Introduction
Hepatocellular carcinoma (HCC) is the sixth most
common type of cancer and the third leading cause of
cancer-associated death in the world (1). HCC development is based on cirrhosis,
and the number of patients is expected to increase in the future
(1-3).
Advances in research regarding the underlying biology and
pathophysiology of HCC is required to develop effective means of
diagnosis and improved treatments for HCC. The surrounding tumor
microenvironment has a notable influence on the development of HCC
and is a target of novel cancer therapies (4,5). However,
elucidating the underlying mechanisms by which the tumor
microenvironment promotes HCC may assist in the development of
improved therapeutics (6,7). The HCC tumor microenvironment consists
primarily of the surrounding blood vessels, non-parenchymal cells,
including fibroblasts, myofibroblasts, macrophages, lymphocytes and
sinusoidal endothelial cells, and extracellular proteins such as
cytokines and chemokines secreted by these cells (8,9). Hepatic
stellate cells (HSCs) are the most abundant type of non-parenchymal
cell present in the tumor microenvironment of HCC. They participate
in modeling the tumor environment through promoting the
transdifferentiation of myofibroblast-like cells, which in turn
induces liver fibrosis (10-12).
Emerging evidence has shown that tumor-associated macrophages
(TAMs) around the tumor lesion facilitate tumor growth (13). TAMs serve important roles in tumor
development and have attracted considerable attention as components
of the tumor microenvironment (14-16).
Most HCCs develop as a consequence of progression of
liver fibrosis (17,18). HSC activation promotes liver fibrosis
through the extracellular production of proteins, such as
transforming growth factor-β, tumor necrosis factor-α and
interleukin-6 (19,20); therefore, numerous studies have
focused on HSC activation (21,22). Based
on this, Yoshimoto et al (23)
proposed that senescent HSCs contribute to the development of HCC
through the expression of proinflammatory cytokines associated with
the senescence-associated secretory phenotype (SASP). Cellular
senescence is thought to be a defense mechanism against tumor
progression, but under certain circumstances may eventually promote
tumor development. However, to the best of our knowledge, the means
by which senescent HSCs contribute to the HCC tumor
microenvironment has not been studied.
Extracellular vesicles (EVs) and cytokines,
participate in extracellular communication in the tumor
microenvironment (24). EVs are
classified as exosomes (40-100 nm), microvesicles (100-1,000 nm) or
apoptotic bodies (1-5 µm) (25-27).
The contents of EVs vary depending on the condition of the cells
and therefore exert differing biological effects (25-29).
Senescent HSCs promote HCC development via pro-inflammatory
cytokines induced by the SASP (23).
However, whether EVs derived from senescent HSCs inhibit or promote
HCC development remains unknown. To attain a more comprehensive
understanding of the HCC tumor microenvironment, it is necessary to
assess the impact that EVs derived from senescent HSCs have on HCC.
The aim of the present study was to elucidate the effects of EVs
derived from senescent HSCs on the HCC tumor microenvironment. The
characteristics of EVs derived from senescent HSCs and their
influence on growth factor secretion from hepatoma cells and
macrophages were assessed.
Materials and methods
Cell culture and reagents
Human hepatic stellate cells (HHSteCs) were obtained
from SteCM; ScienCell Research Laboratories and maintained in
stellate cell medium (ScienCell Research Laboratories) supplemented
with 2% FBS, 1% penicillin/streptomycin solution (ScienCell
Research Laboratories) and 1% stellate cell growth supplement
(ScienCell Research Laboratories). The human HCC cell lines Hep3B
and Huh7 (American Type Culture Collection) were maintained in DMEM
(Wako Pure Chemical Industries Ltd.) supplemented with 10% FBS and
1% PenStrep (Thermo Fisher Scientific, Inc.). The human monocytic
leukemia cell line THP-1 (American Type Culture Collection) was
cultured in RPMI-1640 medium (Wako Pure Chemical Industries Ltd.)
supplemented with 10% FBS and 1% PenStrep (Thermo Fisher
Scientific, Inc.). All cells were maintained in a humidified
incubator with 5% CO2 at 37˚C. THP-1 cells were induced
to differentiate by treating them with 10 mg ml-l
phorbol-12-myristate-13-acetate (Sigma-Aldrich; Merck KGaA) for 3
days. Etoposide (ETP) was purchased from Santa Cruz Biotechnology,
Inc. Erlotinib hydrochloride was purchased from Sigma-Aldrich
(Merck KGaA).
Immunofluorescence assays, EdU
staining and SA-β-gal staining
Cellular senescence was induced by ETP treatment and
confirmed by observing p21 and 53BP1 expression in HHSteCs using
immunofluorescence assays. A total of 5x104 HHSteCs were
mounted on four-chamber slides (Lab-Tek II; Thermo Fisher
Scientific, Inc.) and treated with various concentrations of ETP
for 3 days. Subsequently, cells were fixed with 4% paraformaldehyde
for 30 min at room temperature, permeabilized with ice-cold 70%
ethanol and blocked in 1% BSA for 1 h at room temperature. Primary
antisera, 1:200 rabbit anti-p21 (cat. no. 29475; Cell Signaling
Technology, Inc.) or 1:200 rabbit anti-53BP1 (cat. no. IHC-00001;
Bethyl Laboratories, Inc.) were added and the cells were incubated
for 1 h at 20-25˚C. After washing the cells with PBS, secondary
antisera (AlexaFluor 488-conjugated donkey anti-rabbit IgG;
1:1,000; cat. no. A11008; Molecular Probes; Thermo Fisher
Scientific, Inc.) was added to the cells and incubated for 1 h at
room temperature. The slides were washed, and coverslips were
mounted with DAPI Fluoromount-G (SouthernBiotech). The uptake of
EdU was observed in the HHSteCs treated with ETP for 3 days, and
for cells left to recover, for another 3 days in normal medium
following treatment. EdU staining of the HHSteCs was performed
using a Click-iT EdU AlexaFluor 594 imaging kit (cat. no. C10339;
Thermo Fisher Scientific, Inc.) for 4 h according to the
manufacturer's protocol. Images were acquired using a Keyence
All-in-One fluorescence microscope (Keyence Corporation) at x100
magnification. SA-β-gal staining was performed using a Senescence
β-Galactosidase Staining kit (Cell Signaling Technology, Inc.)
according to the manufacturer's protocol. All assays were performed
at least in duplicate.
Extraction and quantification of EVs
derived from HHSteCs
To collect EVs, 2.5x105 HHSteCs either
untreated or pretreated with ETP were seeded in a 100-mm dish and
grown in medium containing exo-free FBS (System Biosciences) for
7-10 days. The medium was collected and centrifuged at 300 x g for
10 min and at 16,500 x g for 20 min at 4˚C to remove cells and
debris, respectively. After filtration with a 220-nm filter, the
supernatant was ultra-centrifuged at 150,000 x g for 120 min at
4˚C. The EV pellet was washed and resuspended in PBS and
ultra-centrifuged at 150,000 x g for 120 min at 4˚C. EVs derived
from normal cultured HHSteCs and from senescent HHSteCs were termed
‘normal EVs’ and ‘senescent EVs’, respectively. To measure the
particle size and number of normal and senescent EVs,
7.5x105 HHSteCs were seeded in a 60-mm dish and grown
for 3 days. The EVs were extracted in the same manner as described
above and quantified using nanoparticle tracking analysis
(NanoSight; Malvern Panalytical).
Analysis of EV incorporation into
hepatoma cells and macrophage cells
To examine the incorporation of EVs into hepatoma
cells and macrophages, Hep3B and THP-1 cells were treated with EVs
labeled with PKH67. EVs were labeled using a PKH67 Green
Fluorescent Cell Linker Mini kit (cat. no. MIN167-1KT;
Sigma-Aldrich; Merck KGaA) for general cell membrane labeling
according to the manufacturer's protocol. Labeled EVs were pelleted
by ultracentrifugation twice at 150,000 x g for 120 min at 4˚C to
remove excess dye. Subsequently, 5x104 Hep3B and
differentiated THP-1 cells each were seeded on four-chamber slides
and treated with 2x107 labeled EVs daily for 3 days.
PKH67 expression was observed under a Keyence All-in-One
fluorescence microscope at x100 magnification. In addition to the
unlabeled EVs, PBS without EVs based on the same procedure
previously described (to confirm the absence of residual PKH67) was
prepared as a negative control. These experiments were performed in
at least duplicate.
Quantification of secreted growth
factors
Comprehensive quantification of growth factors
secreted by the cells treated with EVs was performed using
multiplex immunoassays. A total of 5x105 Hep3B, Huh7 and
differentiated THP-1 cells each were seeded in a 60-mm culture dish
and treated with 2x108 EV particles daily for 3 days and
the supernatant was subsequently collected. Growth factors were
measured using multiplex immunoassays with a Growth Factor 11-Plex
Human ProcartaPlex panel (Invitrogen; Thermo Fisher Scientific,
Inc.). Epidermal growth factor (EGF) was quantified in the
supernatant with a human EGF ELISA kit (cat. no. DEG00; R&D
Systems, Inc.) according to manufacturer's protocol.
RNA extraction and reverse
transcription-quantitative (RT-q)PCR
EGF mRNA expression in differentiated THP-1 cells
was analyzed using RT-qPCR. The total RNA was extracted from cells
using SuperScript III First-strand Synthesis system (cat. no.
18080051 Thermo Fisher Scientific, Inc.), and cDNA synthesis was
performed for 15 min at 42˚C using 1 µg total RNA as a template, RT
primer and QuantiTect reverse transcriptase (Superscript III;
Thermo Fisher Scientific, Inc.). EGF primers were purchased from
Takara Bio, Inc. (cat. no. HA159157) but the sequences of the
primers were not disclosed. For qPCR, per a reaction, 1 U
LightCycler SYBR Green I Master mix (Roche Diagnostics) was used
and the cycling conditions were as follows: Pre-incubation for 5
min at 95˚C; followed by 30 cycles of 10 sec at 95˚C, 10 sec at
60˚C and 10 sec at 72˚C. GAPDH was used as the endogenous control.
The sequences of the GAPDH primers were: Forward,
5'-AGCCACATCGCTCAGACAC-3' and reverse, 5'-GCCCAATACGACCAAATCC-3'.
Gene expression was calculated using the
2-ΔΔCq method (30). PCR was performed in triplicate.
Cell viability assays
To evaluate the impact of EVs on proliferation of
hepatoma cells, the viability of Hep3B cells treated with EVs was
determined using an MTS assay. A total of 2.5x103 Hep3B
cells were seeded in a 96-well plate and treated with either
1x106 or 3x106 EVs daily for 3 days either
with or without differentiated THP-1 cells. Subsequently, 20 µl
CellTiter96® AQueous One Solution Reagent (Promega
Corporation) was added to each well. Following incubation for 2 h
at 37˚C, the reaction was measured using an automated plate reader
(Bio-Rad Laboratories, Inc.) at 490 nm. To evaluate the
concentration of erlotinib that could be used whilst maintaining
the viability of hepatoma cells, MTS assays were used. A total of
2.5x103 Hep3B cells were seeded in a 96-well plate and
treated with various concentrations of erlotinib for 3 days. MTS
assays were performed in at least duplicate.
Statistical analysis
Data are presented as the mean ± standard deviation
of the mean. Multiple comparisons were performed using an ANOVA
with a post-hoc Tukey's test. P<0.05 was considered to indicate
a statistically significant difference. For multiplex immunoassays
which were used as a comprehensive quantification of growth
factors, >2-fold difference in secretion was used as the
threshold of significance.
Results
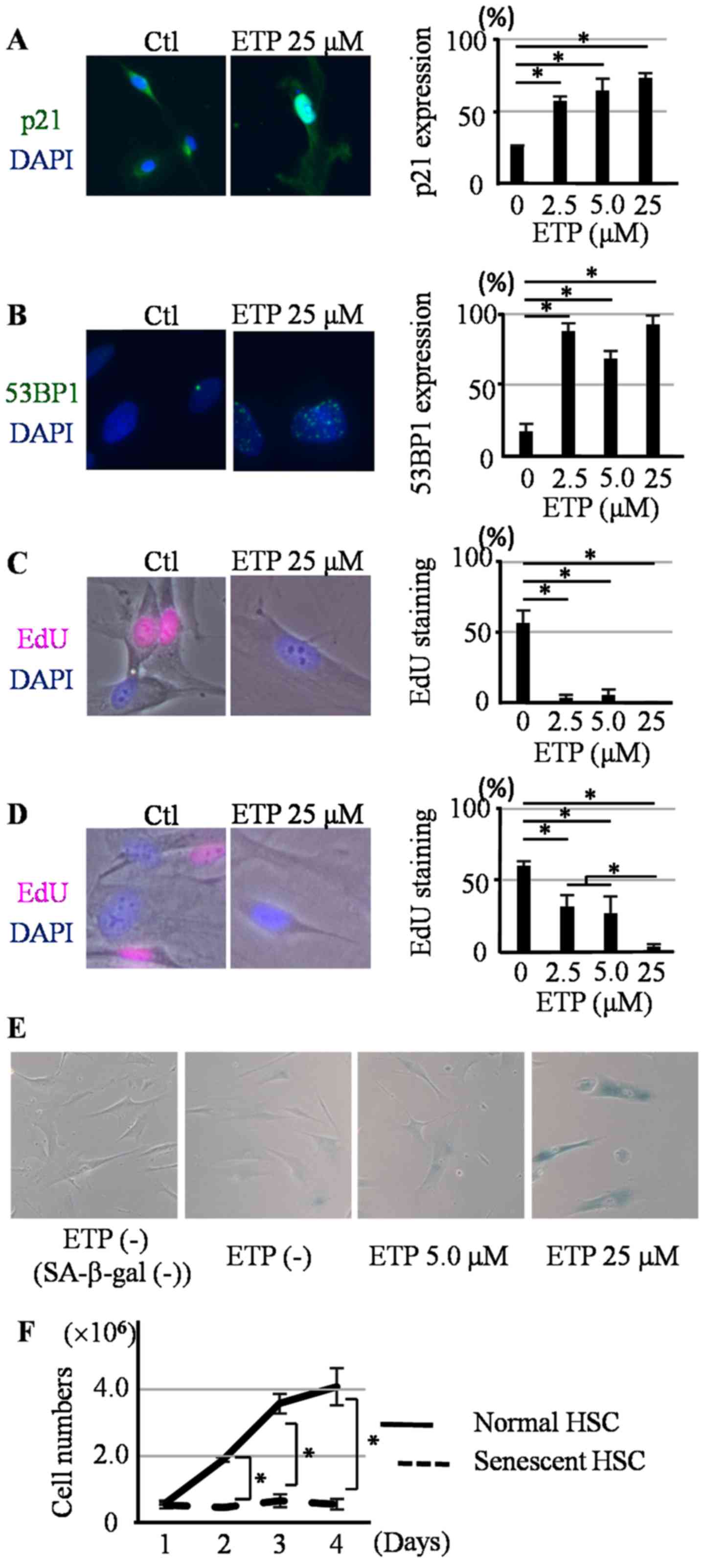
Induction of senescence in HHSteCs
with ETP treatment
HHSteCs were treated with 2.5, 5.0, 25, or 50 µM ETP
for 3 days. Alterations in morphology were observed in the treated
HHSteCs compared with the untreated cells. HHSteCs treated with 50
µM ETP died within 3 days and the number of cells was notably
decreased (Fig. S1). For the HHSteCs
treated with 2.5, 5.0 and 25 µM ETP, the expression of p21 (a cell
cycle arrest marker), 53BP1 (a DNA damage marker) foci and the
uptake of EdU were examined. p21 expression and 53BP1 foci were
significantly increased at all three ETP concentrations (Fig. 1A and B),
whereas EdU uptake decreased (Fig.
1C) compared with the control. To confirm induction of
irreversible cell cycle arrest associated with senescence, HHSteCs
treated with each of the ETP concentrations were grown in fresh
ETP-free recovery medium for 3 days. EdU uptake was still reduced
in HHSteCs treated with 25 µM ETP (Fig.
1D). In contrast, EdU uptake slightly recovered in HHSteCs
treated with 2.5 and 5.0 µM ETP. Therefore, 2.5 and 5.0 µM ETP were
inadequate for the induction of senescence in HHSteCs. Finally,
induction of senescence was confirmed in HHSteCs treated with 25 µM
ETP using SA-β-gal staining (Fig.
1E). The proliferation of HHSteCs treated with 25 µM ETP for 3
days was reduced in the recovery medium (Fig. 1F). Thus, 25 µM ETP was used for all
subsequent experiments for the induction of senescence in
HHSteCs.
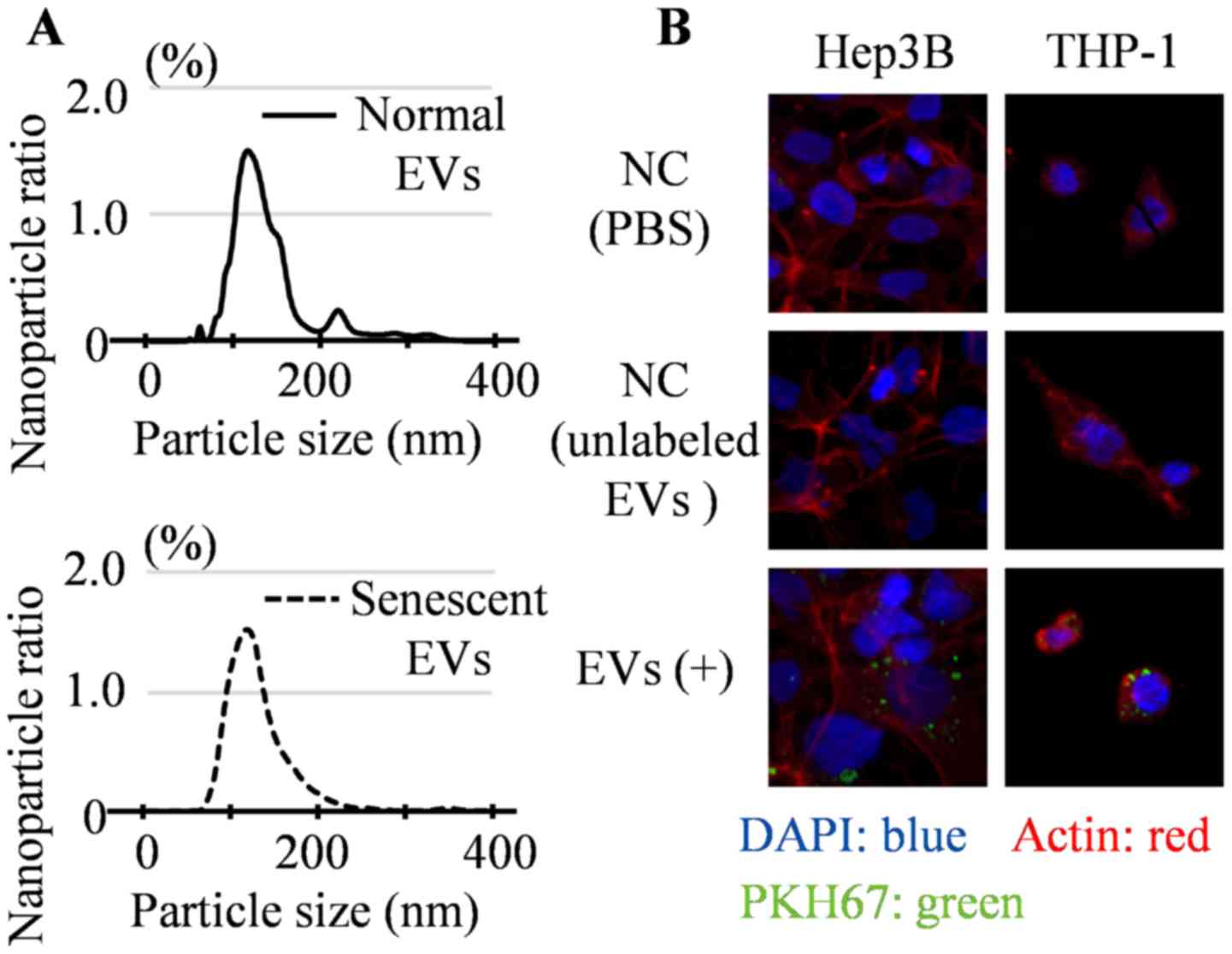
Particle size and number of senescent
HHSteC-derived EVs
After extracting the EVs by ultracentrifugation, the
particle size and number of senescent EVs were compared to those of
normal EVs using nanoparticle tracking analysis. The size of
particles were largely ~120 nm, although some particles were ~200
nm in size. Therefore, it was considered that the extracted EVs
were primarily composed of exosomes with a small number of other
small vesicles (25,26). The median particle size was 126 nm for
normal EVs and 120 nm for senescent EVs (Fig. 2A) and this difference was not
significant. To determine the number of EV particles released per
HHSteC, the area under the curve (AUC) was calculated using a cell
proliferation curve. The cumulative number of EVs in the culture
medium was associated with the cumulative number of HHSteCs,
although the quantities differed notably between the normal and
senescent HHSteC cultures. The AUC of the normal cultured HHSteCs
was 1.23x106 cells day-1, whereas that of the
senescent HHSteCs was 7.06x105 cells day-1
(data not shown). Therefore, it was estimated that there were
2.5x103 particles produced cell-1
day-1 for normal EVs and 4.2x103 particles
cell-1 day-1 for senescent EVs. Senescent
HHSteCs released ~1.7-fold more EVs per cell than normal cultured
HHSteCs, although the significance of this result could not be
analyzed statistically.
Incorporation of EVs derived from
HHSteCs into hepatoma cell lines
To confirm whether EVs secreted by HHSteCs were
incorporated into both hepatoma cells and macrophages, EVs labeled
with PKH67 were added to Hep3B cells and THP-1 cells daily for 3
days. PKH67 expression was observed in both cells (Fig. 2B), which suggests that EVs derived
from HHSteCs were incorporated into cells.
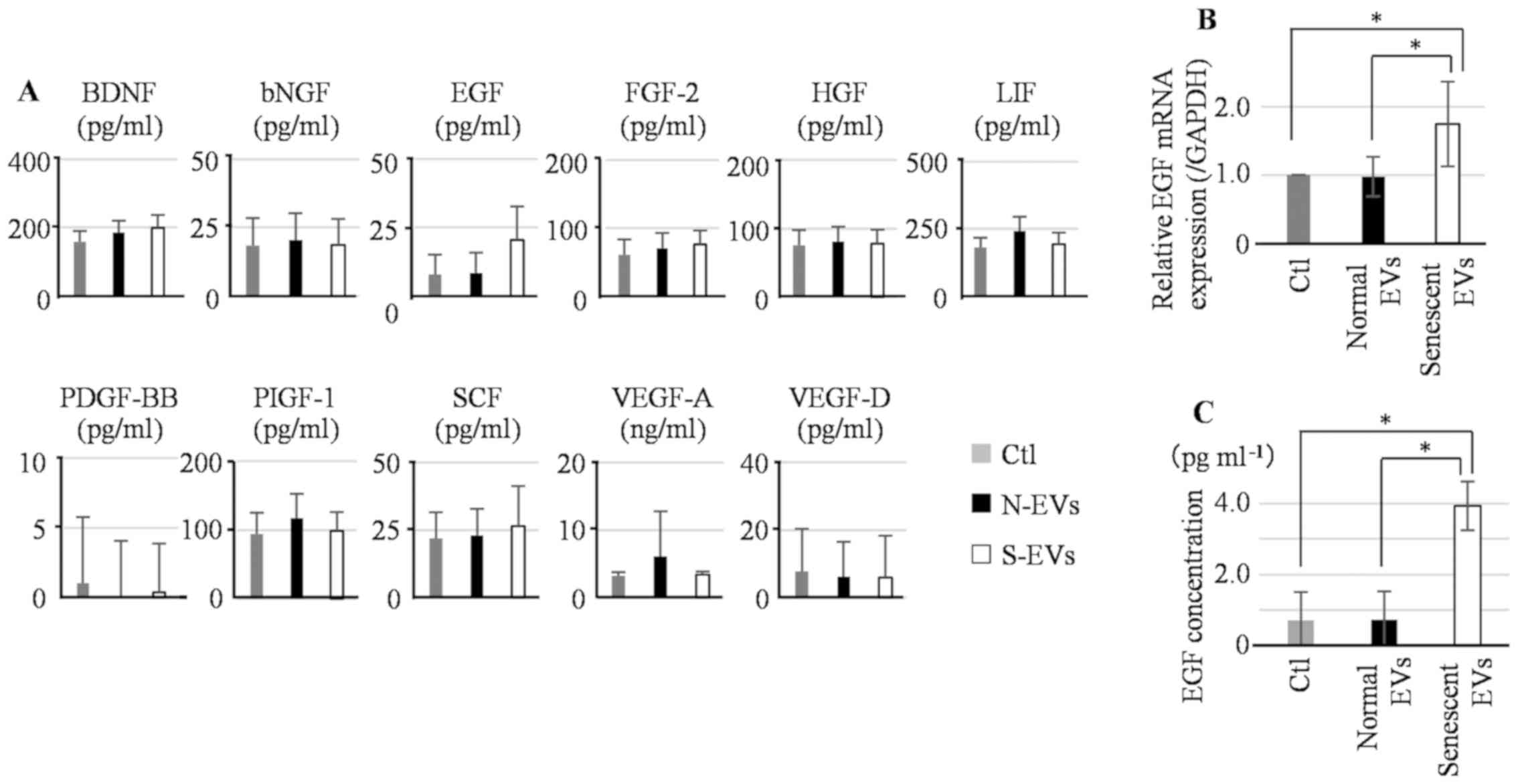
Growth factor secretion from hepatoma
cell lines and differentiated THP-1 cells treated with EVs derived
from senescent HHSteCs
To assess the effect of senescent EVs on growth
factor secretion from hepatoma cells, Hep3B and Huh7 cells were
treated with either normal or senescent EVs daily for 3 days and
the secretion of growth factors into the supernatant was measured
using a panel of multiplex immunoassays. The difference in
secretion of growth factors between normal and senescent EV
treatments was <2-fold (Fig. S2).
Several studies have shown that TAMs participate in hepatoma cell
proliferation via changes in cytokine expression levels (31,32). Thus,
the effect of senescent EVs on growth factor secretion from THP-1
cells was assessed. Differentiated THP-1 cells were treated daily
for 3 days with either normal or senescent EVs and growth factor
secretion was measured. THP-1 cells treated with senescent EVs
secreted significantly more EGF compared with those treated with
normal EVs and the change was >2-fold (Fig. 3A).
EGF expression in THP-1 cells treated with EVs was
further assessed by RT-qPCR and EGF protein-specific ELISA. The
levels of EGF mRNA expression in THP-1 cells treated with senescent
EVs was significantly higher compared with both the THP-1 cells
treated with normal EVs and control (Fig.
3B). To further confirm the results obtained by multiplex
immunoassays, EGF secretion was measured using ELISA. EGF secretion
from THP-1 cells treated with senescent EVs was also increased
compared with THP-1 cells treated with normal EVs and the control
(Fig. 3C).
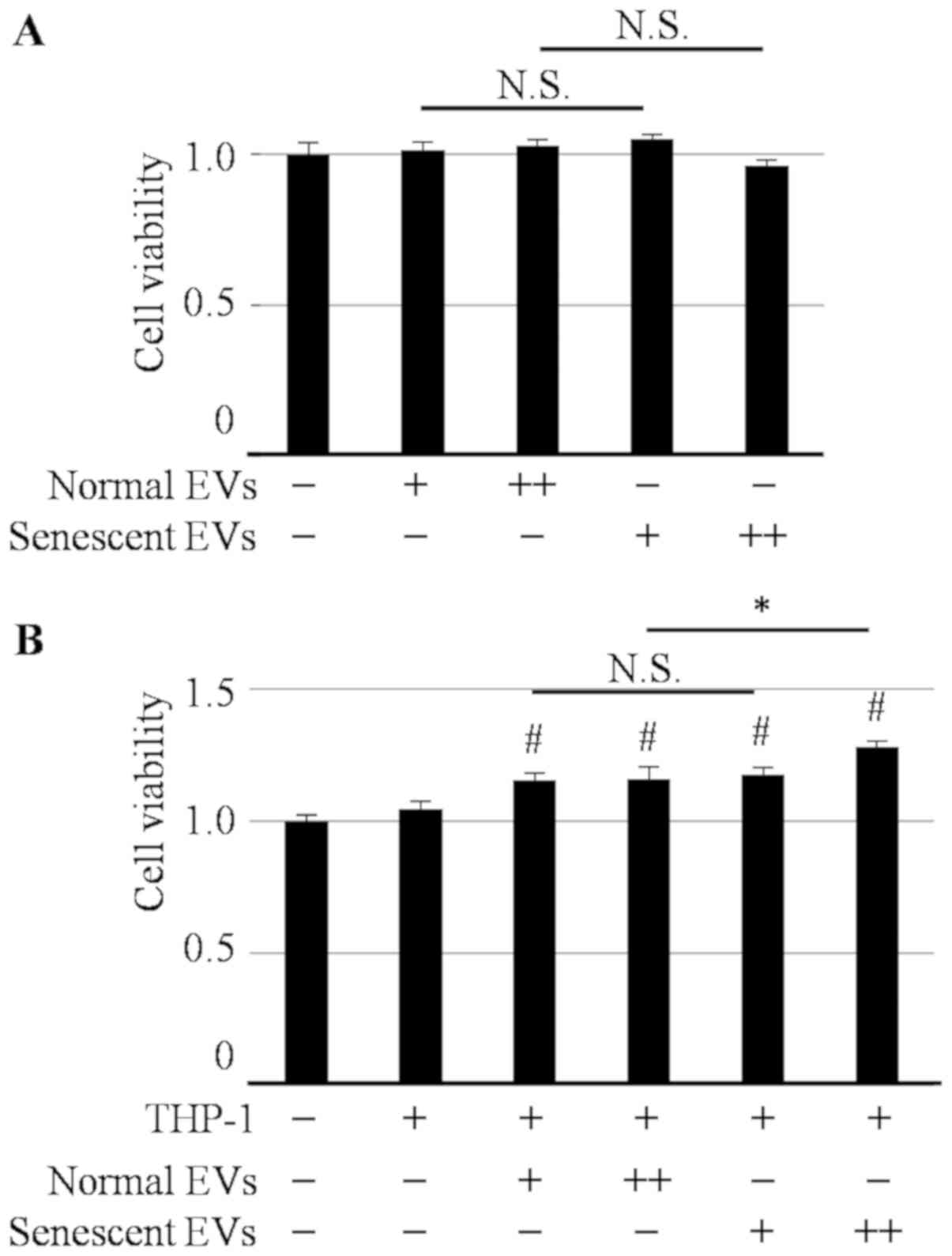
Effect of EVs derived from senescent
HHSteCs on the proliferation of Hep3B cells
The effect of EVs derived from senescent HHSteCs on
hepatoma cell viability was assessed. As shown in Fig. 4A, neither treatment with normal nor
senescent EVs affected the proliferation of Hep3B cells. However,
both EV treatments significantly increased the viability of Hep3B
cells when co-cultured with differentiated THP-1 cells. Notably,
this effect was significantly greater with senescent EVs compared
with normal EVs (Fig. 4B). To
validate the effect of EGF secretion from THP-1 cells on hepatoma
cell lines, the cell viability of hepatoma cells in the presence or
absence of erlotinib, an EGFR tyrosine kinase inhibitor, was
determined. The concentration of erlotinib was set to 2.5 µM to
maintain Hep3B cell viability at >80% (Fig. 5A). As shown in Fig. 5B, senescent EVs were more effective at
enhancing Hep3B cell viability compared with EVs in the absence of
erlotinib; however, this enhancing effect was inhibited in the
presence of erlotinib. This suggests that the effect of senescent
EVs on the proliferation of hepatoma cells co-cultured with THP-1
cells was dependent on EGF secreted from THP-1 cells.
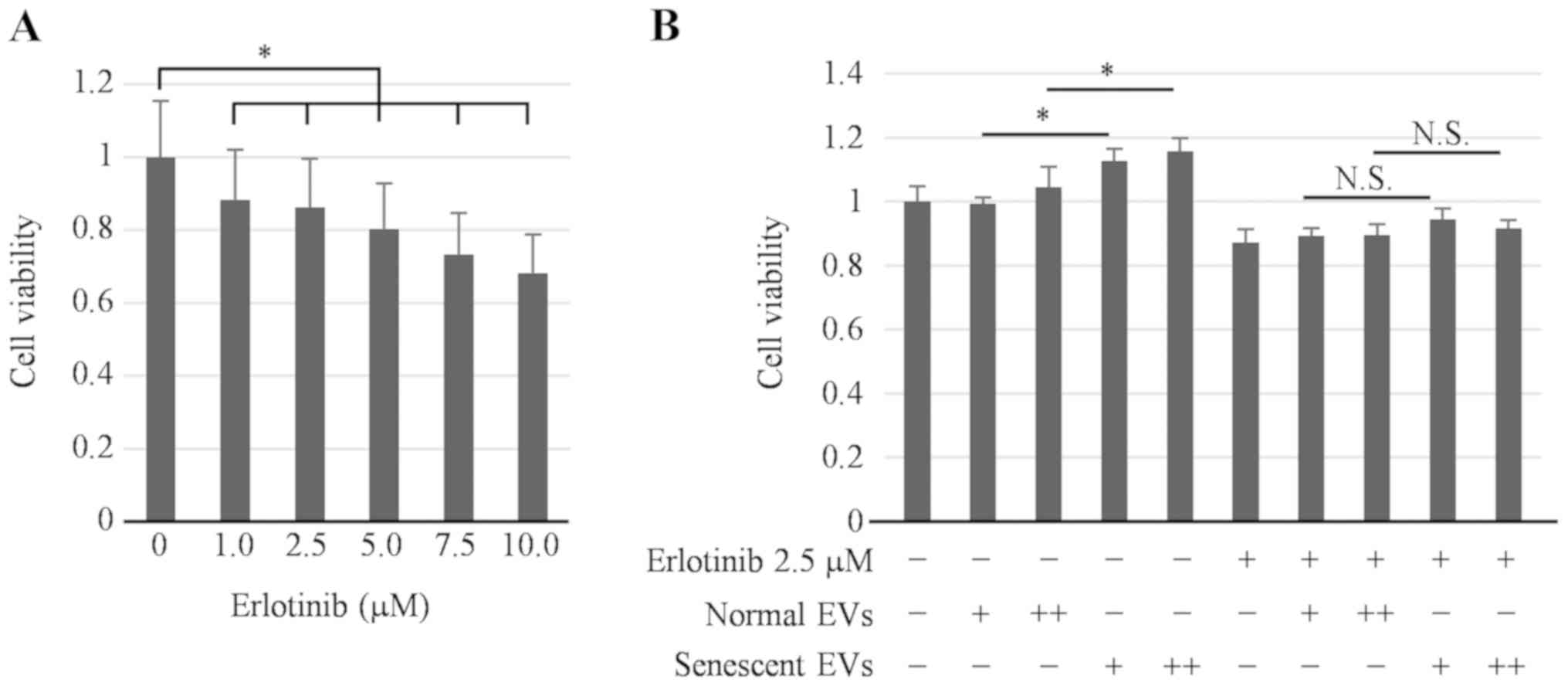 | Figure 5.Effect of EVs derived from senescent
HHSteCs co-cultured with THP-1 cells on the proliferation of
hepatoma cells in the presence of erlotinib, an epidermal growth
factor receptor inhibitor. To determine the involvement of EGF
secreted from senescent HHSteCs in hepatoma cell proliferation upon
co-culture with THP-1 cells, the viability of Hep3B cells
co-cultured with THP-1 cells was evaluated in the absence or
presence of erlotinib using MTS assays. (A) The effect of erlotinib
on the inhibition of cell viability was examined at each
concentration. Cell viability was maintained at >80% with 1.0
and 2.5 µM erlotinib treatment. (B) Cells were treated with two
different quantities of EV particles daily for 3 days. Senescent
EVs were more effective at enhancing Hep3B cell viability compared
with normal EVs in the absence of erlotinib; however this effect
was abrogated in the presence of erlotinib. *P<0.05.
N.S., not significant; EV, extracellular vesicle; HHSteCs, human
hepatic stellate cells; EGF, epidermal growth factor; +,
1x106 added daily; ++, 3x106 added daily;
normal EVs, EVs derived from normal human hepatic stellate cells;
senescent EVs, EVs derived from senescent human hepatic stellate
cells. |
Discussion
In the present study, it was demonstrated that
senescence could be induced in HHSteCs by treatment with ETP, and
that senescent HHSteCs released increased quantities of EV
particles compared with normal HHSteCs. EVs derived from senescent
HHSteCs resulted in increased EGF expression levels in THP-1 cells
compared with EVs derived from normal HHSteCs, which promoted
hepatoma cell viability. Therefore, EVs derived from senescent HSCs
may create a more conducive tumor microenvironment for
proliferation of hepatoma cells.
Senescent HSCs affect their surrounding cells,
inducing alterations to the hepatic microenvironment. Krizhanovsky
et al (33) examined the role
of senescent HSCs in the hepatic microenvironment in detail. They
showed that hepatic fibrosis develops in the absence of HSC
senescence induction and that senescent HSCs promote the activity
of NK cells to eliminate the activated HSCs which cause progression
of hepatic fibrosis. However, they also hypothesized that senescent
HSCs accumulate in the presence of sustainable and excessive liver
damage, caused by viruses or hepatic steatosis, beyond the means of
a physiological immune response. In addition, Yoshimoto et
al (23) showed that senescent
HSCs promote progression of liver cancer. At present, therapy
targeting senescent cells has also been studied. Ogrodnik et
al (34) reported that a
combination of the selective senolytic agents dasatinib and
quercetin reduced hepatic steatosis (34). Senescent cells are now regarded as
attractive targets for novel therapeutic strategies. However, it is
necessary to further elucidate their contributions to the tumor
microenvironment.
To date, there have been numerous reports on the
effects of EVs secreted by hepatocytes on stellate cells (35-37);
however, to the best of our knowledge, there are no report on the
effects of EVs secreted by stellate cells on hepatocytes or
surrounding cells. In addition, it has been reported that the
efficiency of EV uptake varies depending on the type of recipient
cells (38). Accordingly, EV uptake
by hepatoma cells and macrophage cells was initially determined and
confirmed, and EVs affected the secretion of cytokines. Li et
al (39) reported that EVs
containing oncomiRs secreted from hepatoma cells are incorporated
into HSCs, and that EVs secreted from HSCs promote HCC progression
as a positive feedback mechanism. Furthermore, Wan et al
(40) showed that exosomes secreted
from activated HSCs are taken up by Kupffer cells, as well as the
HSCs themselves, which increases the expression of GLUT1 and PKM2.
Therefore, EVs secreted by HSCs are actively taken up by
surrounding cells including hepatocytes and macrophage cells with
functional effects on the hepatic microenvironment.
Several studies have shown that the impact of EVs on
surrounding cells varies by EV content and is regulated by cell
conditions (41,42). Regarding the effect of EVs derived
from senescent cells on cancer cells, Takasugi et al
(43) showed that EVs derived from
senescent cells are absorbed by several breast cancer cell lines
via EphA2, which resulted in SASP factor-like cancer progression.
Similarly, in the present study, it was shown that EVs derived from
senescent HSCs increased secretion of EGF from THP-1 cells, which
in-turn promoted the proliferation of the hepatoma cells
co-cultured with these cells. EVs derived from senescent HSCs may
thus indirectly contribute to the formation of an environment
conducive to the development of HCC.
There are some limitations to the present study. The
primary limitation is that these results are based entirely on
in vitro assays. Additionally, the number of EVs used for
treatment was based on an approximate prediction method. The ratio
of stellate cells in vivo is equivalent to ~10% of the
number of parenchymal cells (21,44). Thus,
the same ratio was used in the present study. As described in the
results section, a single HHSteC secreted 2,500-4,000 particles per
day. Therefore, the number of EVs used for treatment was set to
~400x the number of hepatoma cells. However, it is unclear the
number of EVs secreted from HSCs in the liver microenvironment
in vivo. It is also possible that various changes to the
liver state may result in changes of the number of EVs secreted
from HSCs. In addition, as the experiments in the present study
were only performed in vitro, the conditions are
considerably different from in vivo where immune cells are
also involved. As reported by Krizhanovsky et al (33), senescent HSCs can modulate the immune
system in vivo. Therefore, immunomodulatory signals from EVs
derived from senescent HSCs should be explored in the future.
However, the findings of the present study highlight the role of
EVs derived from senescent HSCs in promoting tumor growth, similar
to that observed with the SASP. Therefore, in our future studies,
the effect of EVs derived from senescent HSCs in vivo will
be assessed. In conclusion, senescent HSCs released increased
quantities of EVs compared with normal HSCs. Similar to, SASP
factors, EVs from senescent HSCs promote HCC development by
upregulating EGF production in macrophages.
Supplementary Material
Alterations in the morphology of
HHSteCs following treatment with different concentrations of ETP.
Alterations in morphology of HHSteCs treated with 2.5, 5.0, 25 or
50 µM ETP was observed daily for 3 days by microscopy.
Magnification, x10. In HHSteCs, treatment with 2.5, 5.0 and 25
µM ETP induced notable alterations in morphology compared
with the untreated cells. Additionally, 50 µM ETP appeared
to kill HHSteCs, as the number of cells appeared to decrease
gradually. ETP, etoposide; HHSteCs, human hepatic stellate
cells.
Quantification of a panel of growth
factors secreted by hepatoma cells treated with EVs. Comprehensive
quantification of growth factors secreted by (A) Hep3B cells and
(B) Huh7 cells treated with EVs, performed using multiplex
immunoassays. There were no differences in secretion of factors
>2-fold between treatment with the normal and senescent EV. EV,
extracellular vesicle; normal EVs, EVs derived from normal human
hepatic stellate cells; senescent EVs, EVs derived from senescent
human hepatic stellate cells.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
All authors have contributed to this work and read
and approved the manuscript. YM and SM designed the study,
performed the experiments, wrote the article and interpreted the
data. YK, SN, RS, MH and HS performed the experiments. TH, HM, NT
and KN interpreted and analyzed the data. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Forner A, Reig M and Bruix J:
Hepatocellular carcinoma. Lancet. 391:1301–1314. 2018. View Article : Google Scholar
|
|
2
|
Baecker A, Liu X, La Vecchia C and Zhang
ZF: Worldwide incidence of hepatocellular carcinoma cases
attributable to major risk factors. Eur J Cancer Prev. 27:205–212.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Katoh M: FGFR inhibitors: Effects on
cancer cells, tumor microenvironment and whole-body homeostasis
(Review). Int J Mol Med. 38:3–15. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Son B, Lee S, Youn H, Kim E, Kim W and
Youn B: The role of tumor microenvironment in therapeutic
resistance. Oncotarget. 8:3933–3945. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bissell MJ and Hines WC: Why don't we get
more cancer? A proposed role of the microenvironment in restraining
cancer progression. Nat Med. 17:320–329. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Maia J, Caja S, Strano Moraes MC, Couto N
and Costa-Silva B: Exosome-based cell-cell communication in the
tumor microenvironment. Front Cell Dev Biol. 6(18)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rani B, Cao Y, Malfettone A, Tomuleasa C,
Fabregat I and Giannelli G: Role of the tissue microenvironment as
a therapeutic target in hepatocellular carcinoma. World J
Gastroenterol. 20:4128–4140. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tahmasebi Birgani M and Carloni V: Tumor
microenvironment, a paradigm in hepatocellular carcinoma
progression and therapy. Int J Mol Sci. 18(E405)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Forbes SJ and Parola M: Liver fibrogenic
cells. Best Pract Res Clin Gastroenterol. 25:207–217.
2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kang N, Gores GJ and Shah VH: Hepatic
stellate cells: Partners in crime for liver metastases? Hepatology.
54:707–713. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ji J, Eggert T, Budhu A, Forgues M, Takai
A, Dang H, Ye Q, Lee JS, Kim JH, Greten TF and Wang XW: Hepatic
stellate cell and monocyte interaction contributes to poor
prognosis in hepatocellular carcinoma. Hepatology. 62:481–495.
2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu Y and Cao X: The origin and function
of tumor-associated macrophages. Cell Mol Immunol. 12:1–4.
2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Qian BZ and Pollard JW: Macrophage
diversity enhances tumor progression and metastasis. Cell.
141:39–51. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Noy R and Pollard JW: Tumor-associated
macrophages: From mechanisms to therapy. Immunity. 41:49–61.
2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Brown JM, Recht L and Strober S: The
promise of targeting macrophages in cancer therapy. Clin Cancer
Res. 23:3241–3250. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Dulai PS, Singh S, Patel J, Soni M, Prokop
LJ, Younossi Z, Sebastiani G, Ekstedt M, Hagstrom H, Nasr P, et al:
Increased risk of mortality by fibrosis stage in nonalcoholic fatty
liver disease: Systematic review and meta-analysis. Hepatology.
65:1557–1565. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Liaw YF and Chu CM: Hepatitis B virus
infection. Lancet. 373:582–592. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Fabregat I and Caballero-Diaz D:
Transforming growth factor-β-induced cell plasticity in liver
fibrosis and hepatocarcinogenesis. Front Oncol.
8(357)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fabregat I, Moreno-Caceres J, Sanchez A,
Dooley S, Dewidar B, Giannelli G and Ten Dijke P: IT-LIVER
Consortium: TGF-β signalling and liver disease. FEBS J.
283:2219–2232. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yin C, Evason KJ, Asahina K and Stainier
DY: Hepatic stellate cells in liver development, regeneration, and
cancer. J Clin Invest. 123:1902–1910. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Thompson AI, Conroy KP and Henderson NC:
Hepatic stellate cells: Central modulators of hepatic
carcinogenesis. BMC Gastroenterol. 15(63)2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yoshimoto S, Loo TM, Atarashi K, Kanda H,
Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et
al: Obesity-induced gut microbial metabolite promotes liver cancer
through senescence secretome. Nature. 499:97–101. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wendler F, Favicchio R, Simon T,
Alifrangis C, Stebbing J and Giamas G: Extracellular vesicles swarm
the cancer microenvironment: From tumor-stroma communication to
drug intervention. Oncogene. 36:877–884. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Borges FT, Reis LA and Schor N:
Extracellular vesicles: Structure, function, and potential clinical
uses in renal diseases. Braz J Med Biol Res. 46:824–830.
2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Raposo G and Stoorvogel W: Extracellular
vesicles: Exosomes, microvesicles, and friends. J Cell Biol.
200:373–383. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Akers JC, Gonda D, Kim R, Carter BS and
Chen CC: Biogenesis of extracellular vesicles (EV): Exosomes,
microvesicles, retrovirus-like vesicles, and apoptotic bodies. J
Neurooncol. 113:1–11. 2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Cheng L, Sharples RA, Scicluna BJ and Hill
AF: Exosomes provide a protective and enriched source of miRNA for
biomarker profiling compared to intracellular and cell-free blood.
J Extracell Vesicles. 3:2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cai S, Cheng X, Pan X and Li J: Emerging
role of exosomes in liver physiology and pathology. Hepatol Res.
47:194–203. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Vansaun MN, Mendonsa AM and Lee Gorden D:
Hepatocellular proliferation correlates with inflammatory cell and
cytokine changes in a murine model of nonalchoholic fatty liver
disease. PLoS One. 8(e73054)2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Shirabe K, Mano Y, Muto J, Matono R,
Motomura T, Toshima T, Takeishi K, Uchiyama H, Yoshizumi T,
Taketomi A, et al: Role of tumor-associated macrophages in the
progression of hepatocellular carcinoma. Surg Today. 42:1–7.
2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Krizhanovsky V, Yon M, Dickins RA, Hearn
S, Simon J, Miething C, Yee H, Zender L and Lowe SW: Senescence of
activated stellate cells limits liver fibrosis. Cell. 134:657–667.
2008.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ogrodnik M, Miwa S, Tchkonia T, Tiniakos
D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, et al:
Cellular senescence drives age-dependent hepatic steatosis. Nat
Commun. 8(15691)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Povero D, Panera N, Eguchi A, Johnson CD,
Papouchado BG, de Araujo Horcel L, Pinatel EM, Alisi A, Nobili V
and Feldstein AE: Lipid-induced hepatocyte-derived extracellular
vesicles regulate hepatic stellate cell via microRNAs targeting
PPAR-γ. Cell Mol Gastroenterol Hepatol. 1:646–663 e4.
2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Devhare PB, Sasaki R, Shrivastava S, Di
Bisceglie AM, Ray R and Ray RB: Exosome-mediated intercellular
communication between hepatitis C virus-infected hepatocytes and
hepatic stellate cells. J Virol. 91:e02225–e02216. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Royo F, Schlangen K, Palomo L, Gonzalez E,
Conde-Vancells J, Berisa A, Aransay AM and Falcon-Perez JM:
Transcriptome of extracellular vesicles released by hepatocytes.
PLoS One. 8(e68693)2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Feng D, Zhao WL, Ye YY, Bai XC, Liu RQ,
Chang LF, Zhou Q and Sui SF: Cellular internalization of exosomes
occurs through phagocytosis. Traffic. 11:675–687. 2010.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Li J, Yan Y, Ang L, Li X, Liu C, Sun B,
Lin X, Peng Z, Zhang X, Zhang Q, et al: Extracellular
vesicles-derived oncomirs mediate communication between cancer
cells and cancer-associated hepatic stellate cells in
hepatocellular carcinoma microenvironment. Carcinogenesis: May 29,
2019 (Epub ahead of print). doi: 10.1093/carcin/bgz096.
|
|
40
|
Wan L, Xia T, Du Y, Liu J, Xie Y, Zhang Y,
Guan F, Wu J, Wang X and Shi C: Exosomes from activated hepatic
stellate cells contain GLUT1 and PKM2: A role for exosomes in
metabolic switch of liver nonparenchymal cells. FASEB J.
33:8530–8542. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Ota Y, Takahashi K, Otake S, Tamaki Y,
Okada M, Aso K, Makino Y, Fujii S, Ota T and Haneda M:
Extracellular vesicle-encapsulated miR-30e suppresses
cholangiocarcinoma cell invasion and migration via inhibiting
epithelial-mesenchymal transition. Oncotarget. 9:16400–16417.
2018.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Kogure T, Yan IK, Lin WL and Patel T:
Extracellular vesicle-mediated transfer of a novel long noncoding
RNA TUC339: A mechanism of intercellular signaling in human
hepatocellular cancer. Genes Cancer. 4:261–272. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Takasugi M, Okada R, Takahashi A, Virya
Chen D, Watanabe S and Hara E: Small extracellular vesicles
secreted from senescent cells promote cancer cell proliferation
through EphA2. Nat Commun. 8(15729)2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Friedman SL: Hepatic stellate cells:
Protean, multifunctional, and enigmatic cells of the liver. Physiol
Rev. 88:125–172. 2008.PubMed/NCBI View Article : Google Scholar
|