Introduction
Parkinson's disease (PD) is one of the most common
neurodegenerative disorders of movement in the aged populations
worldwide. PD is characterized pathologically by the loss of
dopaminergic neurons in the substantia nigra, which results in a
reduction of dopamine, a vital neurotransmitter affecting movement
and balance. Whilst the exact cause of PD is generally unknown, the
development and progression of the disease is considered to be
associated with environmental and genetic factors (1). In total, ~15% of patients have a family
history of PD, and 5-10% of cases are caused by pathogenic
mutations in single genes with monogenic Mendelian inheritance
(2). Mutations in numerous genes,
including SNCA, PARK2 (coding for parkin), DJ-1, PINK1,
LRRK2 and VPS35, are directly linked to PD, whereas
variants in other genes, such as GBA, have been shown to be
strong risk factors for PD (3).
Although extensive research has focused on pathogenic gene
mutations or variants, it is not fully understood how those genetic
changes cause PD or influence the risk of developing this disorder
in the elderly population (4,5).
Parkin is an E3 ubiquitin ligase that serves a
critical role in targeting proteins for degradation. It is also
involved in the clearance of damaged mitochondria via autophagy and
proteasomal mechanisms. Parkin is encoded by the PARK2
(PRKN) gene, which is located on chromosome 6q25.2-q27, and
spans >500 kb. It contains 12 exons, which translate to a
protein of 465 amino acids (6).
PARK2 variants appear to be responsible for PD pathogenesis,
which was first reported in Japanese families in 1998(6). According to the Human Gene Mutation
Database, >300 pathogenic mutations have been identified in the
PARK2 gene (hgmd.cf.ac.uk/ac/gene.php?gene=PRKN; accessed July
2020), most of which are gross deletions, followed by missense
mutations, gross insertions, small deletions, splicing defects and
small insertions. Although the mechanism by which loss of parkin
function results in dopaminergic cell death in PD is unclear, it is
considered to involve the disruption of mitochondrial quality
control (7).
Glucocerebrosidase (GCase; Enzyme Commission
3.2.1.45), also known as acid-β glucosidase (GBA) and
glucosylceramidase, is a lysosomal enzyme that is responsible for
the hydrolysis of glucocerebroside, an intermediate in the
glycolipid metabolism and an abundant sphingolipid present in the
plasma membranes. It is encoded by the GBA gene, which is
located on chromosome 1q21, spans ~7.6 kb and contains 11 exons.
Mutations in the GBA gene cause Gaucher's disease (GD), an
autosomal recessive lysosomal storage disease that is characterized
by the accumulation of glucocerebrosides in macrophages referred to
as ‘Gaucher cells’. There are three clinical subtypes of GD that
have been recognized: Type 1 is non-neuronopathic, whereas types 2
and 3 are acute and chronic neuronopathic, respectively. Based on
the Human Gene Mutation Database, >400 mutations have been
reported in the GBA gene (hgmd.cf.ac.uk/ac/gene.php?gene=GBA; accessed July
2020). The majority of these mutations are missense, followed by
small deletions, splicing defects, complex rearrangements and small
insertions. Of note, a highly homologous pseudogene (GBAP)
is located ~16 kb downstream from the GBA gene and shares
96% sequence identity to the functional GBA gene (8). The presence of this pseudogene at the
same locus causes recombination events between the GBA and
GBAP genes, thus resulting in several different mutations in
GD (9,10).
Several lines of evidence suggest that GBA
homozygous or heterozygous variants are associated with an
increased risk of developing PD. A study on brain samples from 57
subjects clinically diagnosed with PD demonstrated that GBA
variants were present in 12 cases (21%), and these were more
frequent amongst younger subjects (11). Another study on blood samples from 99
Ashkenazi Jewish patients with PD showed that 6 GBA variants
were identified in 31 cases (31%), and that patients carrying the
GBA variants were younger than those who were not carriers
(12). Furthermore, a multicentre
analysis from 16 centres reported that, amongst the GBA
variants of >5,000 patients with PD, 2 known GBA
variants, L444P and N370S, were commonly associated with PD in all
ethnic groups (13). However, each
population also had unique variants (10,12,14,15). In
Thailand, heterozygous GBA variants were reported in 24 out
of 480 patients with PD (5%), including L444P (c.1448T>C),
IVS10-9_10GT>AG, P428S (c.1399C>T), N386K (c.1275C>A),
V398fsX404 (c.1309delG), IVS2+1G>A and IVS9+3G>C (10). Previous studies on various
populations suggested that 5-10% of patients with PD had GBA
variants, although the percentage was much greater in the Ashkenazi
population, whereas the frequency of GBA variants in the control
(non-PD) population was as low as 0.5% (10,16).
Proteomics is a large-scale study of proteins
expressed in cells, mostly detected using a mass spectrometer.
Proteomic findings have been widely used to identify and quantify
proteins involved in the research of drug treatments, biomarkers
and diseases, including PD. For example, a previous study reported
a proteomic approach using 2D gel electrophoresis and tandem mass
spectrometry (MS/MS) to explore the protein expression pattern in
primary skin fibroblasts of patients with PD (17). Another study used non-gel based
proteomics analysis to investigate proteomes in the cerebrospinal
fluid of patients with PD (18).
Furthermore, proteomics analysis has been applied for primary
screening and detection of individuals with neurodegenerative
diseases, and for distinguishing PD from other neurodegenerative
diseases (19). Proteomics,
therefore, is a powerful technique for identification and
quantification of proteins, as well as for protein profiling and
identification of biomarkers of diseases.
Primary skin fibroblasts reflect genetic changes in
patients, and are useful models to study PD (20). Previously, skin fibroblasts from
patients with PD with heterozygous GBA and PARK2
variants, as well as from healthy controls were analysed, and it
was observed that the GBA-PD group showed slightly lower activities
of the complexes II, IV and V of the mitochondrial respiratory
chain than the PARK2-PD group (21).
The present study investigated the GCase activity in primary skin
fibroblasts of 4 patients with PD, including two heterozygous
GBA variants and two heterozygous PARK2 variants, and
this was compared with 4 heathy controls. MS and label-free
quantitative proteomic analysis were performed to identify
potential target proteins associated with the disease.
Materials and methods
Patients and participants
Clinical samples, including primary skin fibroblasts
and peripheral blood samples, were used in this study. Primary skin
fibroblasts were obtained from 4 patients with PD (median age, 46
years; age range, 41-57 years) and 4 healthy age-matched controls
(median age, 45.5 years; age range, 45-53 years). These included
two patients with PD carrying different heterozygous GBA
variants, including c.1309delG and IVS2+1G>A, and two patients
with PD carrying different heterozygous PARK2 variants,
including c.2T>C and exon 8 deletion (exon8del). All
participants were screened and were free of common LRRK2
mutations as well as TBP, SCNA, FBX07 and GCH1
mutations. The age, sex, genotypic and phenotypic characterization
of the investigated individuals are shown in Table I. Written informed consent was
obtained from each participant, and the study protocol approved by
the Institutional Review Board of Ramathibodi Hospital (Bangkok,
Thailand; approval no. ID03-54-22).
 | Table IGenotypic and phenotypic
characterization of the investigated individuals. |
Table I
Genotypic and phenotypic
characterization of the investigated individuals.
| | | | | Mutations | | |
|---|
| Groups | Codes | Sex | Ages of onset,
years | DNA | Protein | Clinical
status | GCase activity,
µmol/h/mg protein, mean ± SD |
|---|
| PD with
heterozygous | PD-GBA-1 | F | 57 | c.1309delG | pV398fsX404
(incomplete protein) | Affected | 127±13 |
| GBA
variants | PD-GBA-2 | F | 49 | IVS2+1G>A | Splicing errors
(incomplete protein) | Affected | 157±7 |
| PD with
heterozygous | PD-PARK2-1 | F | 43 | c.2T>C | pM1T (complete
absence) | Affected | 240±10 |
| PARK2
variants | PD-PARK2-2 | F | 41 | Exon 8
deletion | Incomplete
protein | Affected | 169±10 |
| Controls | Control-1 | F | 45 | None | - | Unaffected | 218±20 |
| | Control-2 | M | 53 | None | - | Unaffected | 242±16 |
| | Control-3 | F | 46 | None | - | Unaffected | 186±16 |
| | Control-4 | F | 45 | None | - | Unaffected | 208±10 |
Preparation and culturing of skin
fibroblasts
Skin samples were obtained from the dorsal region of
the inner upper arm of each participant, maintained in tubes with
DMEM, stored at 4˚C and processed within 4 h. Fibroblast harvesting
was performed by explant, isolating the dermis from the epidermis
with scalpels and scissors. Passaged cells were cultured in DMEM
containing 10% FBS and 1% penicillin/streptomycin. All culture
media and reagents were purchased from Thermo Fisher Scientific,
Inc. Skin fibroblast cultures were maintained in a humidified
incubator with 95% air and 5% CO2 at 37˚C. All
experiments were performed on cells with 6-11 passages. When cell
density reached 80% confluence, the cells were washed and harvested
in 1X PBS using a cell scraper. Next, the cells were washed 2-3
times with PBS by centrifugation at 721 x g for 10 min at 4˚C. The
pelleted cells were stored at -80˚C until required for use in the
enzyme activity and protein identification assays.
GCase activity assay
GCase activity was determined using a fluorometric
assay with 4-methylumbelliferyl β-D-glucopyranoside (4-MU-β-Glc) as
a substrate, as previously described, with certain modifications
(22). All reagents were purchased
from Sigma-Aldrich (Merck KGaA). After cell thawing, the
fibroblasts were mixed with 200 µl 0.9% NaCl containing 1 mM
phenylmethylsulfonyl fluoride, and were homogenized with an
ultrasonic homogenizer (U200H; IKA Labortechnik) at 30% amplitude
and 0.2% cycle. The protein concentrations of the fibroblast
lysates were measured using a Bradford protein assay (Bio-Rad
Laboratories, Inc.) with BSA as a standard (Bio-Rad Protein assay
standard II; cat. no. 5000007, Bio-Rad Laboratories, Inc.). For the
enzyme assay, 10 µl homogenized cell lysate was incubated with 90
µl 5 mM 4-MU-β-Glc in 10 mM sodium taurocholate at 37˚C for 1 h.
After incubation, 200 µl 0.5 M
NaHCO3/Na2CO3 (pH 10.7) was added
to stop the reaction. The clear reaction was transferred into a
96-well microplate, and the fluorescence emission was measured
using a fluorescence spectrophotometer (SpectraMax M2/M2e
Multi-Mode microplate reader; Molecular Devices, LLC) with an
excitation wavelength of 365 nm and an emission wavelength of 450
nm. The GCase activity was calculated as the release of
4-methylumbelliferone (4-MU) per time divided by the quantity of
protein used. One unit was defined as the release of 1 µmole 4-MU
in 1 h per 1 mg protein.
Sample preparation for MS
Skin fibroblasts from patients with PD with
GBA and PARK2 variants as well as from healthy
subjects were grown in three independent passages, and all
fibroblast passages were harvested as pelleted cells and stored at
-80˚C. After thawing, the packed fibroblasts were resuspended in
lysis buffer containing 7 M urea, 2 M thiourea, 4% CHAPS and 1X
protease inhibitor cocktail (Sigma-Aldrich; Merck KGaA), and were
sonicated 3-4 times at 0.3 cycle and 30% amplitude, and then
incubated at 4˚C for 30 min. The cell debris were removed by
centrifugation at 13,000 x g at 4˚C for 10 min. The protein
concentrations of fibroblast lysates were determined using a
Bradford protein assay. After protein determination, equal
quantities of protein (10 µg) from each cell passage were pooled,
and the buffer was exchanged with 50 mM ammonium bicarbonate using
a Micro Bio-Spin Chromatography Column (Bio-Rad Laboratories,
Inc).
In-solution tryptic-digestion was performed as
described previously with certain modifications (23). Briefly, 10 µg protein from fibroblast
lysates (pooled healthy controls and individual patient samples)
was reduced in 10 mM DTT for 5 min at 95˚C and alkylated with 1/10
volume of 20 mM iodoacetamide, prior to incubation for 30 min in
the dark at room temperature. The alkylated proteins were
in-solution digested overnight at 37˚C by adding 1:50 (trypsin:
protein, w/w) of sequencing-grade trypsin (Promega Corporation).
The digestion reaction was stopped by adding 1% formic acid. The
tryptic-digested peptides were cleaned with HiPPR™ Detergent
Removal Resin (Thermo Fisher Scientific, Inc.) to remove the
residual reagents, and then purified using a Ziptip C18
micropipette tip (EMD Millipore). Finally, the peptides were dried
completely using a speed vacuum (Labconco) and stored at -80˚C for
further analysis.
Liquid chromatography (LC)-MS/MS
analysis
A nanoACQUITY UPLC system (Waters Corporation)
coupled with an amaZon speed Ion Trap mass spectrometer (Bruker
Corporation) was used to identify peptides/proteins, as described
previously (23). The
tryptic-digested peptides were dissolved in 0.1% formic acid in
H2O and injected into a nanoACQUITY UPLC column (1.7 µm
BEH, 75 µm x 200 mm C18, Water Corporation) at a flow
rate of 300 nl/min. The column temperature was maintained at 40˚C.
The LC gradient used 0.1% formic acid in H2O as solvent
A and 0.1% formic acid in acetonitrile as solvent B, and was
performed as follows: 1-50% solvent B for 70 min, 50-90% solvent B
for 5 min and 15 min with 90% solvent B. The eluted peptides were
analysed directly via MS/MS on an amaZon speed Ion Trap mass
spectrometer equipped with a captive-electrospray ion source. The
positive mode was used with a spray voltage of 1,300 V, and the
capillary temperature was set at 150˚C. Mass spectra were acquired
from 400 to 1,400 m/z using parameters optimized at 922 m/z with a
target of 500,000 set for ion charge control and a maximum
acquisition time of 100 msec. The scan range was 50-3,000 m/z. The
MS/MS data were processed using Bruker Compass version 1.4 (Bruker
Corporation). Each sample was analysed in triplicate by LC-MS/MS
for the normalization of the quantity of protein injected. Mascot
software version 2.4.0 was used for the identification of peptides
using the Swiss-Prot human protein database (Matrix Science, Ltd.).
The Mascot parameters were as follows: The MS/MS mass tolerance was
set at 0.6 Da; the peptide mass tolerance was set to 1.2 Da;
carbamidomethylation was set as a fixed modification; and 1 missed
cleavage was allowed. A false discovery rate threshold of 1% was
applied.
Data analysis and label-free LC-MS
quantitative profiling
Progenesis label-free LC-MS version 3.1 (Nonlinear
Dynamics, Ltd.) was used to process the raw data obtained from
LC-MS/MS and to compare the significant changes in protein
expression levels. The method was applied to quantify
peptides/proteins, as described previously, with certain
modifications (23). Briefly, two
sets of LC-MS/MS were performed. The first set involved nine runs
with three triplicates of the two different GBA variants and
one pooled healthy control. The second set involved nine runs with
three triplicates of the two different PARK2 variants and
one pooled healthy control. The chromatograms of all the samples of
each set were aligned, and the sample providing the smallest
differences in retention times and MS peaks amongst all samples was
automatically selected as the reference. The ion intensities of the
MS peaks of each sample were then normalized by those of the
established reference. The following criteria were used to filter
all the peptide data prior to exporting the MS/MS output files,
including i) only peptides presenting an ANOVA with Tukey's
post-hoc test difference between the triplicate runs of P<0.05;
ii) only non-conflicted peptides (unique peptides) were used; iii)
at least one run (from triplicate runs of each sample) was
fragmented to generate the peptide sequences for comparison with
those of other samples; and iv) at least two unique peptides with a
MASCOT score of >30 (P<0.05) were accepted for confident
protein identification. The fold changes of protein expression
levels were calculated from the mean values of all accepted
peptides of particular proteins, and were normalized by the mean
number of each protein in the healthy control group.
Heat map analysis of protein
expression levels
The MS ion intensities of each protein (average
peptide ion intensities) were normalized by those in the healthy
control group and counted as 1.00-fold. The expression levels
(normalized intensity ratio) of proteins were represented in a heat
map constructed using R version 3.3.1(24). The proteins were sorted by their
expression levels, starting from high to low, in comparison with
those of the control group. All samples were clustered with the
Euclidean distance computational method and the complete
agglomeration method (24).
Protein-protein interaction (PPI)
analysis
STRING version 11.0(25) was used to predict the potential PPIs
of proteins affected by GBA and PARK2 variants. PPI
networks were constructed by mapping the top 25 proteins with the
highest differential expression levels in patients with PD with
GBA and PARK2 variants compared with those of the
healthy control group. Gene Ontology (GO) functional enrichment
analysis (26,27) was used to identify the biological
processes in which the proteins affected by both variants are
involved. The top three biological processes with the smallest
false discovery rate (FDR) values were selected.
Confirmation of protein expression
levels
The expression levels of certain proteins were
validated by western blot analysis. These included annexin A2
(ANXA2), 60S ribosomal protein L18 (RPL18), tubulin β chain (TUBB)
and collagen α-1 chain (COL1A1). Protein lysates of pooled
fibroblasts from healthy controls and patients with PD carrying
GBA and PARK2 variants were resuspended in the same
lysis buffer, as previously described in the sample preparation for
MS. Equal protein quantities (10 µg) from four pooled healthy
controls and individual patient samples were separated by 10%
SDS-PAGE and then transferred to 0.20-µm PVDF (Pall Life Sciences).
The membranes were blocked with 3% (w/v) BSA in TBST [50 mM Tris,
pH 8.0, 150 mM NaCl and 0.1% Tween-20 (v/v)] for 1 h. Next, the
membranes were incubated at 4˚C overnight in 3% BSA/TBST with the
following antibodies: Anti-ANXA2 (cat. no. ab54771; Abcam;
1:1,000), anti-RPL18 (cat. no. ab241988; Abcam; 1:2,000), anti-TUBB
(cat. no. 2128; Cell Signaling Technology, Inc.; 1:1,000),
anti-COL1A1 (cat. no. ab138492; Abcam; 1:1,000) or anti-β-actin
(cat. no. 3700; Cell Signaling Technology, Inc.; 1:20,000). After
washing with TBST, the membranes were incubated with the
appropriate secondary antibodies conjugated with horseradish
peroxidase (cat. no. P0217, anti-rabbit and cat. no. P0260,
anti-mouse; Dako, Agilent Technologies, Inc.; 1:2,000 except for
anti-β-actin where 1:20,000 was used) at room temperature for 1 h.
After washing with TBST, the bands in the membranes were detected
by chemiluminescence using a WesternBright ECL detection kit
(Advansta, Inc.). β-actin (ACTB) was used as a protein loading
control. The immunoblot signals were detected and analysed using an
ImageQuant LAS 4000 mini system (GE Healthcare). Densitometry
analysis was performed using ImageQuant TL 1D version 7.0 (GE
Healthcare) based on the band intensity of certain proteins divided
by the band intensity of the respective ACTB, and normalized to
those of the pooled healthy control fibroblasts.
Statistical analysis
Protein expression is presented as the mean ±
standard deviation. Statistical differences in protein expression
between the patients and healthy control groups were compared using
a two-tailed unpaired t-test in Microsoft Excel® 2016
(Microsoft Corporation). P<0.05 was considered to indicate a
statistically significant difference.
Results
GCase activity is decreased in
patients with PD with heterozygous GBA variants
The average GCase activity level of the two patients
with PD carrying heterozygous GBA variants (142±21 nmol/h/mg
protein) was 34% lower compared with the healthy controls (201±36
nmol/h/mg protein) and the difference was significant (P<0.05),
whereas the patients carrying heterozygous PARK2 variants
had a mean activity (204±50 nmol/h/mg protein) close to the mean
value of the healthy controls (Table
I).
Protein alterations in patients with
PD carrying heterozygous GBA and PARK2 variants
Proteomics and label-free quantitative analysis were
performed to identify proteins that may be associated with PD. For
this purpose, the protein lysates of four healthy controls were
pooled and compared with those of the individual variants. The
first comparison set was the pool of controls (controls 1-4) and
two individual samples of patients with PD carrying heterozygous
GBA variants; PD-GBA-1 (c.1309delG) and PD-GBA-2
(IVS2+1G>A). The second set was the pool of controls (controls
1-4) and two individual samples of patients with PD carrying
heterozygous PARK2 variants; PD-PARK2-1 (c.2T>C) and
PD-PARK2-2 (exon8del).
According to the criteria used to filter all MS and
MS/MS data, there were 122 proteins in the GBA variants and
119 proteins in the PARK2 variants that could be compared
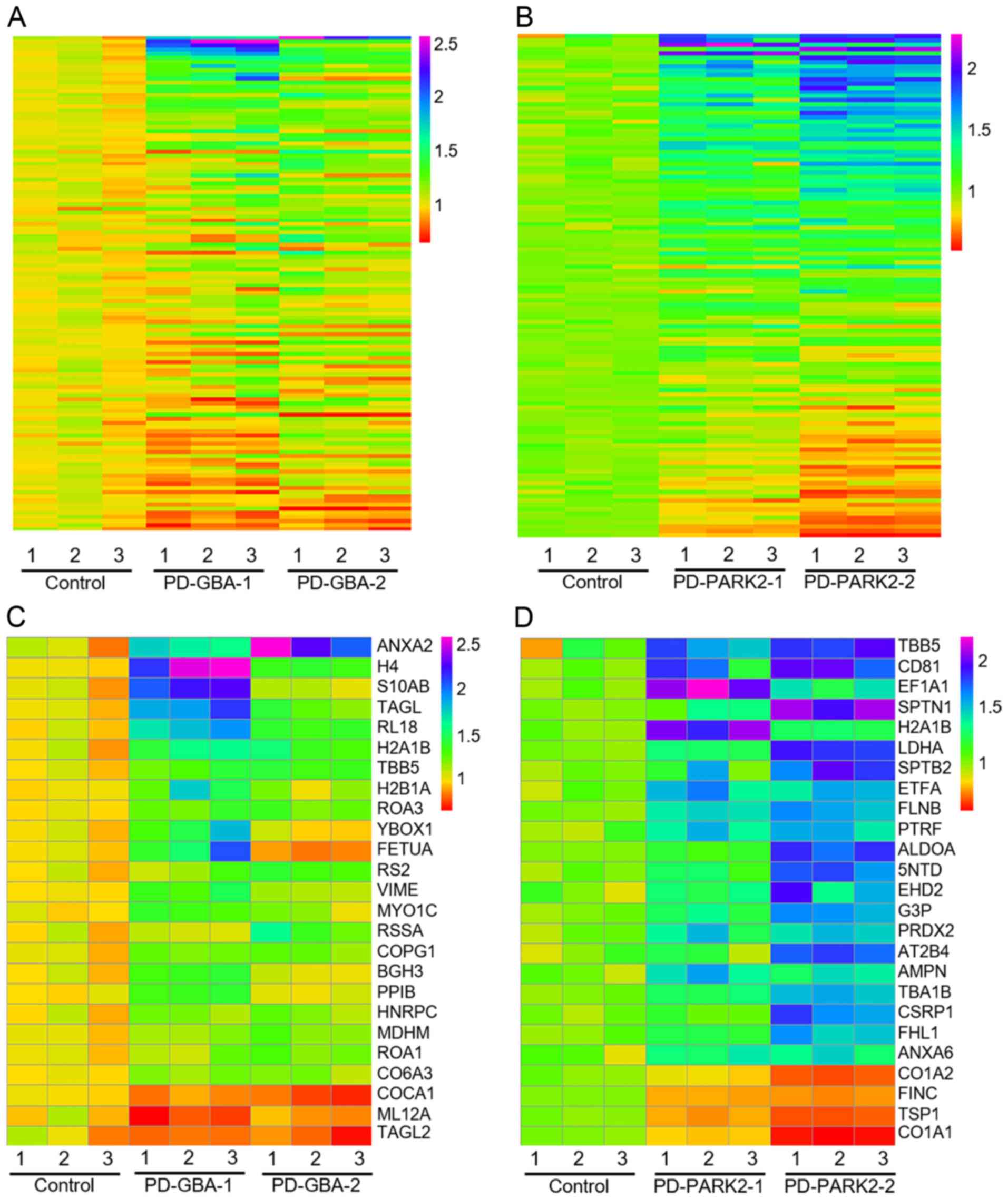
with those found in age-matched healthy controls. Heat map analysis
revealed the differential protein expression levels of fibroblasts
with each GBA and PARK2 variant compared with those
of fibroblasts from healthy controls (Fig. 1A and B). In addition, the top 25 proteins with
the highest differential expression in each variant were selected
and displayed (Fig. 1C and D). Among these, the average expression of 5
and 11 proteins in patients with PD patients with GBA and
PARK2 variants, respectively, was altered by >1.5-fold
compared with that of the controls (Table IIA and B). The levels of ANXA2, histone H4,
protein S100-A11 (S100A11), transgelin (TAGLN) and RPL18 were
higher in the fibroblasts from patients with GBA-PD. The levels of
TUBB, CD antigen 81, elongation factor 1-α (EEF1A1), spectrin α
chain (SPTAN1), histone H2A type 1-B/E (HIST1H2AB), L-lactate
dehydrogenase A chain (LDHA), spectrin β chain (SPTBN1), electron
transfer flavoprotein subunit α (ETFA), filamin-B (FLNB),
polymerase I and transcript release factor (PTRF) and COL1A1 were
altered in fibroblasts from patients with PARK2-PD.
| Table IIList of the top 25 proteins with
differentially altered expression levels from skin fibroblasts of
patients with Parkinson's disease carrying heterozygous GBA
and PARK2 variants. |
Table II
List of the top 25 proteins with
differentially altered expression levels from skin fibroblasts of
patients with Parkinson's disease carrying heterozygous GBA
and PARK2 variants.
| A, Proteins from
skin fibroblasts of patients with PD carrying heterozygous GBA
variants |
|---|
| No. | Uniprot | Accession gene
code | Description | Peptide counts | Protein scores | Fold
changesa | Best peptide
ANOVA |
|---|
| 1 | P07355 | ANXA2 | Annexin A2 | 26 | 1,700.34 | 1.97±0.44 |
1.72x10-7 |
| 2 | P62805 | HIST1H4F | Histone H4 | 6 | 431.19 | 1.88±0.73 |
8.28x10-10 |
| 3 | P31949 | S100A11 | Protein
S100-A11 | 6 | 254.6 | 1.63±0.76 |
6.37x10-7 |
| 4 | Q01995 | TAGLN | Transgelin | 16 | 850.1 | 1.62±0.46 |
1.83x10-8 |
| 5 | Q07020 | RPL18 | 60S ribosomal
protein L18 | 6 | 180 | 1.59±0.30 |
1.72x10-7 |
| 6 | P04908 | HIST1H2AB | Histone H2A type
1-B/E | 7 | 281.52 | 1.47±0.09 |
1.48x10-7b |
| 7 | P07437 | TUBB | Tubulin b
chain | 20 | 866.3 | 1.34±0.03 |
8.28x10-6b |
| 8 | Q96A08 | HIST1H2BA | Histone H2B type
1-A | 8 | 145.32 | 1.32±0.26 |
2.46x10-6 |
| 9 | P51991 | HNRNPA3 | Heterogeneous
nuclear ribonucleoprotein A3 | 14 | 456.83 | 1.27±0.01 |
1.22x10-4 |
| 10 | P67809 | YBX1 | Nuclease-sensitive
element-binding protein 1 | 11 | 252.44 | 1.27±0.37 |
1.53x10-5 |
| 11 | P02765 | AHSG |
α-2-HS-glycoprotein | 6 | 183.46 | 1.26±0.56 |
1.47x10-5 |
| 12 | P15880 | RPS2 | 40S ribosomal
protein S2 | 20 | 328.88 | 1.26±0.11 |
1.22x10-3 |
| 13 | P08670 | VIM | Vimentin | 46 | 2,780.63 | 1.25±0.18 |
2.79x10-8 |
| 14 | O00159 | MYO1C | Unconventional
myosin-Ic | 34 | 336.69 | 1.24±0.14 |
4.32x10-3 |
| 15 | P08865 | RPSA | 40S ribosomal
protein SA | 10 | 379.8 | 1.23±0.23 |
2.72x10-2 |
| 16 | Q9Y678 | COPG1 | Coatomer subunit
g-1 | 31 | 354.38 | 1.23±0.01 |
3.14x10-4 |
| 17 | Q15582 | TGFBI | Transforming growth
factor-b-induced protein ig-h3 | 20 | 546.28 | 1.21±0.22 |
4.71x10-6 |
| 18 | P23284 | PPIB | Peptidyl-prolyl
cis-trans isomerase B | 15 | 550.47 | 1.20±0.23 |
1.30x10-7 |
| 19 | P07910 | HNRNPC | Heterogeneous
nuclear ribonucleoproteins C1/C2 | 18 | 522.89 | 1.19±0.00 |
1.76x10-3 |
| 20 | P40926 | MDH2 | Malate
dehydrogenase, mitochondrial | 17 | 546.5 | 1.19±0.04 |
4.88x10-6 |
| 21 | P09651 | HNRNPA1 | Heterogeneous
nuclear ribonucleoprotein A1 | 14 | 500.11 | 1.19±0.06 |
4.58x10-6 |
| 22 | P12111 | COL6A3 | Collagen α-3(VI)
chain | 102 | 2,434.47 | 1.18±0.01 |
1.07x10-7 |
| 23 | Q99715 | COL12A1 | Collagen α-1(XII)
chain | 82 | 1,558.81 | 0.83±0.06 |
3.19x10-4 |
| 24 | P19105 | MYL12A | Myosin regulatory
light chain 12A | 4 | 298.21 | 0.82±0.10 |
4.88x10-3 |
| 25 | P37802 | TAGLN2 | Transgelin-2 | 12 | 324.07 | 0.82±0.02 |
1.48x10-4 |
| B, Proteins from
skin fibroblasts of patients with PD carrying heterozygous PARK2
variants |
| No. | Uniprot | Accession gene
code | Description | Peptide counts | Protein scores | Fold changesa | Best peptide
ANOVA |
| 1 | P07437 | TUBB | Tubulin b
chain | 16 | 476.78 | 1.77±0.16 |
2.66x10-3b |
| 2 | P60033 | CD81 | CD81 antigen | 6 | 173.21 | 1.76±0.22 |
1.09x10-2 |
| 3 | P68104 | EEF1A1 | Elongation factor
1-α 1 | 15 | 520.37 | 1.75±0.51 |
6.49x10-5 |
| 4 | Q13813 | SPTAN1 | Spectrin α chain,
non-erythrocytic 1 | 91 | 1,606.84 | 1.65±0.56 |
6.85x10-4 |
| 5 | P04908 | HIST1H2AB | Histone H2A type
1-B/E | 4 | 245.77 | 1.64±0.52 |
6.04x10-7a |
| 6 | P00338 | LDHA | L-lactate
dehydrogenase A chain | 15 | 739.16 | 1.58±0.41 |
5.04x10-5 |
| 7 | Q01082 | SPTBN1 | Spectrin b chain,
non-erythrocytic 1 | 71 | 1,350.29 | 1.56±0.40 |
2.89x10-3 |
| 8 | P13804 | ETFA | Electron transfer
flavoprotein subunit α, mitochondrial | 10 | 230.39 | 1.55±0.02 |
2.88x10-3 |
| 9 | O75369 | FLNB | Filamin-B | 89 | 1,800.9 | 1.54±0.11 |
7.31x10-6 |
| 10 | Q6NZI2 | PTRF | Polymerase I and
transcript release factor | 13 | 396.86 | 1.51±0.06 |
6.01x10-3 |
| 11 | P04075 | ALDOA |
Fructose-bisphosphate aldolase A | 16 | 717.61 | 1.49±0.47 |
4.81x10-5 |
| 12 | P21589 | NT5E |
5'-nucleotidase | 11 | 403.14 | 1.49±0.36 |
1.73x10-3 |
| 13 | Q9NZN4 | EHD2 | EH
domain-containing protein 2 | 15 | 422.23 | 1.47±0.24 |
1.04x10-2 |
| 14 | P04406 | GAPDH |
Glyceraldehyde-3-phosphate
dehydrogenase | 16 | 709.19 | 1.46±0.25 |
2.52x10-7 |
| 15 | P32119 | PRDX2 |
Peroxiredoxin-2 | 11 | 245.96 | 1.46±0.07 |
7.45x10-4 |
| 16 | P23634 | ATP2B4 | Plasma membrane
calcium-transporting ATPase 4 | 37 | 678.12 | 1.45±0.47 |
3.24x10-3 |
| 17 | P15144 | ANPEP | Aminopeptidase
N | 33 | 769.39 | 1.44±0.07 |
4.44x10-3 |
| 18 | P68363 | TUBA1B | Tubulin α-1B
chain | 9 | 686.84 | 1.42±0.20 |
3.43x10-5 |
| 19 | P21291 | CSRP1 | Cysteine and
glycine-rich protein 1 | 8 | 340.11 | 1.40±0.44 |
2.42x10-4 |
| 20 | Q13642 | FHL1 | Four and a half LIM
domains protein 1 | 9 | 207.41 | 1.40±0.25 |
9.79x10-5 |
| 21 | P08133 | ANXA6 | Annexin A6 | 37 | 1,269.01 | 1.38±0.03 |
3.17x10-6 |
| 22 | P08123 | COL1A2 | Collagen α-2(I)
chain | 34 | 1,279.13 | 0.72±0.13 |
1.06x10-5 |
| 23 | P02751 | FN1 | Fibronectin | 85 | 4,542.86 | 0.72±0.02 |
2.40x10-7 |
| 24 | P07996 | THBS1 |
Thrombospondin-1 | 36 | 1,290.19 | 0.68±0.07 |
5.24x10-5 |
| 25 | P02452 | COL1A1 | Collagen α-1(I)
chain | 52 | 1,901.37 | 0.65±0.18 |
3.54x10-8 |
PPIs between patients with PD carrying
GBA and PARK2 variants
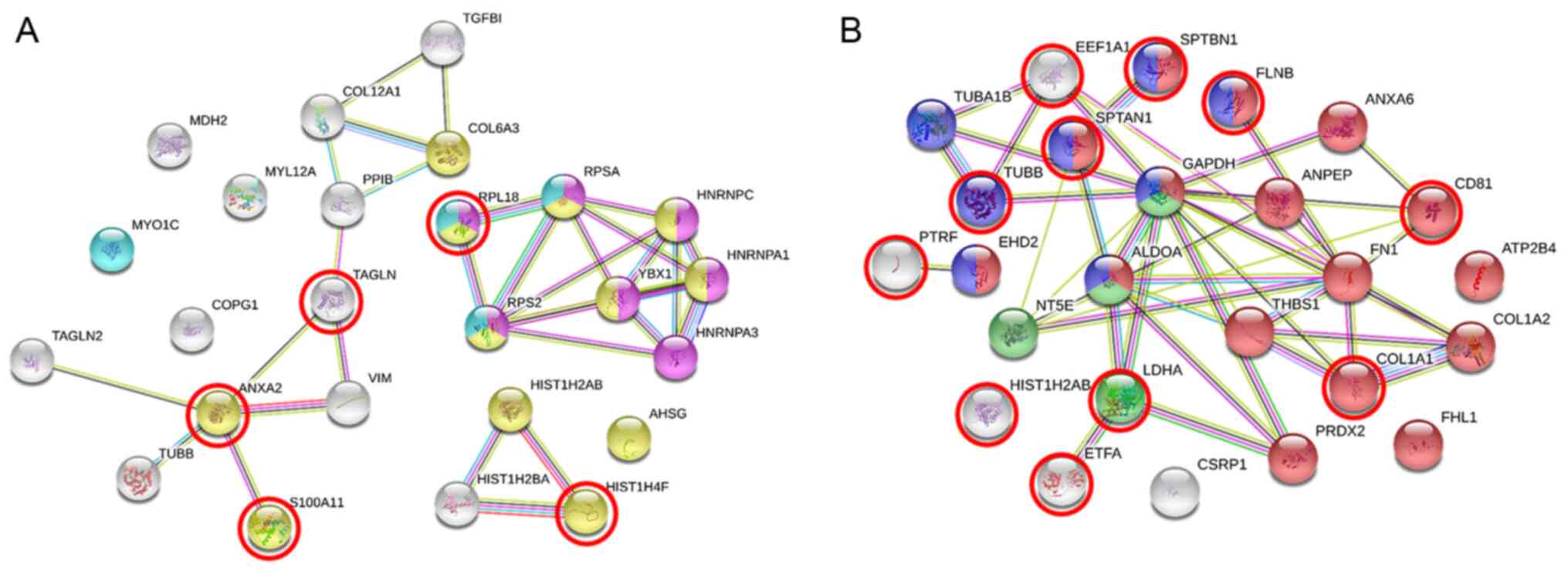
The PPIs of the top 25 proteins with differentially
altered expression in skin fibroblasts of patients with PD
identified by quantitative proteomic analysis were mapped by STRING
analysis (Fig. 2). According to the
biological processes and GO analysis, in the GBA-fibroblasts of
patients with PD, 12 proteins were involved in the negative
regulation of macromolecule metabolic processes (GO:0010605,
FDR=0.0143, yellow), 7 proteins were involved in an mRNA metabolic
process (GO:0016071, FDR=0.0143, purple) and 4 proteins acted in
targeting to the membrane (GO:0006612, FDR=0.0143, cyan) (Fig. 2A). Of note, 4 proteins categorized in
GO:0010605 were upregulated by >1.5-fold in the GBA-fibroblasts
of patients with PD (red circles in Fig.
2A). In the PARK2-fibroblasts of patients with PD, 16 proteins
were involved in the regulation of biological quality (GO:0065008,
FDR=0.0008, red), 4 proteins participated in the nicotinamide
adenine dinucleotide (NAD) metabolic process (GO:0019674,
FDR=0.0013, green) and 8 proteins were involved in cytoskeleton
organization (GO:0007010, FDR=0.0042, blue) (Fig. 2B). Of note, 4 proteins categorized in
GO:0019674, including TUBB, SPTAN1, SPTBN1 and FLNB, LDHA were
categorized in GO:0019674, and another 4 proteins uncategorized in
these GO biological processes, including EEF1A1, PTRF, HIST1H2AB,
and ETFA, were upregulated, whereas COL1A1, categorized in
GO:0065008, was downregulated, by >1.5-fold (red circles in
Fig. 2B).
Validation of protein expression
levels by western blot analysis
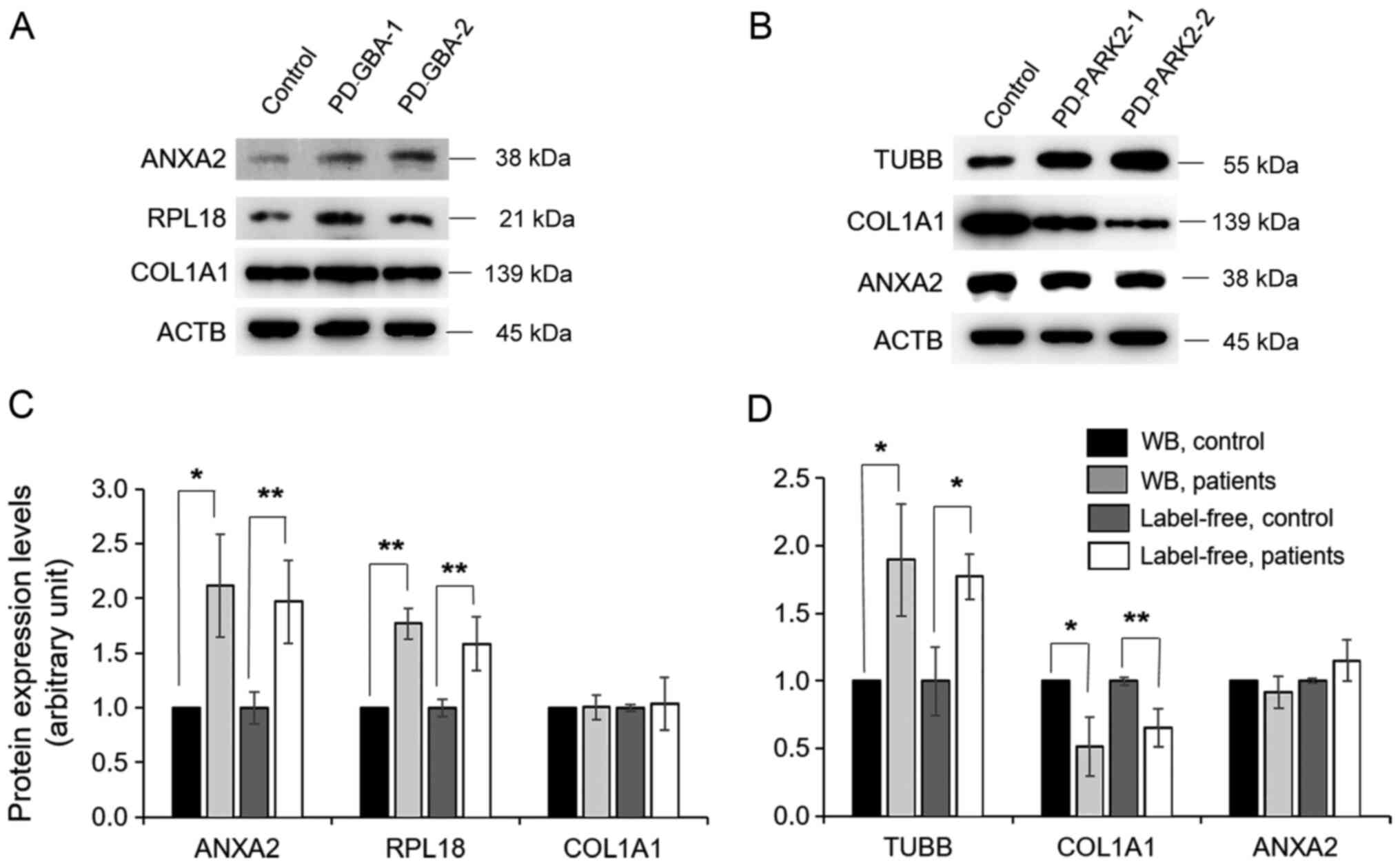
To validate proteins identified by label-free
quantitative proteomic analysis, the levels of certain selected
proteins were determined by western blotting. Specifically, four
proteins, including ANXA2, RPL18, TUBB and COL1A1, whose expression
levels were highly altered based on the bioinformatics analysis,
were selected and analysed in pooled fibroblast samples from
healthy controls and in fibroblasts from individual patients with
PD carrying GBA and PARK2 variants. The levels of
ANXA2 and RPL18 were upregulated, whereas COL1A1 levels were not
different in fibroblasts of patients with PD carrying GBA
variants compared with the levels exhibited by the healthy control
fibroblasts (Fig. 3A). The levels of
TUBB were upregulated, while COL1A1 was downregulated and ANXA2 was
not significantly differentially expressed in patients with PD
carrying PARK2 variants in comparison with the levels
exhibited by the healthy control fibroblasts (Fig. 3B). ACTB was used as a protein loading
control (Fig. 3A and B). The expression levels of selected
proteins based on densitometry analysis were consistent with those
obtained by the label-free quantitative proteomic analysis
(Fig. 3C and D).
Discussion
Abnormalities in GBA and PARK2 genes
have been reported previously to be strong risk factors for
developing PD (28). In the present
study, the GCase activity level was decreased in fibroblasts of
patients with PD with GBA variants [c.1309delG (pV398fsX404)
and IVS2+1G>A] compared with that found in fibroblasts of
healthy controls. In addition, the GCase activity from peripheral
blood leucocytes of patients with PD carrying GBA variants
was lower than that observed in patients with PD without
heterozygous GBA variants and in healthy subjects (data not
shown). Low GCase enzymatic activity has been previously reported
in the cerebrospinal fluid of patients with PD and Lewy body
dementia (29,30), and in two brain autopsy studies
(31,32). The GCase activity in peripheral blood
samples of GBA-PD was reported to be lower than that of PD
patients without GBA variants from the Ashkenazi Jewish
population (33). These findings are
consistent with the observations of the present study.
In addition to GBA variants, there is growing
evidence to suggest that heterozygous pathogenic variants in the
gene that encodes the parkin protein are also susceptibility
factors for PD with a lower age of onset (34,35). It
was previously reported that heterozygosity for a single
PARK2 variant in patients with deletions may contribute to
PD (36). In the present study, two
cases of heterozygous PARK2 variants were studied. The
c.2T>C (p.M1T) variant was previously described in a Chinese
patient (37). This variant showed a
change in the start codon from ATG to ACG, resulting in the
complete absence of the parkin protein. It was reported that
PARK2 point variants are not exclusive to PD, and the
presence of a single point variant in a patient in the absence of a
second variant should not be considered a cause of disease unless
it is corroborated by family data and functional studies (38). However, another report revealed that
a large proportion of patients with a sporadic form of PD (21 of
327 patients with PD, 6.4%) were carriers of heterozygous deletions
or duplications in PARK2 (39). The authors suggested that these
heterozygous forms may be considered dominant mutations with low
penetrance (39). Therefore, further
studies are required to identify the PD-associated effects of
disease caused by heterozygous PARK2 variants.
Primary skin fibroblast cultures exhibit a
patient-specific phenotype that recapitulates the PD chronological
and epigenetic aging pattern, and are used as study models of
several gene variants associated with PD (17,20,40).
Therefore, skin fibroblast cultures of patients with PD and healthy
controls were used in the present study to explore proteins
affected by these two variants. Based on quantitative proteomics
and PPI analysis, the top 25 proteins with expression levels
affected by GBA variants belong to three categories,
including negative regulation of macromolecule metabolic processes
(12 proteins, GO:0010605), an mRNA metabolic process (7 proteins,
GO:0016071) and protein targeting to the membrane (4 proteins,
GO:0006612). ANXA2 was the most highly upregulated gene in the
fibroblasts of patients with PD exhibiting GBA variants.
ANXA2 is involved in calcium signalling and is associated with
S100A11 and TAGLN, which were also highly upregulated in the GBA
fibroblasts of patients with PD. ANXA2 and S100A11 are
Ca2+-dependent binding proteins, whereas TAGLN is an
actin cross-linking protein involved in Ca2+
interactions and regulates contractile properties (41). It has been reported that several
PD-associated proteins, including GBA, are associated with
Ca2+ homeostasis (42).
Kilpatrick et al (43)
demonstrated that the GBA fibroblasts of patients with PD exhibited
an increase in cytosolic Ca2+, whereas the lysosomal
Ca2+ store content was reduced. The authors suggested
that accelerated remodelling of Ca2+ stores by GBA may
be involved in PD pathogenesis. ANXA2 is a multicompartmental
protein that serves important roles in a range of intracellular
membrane-related functions, including organization of specialized
membrane microdomains, recruitment of peripheral membrane proteins,
and regulation of membrane fusion and repair events (44). It has been reported that single
nucleotide polymorphisms (SNPs) of ANXA2 were associated with an
increased risk of stroke (45), and
were a risk factor of avascular necrosis of the bone (osteonecrosis
(46). ANXA2 is involved in numerous
biological processes, including stress responses. It was reported
that ANXA2 expression is increased after starvation, and this
increase is associated with autophagy (47). ANXA2 is considered to be a brain
pathology-associated protein, since its expression is increased
under pathological conditions in various brain diseases (48,49), but
it was undetectable in the neurons and glial cells of a normal
healthy brain (50). Furthermore,
another study showed that skin fibroblasts of patients with PD
exhibiting GBA variants have altered GCase activity and
impaired autophagic flux (40).
S100A11 is implicated in membrane and cytoskeletal dynamics. It can
interact with multiple cytoskeletal proteins, including tubulin,
actin, ANXA1 and ANXA2(51).
Scherzer et al (52) reported
that the S100A11 level was upregulated in peripheral blood samples
of patients with PD. In addition, the S100A11 level was increased
in amyotrophic lateral sclerosis, a neurodegenerative disease
affecting motor neurons in the spinal cord and motor cortex
(53). These data suggest that
aberrant expression of ANXA2 and S100A11 may contribute to PD
pathogenesis; however, further studies are required to clarify the
mechanism(s) by which decreased GCase activity affects autophagic
flux and the roles of Ca2+ in the function of ANXA2 and
S100A11 in the cells of patients with PD.
It has been reported that numerous genes mutated in
familial PD alter RNA metabolism, particularly mRNA translation,
which may contribute to PD pathogenesis (54). Garcia-Esparcia et al (55) showed that the machinery of protein
synthesis was altered, and several ribosomal protein (RP) subunits
were abnormally regulated in the brain of patients with PD. The
present study showed that the expression levels of several proteins
involved in RNA metabolism, including RPL18, RPSA, RPS2, HNRNPC,
HNRNPA1, HNRNPA3 and YBX1, were altered in the fibroblasts of
patients with PD carrying GBA variants. All these proteins
are categorized in the group of mRNA metabolic process
(GO:0016071). Among these, the level of RPL18 exhibited the
greatest degree of upregulation in the GBA fibroblasts of patients
with PD, and it was involved in three GO processes. It is likely
that GBA variants may contribute to alterations of RNA
metabolism; however, further studies are needed to investigate how
GBA affects RNA metabolism.
Another major group of proteins affected by
GBA variants is the histone family. Based on PPI analysis,
it was found that the levels of three histone family members,
histones H4, H2A and H2B, were strongly upregulated. It was
reported that the expression levels of histone proteins and their
acetylation were altered in PD brains (56). Therefore, it is necessary to clarify
how GBA variants modulate the expression of these
histones.
PARK2 variants are involved in the alteration
of proteins via three mechanisms, including regulation of
biological quality (16 proteins, GO:0065008), cytoskeleton
organization (8 proteins, GO:0019674) and NAD metabolic process (4
proteins, GO:0019674). The levels of TUBB showed the highest degree
of upregulation in the PARK2 group. TUBB was directly linked to
TUBA1B and GAPDH, which are involved in cytoskeleton organization
(GO:0019674). It has been reported that the expression of several
cytoskeletal proteins is altered in neurodegenerative diseases
(17,57,58).
Microtubules, composed of α and β forms, are essential components
of neurons, and defects in tubulin genes are likely to cause
neuronal diseases (59). It was
reported that de novo mutations in TUBB5 (M299V, V353I and
E401K) were found in microcephalic patients with structural brain
abnormalities. These mutants were observed to affect the
chaperone-dependent assembly of tubulin heterodimers, and to
disrupt neurogenic division and/or migration in vivo
(60). Generally, α and β-tubulins
can polymerize as heterodimers that join to form microtubules. The
correct folding of tubulin monomers and the formation of functional
α/β heterodimers require a series of cellular chaperonins and
co-factors (61). It was reported
that parkin protein binds to α/β tubulin, and increases their
ubiquitination as well as the degradation of misfolded tubulins in
order to prevent cytotoxicity (62).
Cartelli et al (57) showed
an increased level of β-tubulin and actin in skin fibroblasts of
patients with PD carrying PARK2 variants. These data are
consistent with our results. Nevertheless, further investigation is
required to determine why the β-tubulin found in skin fibroblasts
of patients with PD carrying PARK2 variants is upregulated,
and whether these increased levels are related to misfolding and
malfunctions of ubiquitination.
Another major protein group affected by PARK2
variants is the group of proteins involved in the regulation of
biological quality and extracellular matrix (ECM). The ECM is a
network of extracellular macromolecules, including collagen,
fibronectin (FN1) and thrombospondin-1 (THBS1), which regulate a
range of cellular functions such as proliferation, migration and
differentiation (63). It has been
reported that alterations of several proteins in the ECM may be
involved in the pathogenesis of neurodegenerative disorders
(64). The present results showed
that the levels of ECM proteins, including the collagen family
members COL1A1 and COL1A2, FN1 and THBS1 were all downregulated in
the patients with PD carrying PARK2 variants. The COL1A1
level was reported to be altered in fibroblasts of patients with PD
(17). FN1 was shown to have a
neuroprotective role in neuron-glial extrasynaptic transmission
(65). THBS1 was reported to be a
critical factor in the maintenance of adult neural progenitor cells
(66). These data suggest that ECM
proteins are involved in PD pathogenesis; however, further
investigation is needed on how PARK2 variants affect ECM
modelling.
The present study has various limitations. Firstly,
the proteins were identified without consideration of protein
isoforms or processing, such as cleaved, mature and truncated forms
or other post-translational modifications. Secondly, although the
present study provides potential proteins associated with the
disease, the numbers of patients with PD exhibiting GBA and
PARK2 variants from whom fibroblasts were acquired was
small. Therefore, larger numbers of samples need to be analysed to
represent phenotype and variant correlations. Lastly, the
identified proteins obtained from skin fibroblasts of patients may
not be the same as those of a patient's neuronal cells, which are
more likely to demonstrate a direct clinical correlation with the
variants. The results from studies focused on induced dopaminergic
neurons derived from pluripotent stem cells from patients should
reveal more precisely the molecular defects that occur in those
neuronal cells (67,68). Nevertheless, the present study on
primary skin fibroblasts of patients with PD with different
variants of GBA and PARK2 genes provides unique
proteome profiles, which suggest that the proteins identified may
serve important roles in the disease status.
In conclusion, the present study used a label-free
MS-based proteomic approach to study differentially expressed
proteins in fibroblasts of patients with PD and healthy controls.
Several proteins were identified, and a few predominant proteins
were validated to confirm their expression levels. It was
demonstrated that the proteome profiles of skin fibroblasts from
patients with PD carrying heterozygous GBA and PARK2
variants and those from healthy controls are different and unique.
Heterozygous GBA variants reveal a signature of protein
alterations related to the regulation of macromolecular processes.
Among these proteins, the ANXA2 level is most highly upregulated,
and this increase is considered to be involved in autophagy
regulation and Ca2+ homeostasis. Although heterozygous
PARK2 variants are rare in patients with PD, this
heterozygous form shows a signature of protein alterations related
to the regulation of cytoskeleton organization and biological
quality. Among them, the TUBB level is most highly upregulated, and
this increase is likely to be involved in cytoskeleton
organization. However, further studies are required to clarify the
functional roles of these proteins in the cells of patients with PD
carrying GBA and PARK2 variants. Taken together, this
study demonstrated the value of the proteomic approach to identify
proteome profiles and to compare their expression levels that may
be associated with the physiological changes occurring in the
cells. These alterations may be related to disease progression and
development.
Acknowledgements
We would like to thank Dr Somsak Tanrattanakorn,
Department of Medicine, Faculty of Medicine, Ramathibodi Hospital,
Mahidol University, for his help with the skin biopsy.
Funding
Funding: This study was supported by the Chulabhorn Research
Institute (grant no. BC-2020-05) and the Faculty of Medicine,
Ramathibodi Hospital, Mahidol University.
Availability of data and materials
The datasets used and/or analysed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
LN and VC conceived and designed the study. LN, PS,
DC and KK performed the experiments. KS performed the
bioinformatics analysis and statistical analysis. CS analysed and
interpreted the results, and reviewed the manuscript. PS, LN, and
VC wrote and drafted the manuscript. JS, JRKC, PD, and TP reviewed
and edited the manuscript and were involved in the conception of
the study. All authors read and approved the final manuscript. PS,
LN and VC confirmed the authenticity of all the raw data.
Ethics approval and consent to
participate
This study was approved by the Ethical Clearance
Committee on Human Rights Related to Research Involving Human
Subjects, Faculty of Medicine, Ramathibodi Hospital, Mahidol
University (approval no. ID03-54-22).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kalia LV and Lang AE: Parkinson's disease.
Lancet. 386:896–912. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Deng H, Wang P and Jankovic J: The
genetics of Parkinson disease. Ageing Res Rev. 42:72–85.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hernandez DG, Reed X and Singleton AB:
Genetics in Parkinson disease: Mendelian versus non-Mendelian
inheritance. J Neurochem. 139 (Suppl 1):59–74. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kuusimäki T, Korpela J, Pekkonen E,
Martikainen MH, Antonini A and Kaasinen V: Deep brain stimulation
for monogenic Parkinson's disease: A systematic review. J Neurol.
267:883–897. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Billingsley KJ, Bandres-Ciga S,
Saez-Atienzar S and Singleton AB: Genetic risk factors in
Parkinson's disease. Cell Tissue Res. 373:9–20. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kitada T, Asakawa S, Hattori N, Matsumine
H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y and Shimizu N:
Mutations in the parkin gene cause autosomal recessive juvenile
parkinsonism. Nature. 392:605–608. 1998.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Hattori N and Mizuno Y: Twenty years since
the discovery of the parkin gene. J Neural Transm (Vienna).
124:1037–1054. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Horowitz M, Wilder S, Horowitz Z, Reiner
O, Gelbart T and Beutler E: The human glucocerebrosidase gene and
pseudogene: Structure and evolution. Genomics. 4:87–96.
1989.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hruska KS, LaMarca ME, Scott CR and
Sidransky E: Gaucher disease: Mutation and polymorphism spectrum in
the glucocerebrosidase gene (GBA). Hum Mutat. 29:567–583.
2008.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pulkes T, Choubtum L, Chitphuk S,
Thakkinstian A, Pongpakdee S, Kulkantrakorn K, Hanchaiphiboolkul S,
Tiamkao S and Boonkongchuen P: Glucocerebrosidase mutations in Thai
patients with Parkinson's disease. Parkinsonism Relat Disord.
20:986–991. 2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lwin A, Orvisky E, Goker-Alpan O, LaMarca
ME and Sidransky E: Glucocerebrosidase mutations in subjects with
parkinsonism. Mol Genet Metab. 81:70–73. 2004.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Aharon-Peretz J, Rosenbaum H and
Gershoni-Baruch R: Mutations in the glucocerebrosidase gene and
Parkinson's disease in Ashkenazi Jews. N Engl J Med. 351:1972–1977.
2004.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sidransky E, Nalls MA, Aasly JO,
Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J,
Brice A, et al: Multicenter analysis of glucocerebrosidase
mutations in Parkinson's disease. N Engl J Med. 361:1651–1661.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Malek N, Weil RS, Bresner C, Lawton MA,
Grosset KA, Tan M, Bajaj N, Barker RA, Burn DJ, Foltynie T, et al:
PRoBaND clinical consortium: Features of GBA-associated Parkinson's
disease at presentation in the UK Tracking Parkinson's study. J
Neurol Neurosurg Psychiatry. 89:702–709. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ziegler SG, Eblan MJ, Gutti U, Hruska KS,
Stubblefield BK, Goker-Alpan O, LaMarca ME and Sidransky E:
Glucocerebrosidase mutations in Chinese subjects from Taiwan with
sporadic Parkinson disease. Mol Genet Metab. 91:195–200.
2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Schapira AH: Glucocerebrosidase and
Parkinson disease: Recent advances. Mol Cell Neurosci. 66 (Pt
A):37–42. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Lippolis R, Siciliano RA, Pacelli C,
Ferretta A, Mazzeo MF, Scacco S, Papa F, Gaballo A, Dell'Aquila C,
De Mari M, et al: Altered protein expression pattern in skin
fibroblasts from parkin-mutant early-onset Parkinson's disease
patients. Biochim Biophys Acta. 1852:1960–1970. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Rotunno MS, Lane M, Zhang W, Wolf P, Oliva
P, Viel C, Wills AM, Alcalay RN, Scherzer CR, Shihabuddin LS, et
al: Cerebrospinal fluid proteomics implicates the granin family in
Parkinson's disease. Sci Rep. 10(2479)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
O'Bryant SE, Edwards M, Zhang F, Johnson
LA, Hall J, Kuras Y and Scherzer CR: Potential two-step proteomic
signature for Parkinson's disease: Pilot analysis in the Harvard
Biomarkers Study. Alzheimers Dement (Amst). 11:374–382.
2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Auburger G, Klinkenberg M, Drost J, Marcus
K, Morales-Gordo B, Kunz WS, Brandt U, Broccoli V, Reichmann H,
Gispert S, et al: Primary skin fibroblasts as a model of
Parkinson's disease. Mol Neurobiol. 46:20–27. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Khwanraj K, Choubtum L, Taweewongsounton
A, Tanrattanakorn S, Pulkes T and Dharmasaroja P: Mitochondrial
Respiratory Chain Enzymatic Activities on Skin Fibroblasts in
Patients With Mutant Glucocerebrosidase and PARK2 Genes. J Neurol
Res. 6:12–17. 2016.
|
|
22
|
Peters SP, Coyle P and Glew RH:
Differentiation of beta-glucocerebrosidase from beta-glucosidase in
human tissues using sodium taurocholate. Arch Biochem Biophys.
175:569–582. 1976.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chokchaichamnankit D, Watcharatanyatip K,
Subhasitanont P, Weeraphan C, Keeratichamroen S, Sritana N,
Kantathavorn N, Diskul-Na-Ayudthaya P, Saharat K, Chantaraamporn J,
et al: Urinary biomarkers for the diagnosis of cervical cancer by
quantitative label-free mass spectrometry analysis. Oncol Lett.
17:5453–5468. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Team RC: A language and environment for
statistical computing. R Foundation for Statistical Computing,
Vienna, Austria. www.r-project.org.
|
|
25
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47D:D607–D613.
2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: The Gene Ontology Consortium: Gene ontology: Tool for the
unification of biology. Nat Genet. 25:25–29. 2000.PubMed/NCBI View Article : Google Scholar
|
|
27
|
The Gene Ontology Consortium. The Gene
Ontology Resource: 20 years and still GOing strong. Nucleic Acids
Res. 47D:D330–D338. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Delamarre A and Meissner WG: Epidemiology,
environmental risk factors and genetics of Parkinson's disease.
Presse Med. 46:175–181. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Balducci C, Pierguidi L, Persichetti E,
Parnetti L, Sbaragli M, Tassi C, Orlacchio A, Calabresi P, Beccari
T and Rossi A: Lysosomal hydrolases in cerebrospinal fluid from
subjects with Parkinson's disease. Mov Disord. 22:1481–1484.
2007.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Borroni B, Gardoni F, Parnetti L, Magno L,
Malinverno M, Saggese E, Calabresi P, Spillantini MG, Padovani A
and Di Luca M: Pattern of Tau forms in CSF is altered in
progressive supranuclear palsy. Neurobiol Aging. 30:34–40.
2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gegg ME, Burke D, Heales SJ, Cooper JM,
Hardy J, Wood NW and Schapira AH: Glucocerebrosidase deficiency in
substantia nigra of parkinson disease brains. Ann Neurol.
72:455–463. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Murphy KE, Gysbers AM, Abbott SK, Tayebi
N, Kim WS, Sidransky E, Cooper A, Garner B and Halliday GM: Reduced
glucocerebrosidase is associated with increased α-synuclein in
sporadic Parkinson's disease. Brain. 137:834–848. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ortega RA, Torres PA, Swan M, Nichols W,
Boschung S, Raymond D, Barrett MJ, Johannes BA, Severt L, Shanker
V, et al: Glucocerebrosidase enzyme activity in GBA mutation
Parkinson's disease. J Clin Neurosci. 28:185–186. 2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Sun M, Latourelle JC, Wooten GF, Lew MF,
Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, et
al: Influence of heterozygosity for parkin mutation on onset age in
familial Parkinson disease: The GenePD study. Arch Neurol.
63:826–832. 2006.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Klein C, Lohmann-Hedrich K, Rogaeva E,
Schlossmacher MG and Lang AE: Deciphering the role of heterozygous
mutations in genes associated with parkinsonism. Lancet Neurol.
6:652–662. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wu YR, Wu CH, Chao CY, Kuan CC, Zhang WL,
Wang CK, Chang CY, Chang YC, Lee-Chen GJ and Chen CM: Genetic
analysis of Parkin in early onset Parkinson's disease (PD): Novel
intron 9 g > a single nucleotide polymorphism and risk of
Taiwanese PD. Am J Med Genet B Neuropsychiatr Genet. 153B:229–234.
2010.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhang BR, Hu ZX, Yin XZ, Cai M, Zhao GH,
Liu ZR and Luo W: Mutation analysis of parkin and PINK1 genes in
early-onset Parkinson's disease in China. Neurosci Lett. 477:19–22.
2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kay DM, Moran D, Moses L, Poorkaj P,
Zabetian CP, Nutt J, Factor SA, Yu CE, Montimurro JS, Keefe RG, et
al: Heterozygous parkin point mutations are as common in control
subjects as in Parkinson's patients. Ann Neurol. 61:47–54.
2007.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Shulskaya MV, Shadrina MI, Fedotova EY,
Abramycheva NY, Limborska SA, Illarioshkin SN and Slominsky PA:
Second mutation in PARK2 is absent in patients with sporadic
Parkinson's disease and heterozygous exonic deletions/duplications
in parkin gene. Int J Neurosci. 127:781–784. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Collins LM, Williams-Gray CH, Morris E,
Deegan P, Cox TM and Barker RA: The motor and cognitive features of
Parkinson's disease in patients with concurrent Gaucher disease
over 2 years: A case series. J Neurol. 265:1789–1794.
2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Assinder SJ, Stanton JA and Prasad PD:
Transgelin: An actin-binding protein and tumour suppressor. Int J
Biochem Cell Biol. 41:482–486. 2009.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zaichick SV, McGrath KM and Caraveo G: The
role of Ca2+ signaling in Parkinson's disease. Dis Model
Mech. 10:519–535. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kilpatrick BS, Magalhaes J, Beavan MS,
McNeill A, Gegg ME, Cleeter MW, Bloor-Young D, Churchill GC, Duchen
MR, Schapira AH, et al: Endoplasmic reticulum and lysosomal
Ca²+ stores are remodelled in GBA1-linked Parkinson
disease patient fibroblasts. Cell Calcium. 59:12–20.
2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Gerke V, Creutz CE and Moss SE: Annexins:
Linking Ca2+ signalling to membrane dynamics. Nat Rev
Mol Cell Biol. 6:449–461. 2005.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sebastiani P, Ramoni MF, Nolan V, Baldwin
CT and Steinberg MH: Genetic dissection and prognostic modeling of
overt stroke in sickle cell anemia. Nat Genet. 37:435–440.
2005.PubMed/NCBI View
Article : Google Scholar
|
|
46
|
Baldwin C, Nolan VG, Wyszynski DF, Ma QL,
Sebastiani P, Embury SH, Bisbee A, Farrell J, Farrer L and
Steinberg MH: Association of klotho, bone morphogenic protein 6,
and annexin A2 polymorphisms with sickle cell osteonecrosis. Blood.
106:372–375. 2005.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Moreau K, Ghislat G, Hochfeld W, Renna M,
Zavodszky E, Runwal G, Puri C, Lee S, Siddiqi F, Menzies FM, et al:
Transcriptional regulation of Annexin A2 promotes
starvation-induced autophagy. Nat Commun. 6(8045)2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Nygaard SJ, Haugland HK, Kristoffersen EK,
Lund-Johansen M, Laerum OD and Tysnes OB: Expression of annexin II
in glioma cell lines and in brain tumor biopsies. J Neurooncol.
38:11–18. 1998.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Eberhard DA, Brown MD and VandenBerg SR:
Alterations of annexin expression in pathological neuronal and
glial reactions. Immunohistochemical localization of annexins I, II
(p36 and p11 subunits), IV, and VI in the human hippocampus. Am J
Pathol. 145:640–649. 1994.PubMed/NCBI
|
|
50
|
de la Monte SM, Bhavani K, Xu YY, Puisieux
A and Wands JR: Modulation of p36 gene expression in human neuronal
cells. J Neurol Sci. 128:122–133. 1995.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Réty S, Osterloh D, Arié JP, Tabaries S,
Seeman J, Russo-Marie F, Gerke V and Lewit-Bentley A: Structural
basis of the Ca(2+)-dependent association between S100C (S100A11)
and its target, the N-terminal part of annexin I. Structure.
8:175–184. 2000.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Scherzer CR, Gullans SR and Jensen R:
Prediction of Parkinson's disease using gene expression levels of
peripheral blood samples. US Patent 7595159B2 2009. Filed: November
3,2005; issued: September 29, 2009.
|
|
53
|
Iridoy MO, Zubiri I, Zelaya MV, Martinez
L, Ausín K, Lachen-Montes M, Santamaría E, Fernandez-Irigoyen J and
Jericó I: Neuroanatomical Quantitative Proteomics Reveals Common
Pathogenic Biological Routes between Amyotrophic Lateral Sclerosis
(ALS) and Frontotemporal Dementia (FTD). Int J Mol Sci.
20(20)2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Lu B, Gehrke S and Wu Z: RNA metabolism in
the pathogenesis of Parkinson's disease. Brain Res. 1584:105–115.
2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Garcia-Esparcia P, Hernández-Ortega K,
Koneti A, Gil L, Delgado-Morales R, Castaño E, Carmona M and Ferrer
I: Altered machinery of protein synthesis is region- and
stage-dependent and is associated with α-synuclein oligomers in
Parkinson's disease. Acta Neuropathol Commun. 3(76)2015.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Toker L, Tran GT, Sundaresan J, Tysnes
O-B, Alves G, Haugarvoll K, Nido GS, Dolle C and Tzoulis C:
Genome-wide dysregulation of histone acetylation in the Parkinson's
disease brain. BioRxiv: Apr 2, 2020 (Epub ahead of print). doi:
https://doi.org/10.1101/785550.
|
|
57
|
Cartelli D, Goldwurm S, Casagrande F,
Pezzoli G and Cappelletti G: Microtubule destabilization is shared
by genetic and idiopathic Parkinson's disease patient fibroblasts.
PLoS One. 7(e37467)2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Cairns NJ, Lee VM and Trojanowski JQ: The
cytoskeleton in neurodegenerative diseases. J Pathol. 204:438–449.
2004.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Tischfield MA, Baris HN, Wu C, Rudolph G,
Van Maldergem L, He W, Chan WM, Andrews C, Demer JL, Robertson RL,
et al: Human TUBB3 mutations perturb microtubule dynamics, kinesin
interactions, and axon guidance. Cell. 140:74–87. 2010.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Breuss M, Heng JI, Poirier K, Tian G,
Jaglin XH, Qu Z, Braun A, Gstrein T, Ngo L, Haas M, et al:
Mutations in the β-tubulin gene TUBB5 cause microcephaly with
structural brain abnormalities. Cell Rep. 2:1554–1562.
2012.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Lewis SA, Tian G and Cowan NJ: The alpha-
and beta-tubulin folding pathways. Trends Cell Biol. 7:479–484.
1997.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Ren Y, Zhao J and Feng J: Parkin binds to
alpha/beta tubulin and increases their ubiquitination and
degradation. J Neurosci. 23:3316–3324. 2003.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Bonnans C, Chou J and Werb Z: Remodelling
the extracellular matrix in development and disease. Nat Rev Mol
Cell Biol. 15:786–801. 2014.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Bielefeld KA, Amini-Nik S, Whetstone H,
Poon R, Youn A, Wang J and Alman BA: Fibronectin and beta-catenin
act in a regulatory loop in dermal fibroblasts to modulate
cutaneous healing. J Biol Chem. 286:27687–27697. 2011.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Wang J, Yin L and Chen Z: Neuroprotective
role of fibronectin in neuron-glial extrasynaptic transmission.
Neural Regen Res. 8:376–382. 2013.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Lu Z and Kipnis J: Thrombospondin 1 - a
key astrocyte-derived neurogenic factor. FASEB J. 24:1925–1934.
2010.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Shuvalova LD, Eremeev AV, Bogomazova AN,
Novosadova EV, Zerkalenkova EA, Olshanskaya YV, Fedotova EY,
Glagoleva ES, Illarioshkin SN, Lebedeva OS, et al: Generation of
induced pluripotent stem cell line RCPCMi004-A derived from patient
with Parkinson's disease with deletion of the exon 2 in PARK2 gene.
Stem Cell Res (Amst). 44(101733)2020.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Gustavsson N, Marote A, Pomeshchik Y, Russ
K, Azevedo C, Chumarina M, Goldwurm S, Collin A, Pinto L, Salgado
AJ, et al: Generation of an induced pluripotent stem cell line
(CSC-46) from a patient with Parkinson's disease carrying a novel
p.R301C mutation in the GBA gene. Stem Cell Res (Amst).
34(101373)2019.PubMed/NCBI View Article : Google Scholar
|