quantitative polymerase chain reaction
(qPCR)
Confirmation of the CMA result and analysis of the
unaffected sisters and the parents were performed by qPCR with
Genomic DNA extracted from the uncultured blood samples. qPCR
experiments were conducted on an ABI Prism 9700TH Sequence
Detection System (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) using SYBR™ Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The authors selected
non-polymorphic fragments locating in the LARGE gene. The
used primers for qPCR were as follow: Forward,
5′-CAACCACTCCAAGACCTACTC-3′ and reverse, 5′-CGCATTTCTCCACGACCG-3′.
GADPH was used as internal reference (forward, GGAGTCAACGGATTTGGTCG
and reverse, TCCTGGAAGATGGTGATGGG) and the reaction conditions were
as follows: 95°C for 10 min, 40 cycles of 95°C for 10 sec, 60°C for
30 sec and 72°C for 30 sec). The resultant crossing thresholds were
analyzed using the Cq method. The final data were compared with a
normal control in the same loci using 2−ΔΔCq (11).
LARGE gene sequencing
Overlapping amplicons covering the entire coding
region of 15 exons from all isoforms of LARGE (MIM no.
603590) were amplified. PCR was performed in 10 reactions using 100
ng genomic DNA or 1 ng cloned DNA in standard buffer (Boehringer
Mannheim GmbH, Mannheim, Germany): 10 mM Tris-HCl, 1.5 mM
Mg2+, 50 mM KCl (pH 8.3), with 0.5 U Taq polymerase
(Boehringer Mannheim GmbH, Mannheim, Germany) and 2 nM primers.
Templates were denatured at 95°C for 3 min, followed by 12 cycles
of 95°C for 2 min, 65°C for 40 sec, and 72°C for 1 min, then
followed by 45 cycles of 95°C for 2 min, 55°C for 40 sec, and 72°C
for 1 min. A final extension was performed for 10 min at 72°C.
Reactions were performed using a Perkin Elmer 9600 thermal cycler.
Individual primer sequences were listed in Table I. PCR products were analyzed using gel
electrophoresis with 1.5% agarose. PCR products for sequencing were
isolated from low melting point agarose using the Spinbind
purification kit (FMC Bioproducts, Rockland, ME, USA), and
sequenced in two directions using a BigDye Terminator v3.1 Cycle
Sequencing kit (cat. no. 4337455; Applied Biosystems; Thermo Fisher
Scientific, Inc.) and an ABI 3100 Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). DNA sequences were
analyzed by comparison to GRCh37.
 | Table I.Primer sequences for sequencing of
LARGE gene. |
Table I.
Primer sequences for sequencing of
LARGE gene.
| Primer | Sequence (5′-3′) |
|---|
| 1 |
F:GCTGTGTGTAAGTGTGTTTTATATC |
|
|
R:GCAAGCCAGTGGAGAG |
| 2 |
F:GTTTACGCCTCATGGATTTA |
|
|
R:GGGCACACAGTCCCAA |
| 3 |
F:TCAAAGACCCATATCAACCA |
|
|
R:GTGCTGAAAAGCGACACTTA |
| 4 |
F:AGCTTCATACACTGAAATTGTTG |
|
|
R:CACCGGGAAACCTTGAT |
| 5 |
F:CTCTGAGACCACTTTCAAGC |
|
|
R:ATGTTACCCATTTGTGGAGA |
| 6 |
F:AGTACCTGAAAGGGTGGG |
|
|
R:GATCAGTGTAGGCTCCAAA |
| 7 |
F:CTTAATGGTTTGGCAGGATA |
|
|
R:AGCTTAAAANCAAAATCTCCC |
Results
Cytogenetic investigations performed on peripheral
blood lymphocytes in all members of the family exhibited a normal
karyotype. Genome-wide array analysis revealed an interstitial
575-kb duplication of chromosome 22p12.3 from 34,151,455 to
34,726,809 bp (Fig. 1B). FISH analysis
of the proband and his parents confirmed the rearrangements and
excluded translocation with other chromosomes at this duplication
region (Fig. 1C). qPCR performed on
the gene within the borders of the identified duplication evidenced
the overlap of the LARGE gene (intron 1). However, no
significant mutations were identified by sequencing of the
LARGE gene.
Discussion
The current study describes a three-generation
Chinese family (six members) with an interstitial 575-kb
duplication of chromosome 22p12.3 that involved the LARGE gene. The
family comprised three healthy female carriers and three affected
males, including a 9-year-old boy and two male fetuses. In
addition, in the Decipher database (https://decipher.sanger.ac.uk/) numerous patients
exhibited overlapping duplication on chromosome 22q12.3 similar to
the current patient. Patient no. 290286 had a 630-kb duplication at
chr22:33,440,702–34,073,409 and exhibited a phenotype of global
developmental delay and delayed gross motor development. In
addition, complex rearrangement or duplication may cause gene
mutations, which lead to cognitive disorders, such as ASD if such
genes are important in the development and modulation of synaptic
connectivity (12). While sequencing
the 15 exons of this gene did not identify any significant
mutations, it is suggested that the duplication associated with ASD
is more frequent in males.
The common 575-kb duplication interval of 22q12.3
observed in these patients includes only one known gene,
LARGE (MIM no. 603590) and the breakpoint between exon 1 and
exon 2. The duplication region overlapped the 5′ untranslated
region (5′UTR) within exon 1 of the LARGE gene. LARGE
encodes one of a number of proteins that are critical in the
development of the neuromuscular junction. The encoded protein
contains laminin G, Kazal-type serine protease inhibitor and
epidermal growth factor domains. Additional post-translational
modifications occur to add glycosaminoglycans and disulfide bonds.
Mutation of the LARGE gene is the rarest of the six known
genetic causes (protein O-mannosyltransferase 1, protein
O-mannosyltransferase 2, O-mannose
β-1,2-N-acetylglucosaminyltransferase, Fukuyama type congenital
muscular dystrophy protein, Fukutin-related protein and
LARGE) of α-dystroglycanopathy (αDG). Abnormality in the
glycosylation of αDG is the hallmark histological abnormality and
the likely pathogenic mechanism for a group of congenital muscular
dystrophies, collectively termed αDGs (13). Affected children exhibit typical
neurological and muscular abnormalities associated with the αDGs,
but with very different severities. For example, one family member
had mild muscle-eye-brain disease (and the other two had typical
Walker-Warburg syndrome (WWS). In addition, a WWS patient has been
reported to have a single heterozygous nonsense mutation in
LARGE (14,15). In one family with congenital myasthenic
syndrome affecting limb-girdle muscles, a mutation in LARGE
was observed (16). Patients with WWS
frequently demonstrate a complete lack of psychomotor development,
severe eye malformations, cobblestone lissencephaly and a
hypoplastic cerebellum and brainstem, seizures, hydrocephalus and
poor prognosis (17). Previously,
Clarke et al (18) described a
family with mental retardation, and identified a large
intra-chromosomal duplication inserted into intron 10 of
LARGE in a homozygous state. As a result, it is proposed
that mutations of the LARGE gene may cause a wide spectrum
of clinical phenotypes. In the patient investigated in the current
study, disruption of the LARGE gene was anticipated to
result in a different protein and may account for ASD and
associated clinical features in the patient, as it is highly
expressed in the brain.
The current results imply high diversity of clinical
presentation as well as sex-bias, such as the three normal females
and three affected males in the present study. The high diversity
of clinical presentation may also be caused by the influence of
interactions between genetic and environmental factors on clinical
manifestation or differential environmental exposures experienced
by different individuals (19,20). As for the striking sex-bias in autism,
early exposure to androgenic hormones and early maternal immune
activation are environmental factors, and have been proposed to
affect the sex-specific susceptibility to ASD (21). Baron-Cohen's hypothesis that autism
results from exposure to high intrauterine testosterone levels is
considered in the context of a hormonal hypothesis of sex ratio and
the notion of multifactorial inheritance (22). This hypothesis yields three suggestions
as follows: i) Female cases of autism may be the product of higher
genetic loading combined with moderate environmental exposure and
male cases of high environmental exposure combined with moderate
genetic loading; ii) notably, one of the environmental agents is
intrauterine testosterone; and iii) the mother is the major source
of that testosterone. A gender-atypical pattern for these types of
feature is indicated in ASD (23).
These suggestions may help to explain the majority of the major
established epidemiological risk factors for autism.
In conclusion, the present study identified a role
for 22q12.3 duplication in the diversity of phenotypes with ASD and
brain deformation in patients exhibiting a 22q12.3 duplication. In
addition, it is hypothesized, but as yet unproven, that the
specific 22q12.3 duplication overlapping the LARGE gene may
be within the male-only loci that are responsible for increasing
ASD risk. However, further in-depth evaluation of other ASD
patients with defects in the LARGE gene are required to
elucidate the genetic mechanism underlying ASD.
Acknowledgements
The authors would like to thank the Clinical
Cytogenetics Laboratory (the First Affiliated Hospital of Sun
Yat-sen University, Guangzhou, China) for facilitating with data
collection. The authors would also like to thank the individuals
included in the current study and their families.
References
|
1
|
Cho SC, Yim SH, Yoo HK, Kim MY, Jung GY,
Shin GW, Kim BN, Hwang JW, Kang JJ, Kim TM, et al: Copy number
variations associated with idiopathic autism identified by
whole-genome microarray-based comparative genomic hybridization.
Psychiatr Genet. 19:177–185. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Horiuchi F, Oka Y, Uno H, Kawabe K, Okada
F, Saito I, Tanigawa T and Ueno S: Age- and sex-related emotional
and behavioral problems in children with autism spectrum disorders:
Comparison with control children. Psychiatry Clin Neurosci.
68:542–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosenfeld JA, Ballif BC, Torchia BS, Sahoo
T, Ravnan JB, Schultz R, Lamb A, Bejjani BA and Shaffer LG: Copy
number variations associated with autism spectrum disorders
contribute to a spectrum of neurodevelopmental disorders. Genet
Med. 12:694–702. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Griswold AJ, Ma D, Cukier HN, Nations LD,
Schmidt MA, Chung RH, Jaworski JM, Salyakina D, Konidari I,
Whitehead PL, et al: Evaluation of copy number variations reveals
novel candidate genes in autism spectrum disorder-associated
pathways. Hum Mol Genet. 21:3513–3523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sebat J, Lakshmi B, Malhotra D, Troge J,
Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et
al: Strong association of de novo copy number mutations with
autism. Science. 316:445–449. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cook EHJ Jr and Scherer SW: Copy-number
variations associated with neuropsychiatric conditions. Nature.
455:919–923. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Prasad A, Merico D, Thiruvahindrapuram B,
Wei J, Lionel AC, Sato D, Rickaby J, Lu C, Szatmari P, Roberts W,
et al: A discovery resource of rare copy number variations in
individuals with autism spectrum disorder. G3 (Bethesda).
2:1665–1685. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Verma D, Chakraborti B, Karmakar A,
Bandyopadhyay T, Singh AS, Sinha S, Chatterjee A, Ghosh S,
Mohanakumar KP, Mukhopadhyay K, et al: Sexual dimorphic effect in
the genetic association of monoamine oxidase A (MAOA) markers with
autism spectrum disorder. Prog Neuropsychopharmacol Biol
Psychiatry. 50:11–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mouridsen SE, Rich B and Isager T: The Sex
Ratio of Full and Half Siblings of People Diagnosed With ADHD in
Childhood and Adolescence: A Danish Nationwide Register-Based
Cohort Study. J Atten Disord. 20:1017–1022. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Werling DM, Lowe JK, Luo R, Cantor RM and
Geschwind DH: Replication of linkage at chromosome 20p13 and
identification of suggestive sex-differential risk loci for autism
spectrum disorder. Mol Autism. 5:132014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Knight D, Xie W and Boulianne GL:
Neurexins and neuroligins: Recent insights from invertebrates. Mol
Neurobiol. 44:426–440. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martin PT: Mechanisms of disease:
Congenital muscular dystrophies-glycosylation takes center stage.
Nat Clin Pract Neurol. 2:222–230. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mercuri E, Messina S, Bruno C, Mora M,
Pegoraro E, Comi GP, D'Amico A, Aiello C, Biancheri R, Berardinelli
A, et al: Congenital muscular dystrophies with defective
glycosylation of dystroglycan: A population study. Neurology.
72:1802–1809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Godfrey C, Clement E, Mein R, Brockington
M, Smith J, Talim B, Straub V, Robb S, Quinlivan R, Feng L, et al:
Refining genotype phenotype correlations in muscular dystrophies
with defective glycosylation of dystroglycan. Brain. 130:2725–2735.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maselli RA, Fernandez JM, Arredondo J,
Navarro C, Ngo M, Beeson D, Cagney O, Williams DC, Wollmann RL,
Yarov-Yarovoy V, et al: LG2 agrin mutation causing severe
congenital myasthenic syndrome mimics functional characteristics of
non-neural (z-) agrin. Hum Genet. 131:1123–1135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Czeschik JC, Hehr U, Hartmann B, Lüdecke
HJ, Rosenbaum T, Schweiger B and Wieczorek D: 160 kb deletion in
ISPD unmasking a recessive mutation in a patient with
Walker-Warburg syndrome. Eur J Med Genet. 56:689–694. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clarke NF, Maugenre S, Vandebrouck A,
Urtizberea JA, Willer T, Peat RA, Gray F, Bouchet C, Manya H,
Vuillaumier-Barrot S, et al: Congenital muscular dystrophy type 1D
(MDC1D) due to a large intragenic insertion/deletion, involving
intron 10 of the LARGE gene. Eur J Hum Genet. 19:452–457. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lai MC, Lombardo MV and Baron-Cohen S:
Autism. Lancet. 383:896–910. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cuccaro ML, Shao Y, Bass MP, Abramson RK,
Ravan SA, Wright HH, Wolpert CM, Donnelly SL and Pericak-Vance MA:
Behavioral comparisons in autistic individuals from multiplex and
singleton families. J Autism Dev Disord. 33:87–91. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schaafsma SM and Pfaff DW: Etiologies
underlying sex differences in Autism Spectrum Disorders. Front
Neuroendocrinol. 35:255–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
James WH: An update on the hypothesis that
one cause of autism is high intrauterine levels of testosterone of
maternal origin. J Theor Biol. 355:33–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bejerot S and Eriksson JM: Sexuality and
gender role in autism spectrum disorder: A case control study. PLoS
One. 9:e879612014. View Article : Google Scholar : PubMed/NCBI
|