Introduction
Genetic predispositions and environmental gene
interactions serve crucial roles in the pathogenesis of various
interstitial lung diseases, including idiopathic pulmonary fibrosis
(IPF) (1-3).
The most often accepted concept is related to genetically based
susceptibility, which may or may not lead to impairment of the
alveolar epithelium, followed by the development of lung fibrosis
due to the effect of extrinsic factors, such as smoking and
inhalation of pollutants in the environment (4).
Various studies have demonstrated that polymorphisms
of several genes, including mucin 5B (MUC5B), genes associated with
telomere integrity and genes encoding surfactants increase the risk
of IPF (5-7).
However, a genetic predisposition has been identified in 30% of
cases, whereas in the remainder of cases, this information is
lacking (8). The course of IPF is as
heterogeneous as the genetic susceptibility to the development of
the disease. Thus, the individual genetic background may affect
both the predisposition to IPF and its course (9).
The association between the MUC5B rs 35705950 minor
allele and IPF risk in the non-Hispanic white population has been
published elsewhere (10). The
effect of the MUC5B polymorphism on IPF outcomes has been also
debated in the literature. While surfactant protein A (SFTPA) plays
an important role in pulmonary host defense and repair processes in
the lung parenchyma, transforming growth factor-β (TGF-β) plays a
crucial role in different types of fibrotic interstitial pneumonia,
including IPF (11,12). Toll interacting protein (TOLLIP) is
an adaptor protein that acts as an inhibitory factor in TLR
signaling. Noth et al (13)
identified three TOLLIP SNPs associated with IPF risk. Transporters
associated with antigen processing (TAP)-1 and TAP-2 gene
polymorphisms are susceptibility factors in patients with
hypersensitivity pneumonitis (14).
TAP is crucial for antigen processing, delivering cytosolic
proteins into the endoplasmic reticulum, where they bind to nascent
major histocompatibility complex I (15). For both IL-4 rs 2070874 and rs
2243250, the T allele has been reported less frequently than the C
allele in the European population (16). The T allele was reported to be
associated with enhanced IL-4 production and was found to be
associated with the response to infections, tumor susceptibility
and immune-mediated disorders (17).
IL-4 plays an important role in both the innate and adaptive immune
responses. Both basophils and natural killer T-cells have been
suggested as primary sources of IL-4 in humans (18). Impairment of alveolar epithelial
cells leads to the induction of alarmin production, stimulating
basophils and leading to IL-4 secretion. The major role of IL-4
concerns not only the induction of a Th2 response, but also the
stimulation of dendritic cells to present antigens to other immune
cells and alternative activation of macrophages. These
IL-4-activated macrophages produce matrix metalloproteinase 12, a
collagenolytic factor that may induce a fibrotic response to injury
(19).
In the present study, whether gene variants of
interleukin-4 (IL-4), MUC5B, toll interacting protein (TOLLIP),
surfactant protein A (SFPTA), transforming growth factor-β (TGF-β)
and transporters associated with antigen processing (TAP1 and TAP2)
play a role in the IPF course was investigated.
Materials and methods
Patients and study design
A total of 50 consecutive patients (median age 69.3;
age range, 57-82 years); 24 males and 26 females, 38 of which were
non-smokers) with IPF were enrolled in the present prospective
observational study after providing signed informed content;
refusal to take part in the study and refusal or unable to
sign/obtain informed consent were the only exclusion criteria. The
study was approved by the ethics committee of Thomayer Hospital and
the Institute of Clinical and Experimental Medicine, Prague, Czech
Republic. The diagnosis of IPF was based on the ERS/ATS/ALAT/JRS
guidelines (20). All of the
patients underwent a historical assessment focused on possible
extrinsic exposures, a physical examination, blood tests including
autoantibody tests, high-resolution computed tomography of the
chest, including the quantification of inflammatory and fibrotic
changes (high-resolution computed tomography alveolar score
0.1±0.3, high-resolution computed tomography interstitial score
2.4±0.8), lung function tests, bronchoscopy with bronchoalveolar
lavage (cell percentages in retrieved fluid: Alveolar macrophages
70.4±19.7; lymphocytes 10.7±10.0; neutrophils 12.1±11.8; and
eosinophils 6.5±8.6) and multidisciplinary team consultation, as is
routine (21). Either surgical lung
biopsy or transbronchial cryobiopsy was performed in select cases
when the multidisciplinary team could not reach a diagnosis and the
patient was willing and able to undergo any of these
procedures.
Patients diagnosed with IPF provided blood for DNA
analysis at the time of enrollment in the study. Participation in
this study did not affect the management of the patients, and all
patients received appropriate treatment for IPF according to their
baseline lung functions, comorbidities and patient preferences. IPF
treatment guidelines were followed (20).
Lung function and outcome
assessment
The diffusing capacity for carbon monoxide (Dlco)
was investigated using a ZAN 300 CO diffusion instrument (nSpire
Health GmbH). The Dlco was measured using the single-breath method
(22). Values are presented as
percentages of the predicted values (23).
The patients' health status, including lung
function, were re-assessed after 6, 12 and 18 months. At these time
points, the patients' lung function was assessed, and data
concerning the occurrence of acute exacerbations were
collected.
IPF progression was defined as either a drop in the
forced vital capacity (FVC) >10% and/or a drop in the Dlco
>15% and/or an acute exacerbation and/or death within 6 months.
The percentages of change in the FVC/Dlco were determined based on
the absolute values of the previous results. Each patient with a
>10% FVC decline and/or a >15% Dlco decline underwent a
complete evaluation to exclude a non-IPF based etiology of
deterioration (24).
Genotyping
A panel of single-nucleotide polymorphisms (SNPs) in
genes associated with immune function and candidate loci for IPF
susceptibility were selected for the present study (Table SI). Selection was based on previous
studies published by our study team and a literature review with an
emphasis on antigen processing (25). The selected SNPs were investigated in
patients with IPF using MALDI-TOF MS-based MassARRAY (Agena
Bioscience, Inc.) or TaqMan (Thermo Fisher Scientific, Inc.)
genotyping assays. For positive controls, DNA samples were obtained
from the NIGMS Human Genetic Cell Repository at the Coriell
Institute for Medical Research (Coriell DNA nos. NA16689, NA17084
and NA17252). Primers were designed using Assay Design Suite
version 2.2 (Agena Bioscience, Inc.), available only for registered
users of the MassARRAY system.
The detailed procedures for the MassARRAY-based
multiplexed assay design and iPLEX-based genotyping have been
described previously (26). Briefly,
the DNA template (10 ng) with multiplex primers and PCR mix
(iPLEX® Gold Reagent Set; Agena Bioscience) were used
for single-base extension-based PCR amplification of the target
region. The iPLEX-extended amplicons were spotted on the
SpectroCHIP Array using the MassARRAY® Nanodispenser RS
1000 station and were genotyped using the MassARRAY platform. The
distinct mass of the extended primer for the alternative alleles
was traced using MassARRAY Typer version 4.0.20. For TaqMan-based
genotyping, custom TaqMan® primer probes (labeled with
either VIC® or 6-FAM™ dye) were designed for the SNPs
TOLLIP rs111521887 and TOLLIP rs5743894 using the
online Custom TaqMan® Assay Design Tool (Thermo Fisher
Scientific, Inc.), and predesigned assays for C_16176216_10 and
C_11464118_30 were used to genotype IL-4 rs2243250 and
TGF-β1 rs1800471 (Thermo Fisher Scientific, Inc.),
respectively, in the subjects using standard protocols.
Specifically, 10 ng template DNA, LightCycler 480 Probes Master
(Roche Diagnostics) and the custom TaqMan SNP genotyping assay were
used. The thermocycling conditions on the LightCycler 480 system
(Roche Diagnostics) were: 5 min at 95˚C; followed by 45 cycles of
95˚C for 10 sec, 60˚C for 45 sec and 72˚C for 1 sec (acquisition
was performed each cycle) and then 40˚C for 30 sec. The data was
obtained by endpoint analysis (27).
For assessment of the quality of the genotyping
protocol, positive and negative template control samples were
included in each assay plate. For each SNP, the genotype data were
manually verified using the call cluster plot (26). Measurements were performed in
duplicates. The datasets generated and analyzed during the current
study are available in the European Variation Archive (EVA)
repository (ebi.ac.uk/ena/data/view/PRJEB44734).
Statistical analysis
A repeated measures ANOVA (with data exhibiting
sphericity) and grouping factors was used to test the association
between the gene polymorphisms and lung function decline at the 6-,
12- and 18-month follow-up assessments (FVC and Dlco). Bonferroni
corrections were applied for multiple comparisons. Two-way
frequency tables were analyzed using Fisher's exact test to assess
an effect of nucleotide polymorphisms on patient outcomes. Relative
risk (RR, calculated by dividing the percentage of subjects with
progression for each allele) with 95% confidence intervals (CI)
were calculated for ‘risk’ alleles in comparison with ‘protective’
alleles (MedCalc Software, Ltd., version 14.8.1). All tests were
two-sided, and P<0.05 was considered to indicate a statistically
significant difference. Comparisons with P<0.06 are considered
‘clinically significant’. Associations between gene polymorphisms
and lung functions are graphically illustrated, documenting FVC and
Dlco at the time of diagnosis [mean ± standard deviation(SD)] and
FVC and Dlco at the time for which effect of gene polymorphisms
were found (mean ± SD).
Results
The lung function at the time of diagnosis, and
after 6, 12 and 18 months, as well as the counts of acute
exacerbations and deaths in each observation period are summarized
in Table I. An acute exacerbation
was defined following criteria published by Collard et al
(28).
 | Table ILung function at the time of
diagnosis, and after 6, 12 and 18 months, as well as counts of
acute exacerbations and deaths in each observation period. |
Table I
Lung function at the time of
diagnosis, and after 6, 12 and 18 months, as well as counts of
acute exacerbations and deaths in each observation period.
| Time after
diagnosis, months | Patient, n | FVC, la | FVC, %
pva | Dlco,
mmol/kPa/mina | Dlco, %
pva | Acute exacerbation,
n | Death, n |
|---|
| 0 | 50 | 2.8±0.7 | 81.0±14.8 | 3.7±1.6 | 44.2±11.8 | 0 | 0 |
| 6 | 28 | 2.8±0.8 | 83.4±17.3 | 3.8±1.0 | 46.0±16.9 | 0 | 0 |
| 12 | 27 | 2.7±0.7 | 79.8±14.4 | 3.65±4.6 | 43.6±13.5 | 2 | 4 |
| 18 | 16 | 2.8±0.7 | 77.6±14.4 | 3.09±1.8 | 46.8±11.8 | 0 | 2 |
No effect of the selected gene polymorphisms during
the observation period was found on the patient outcomes in the
first 6 months. However, 12 months after diagnosis, an effect of
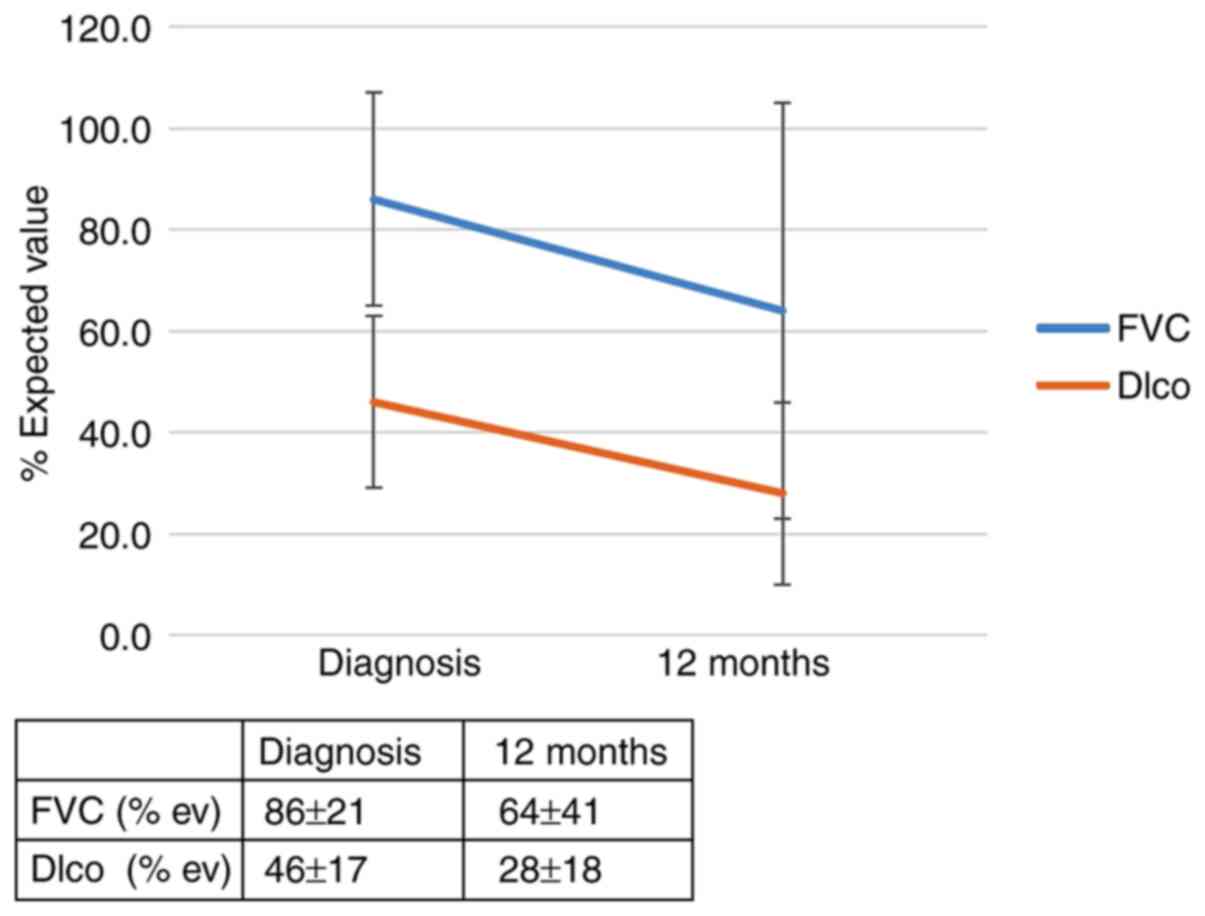
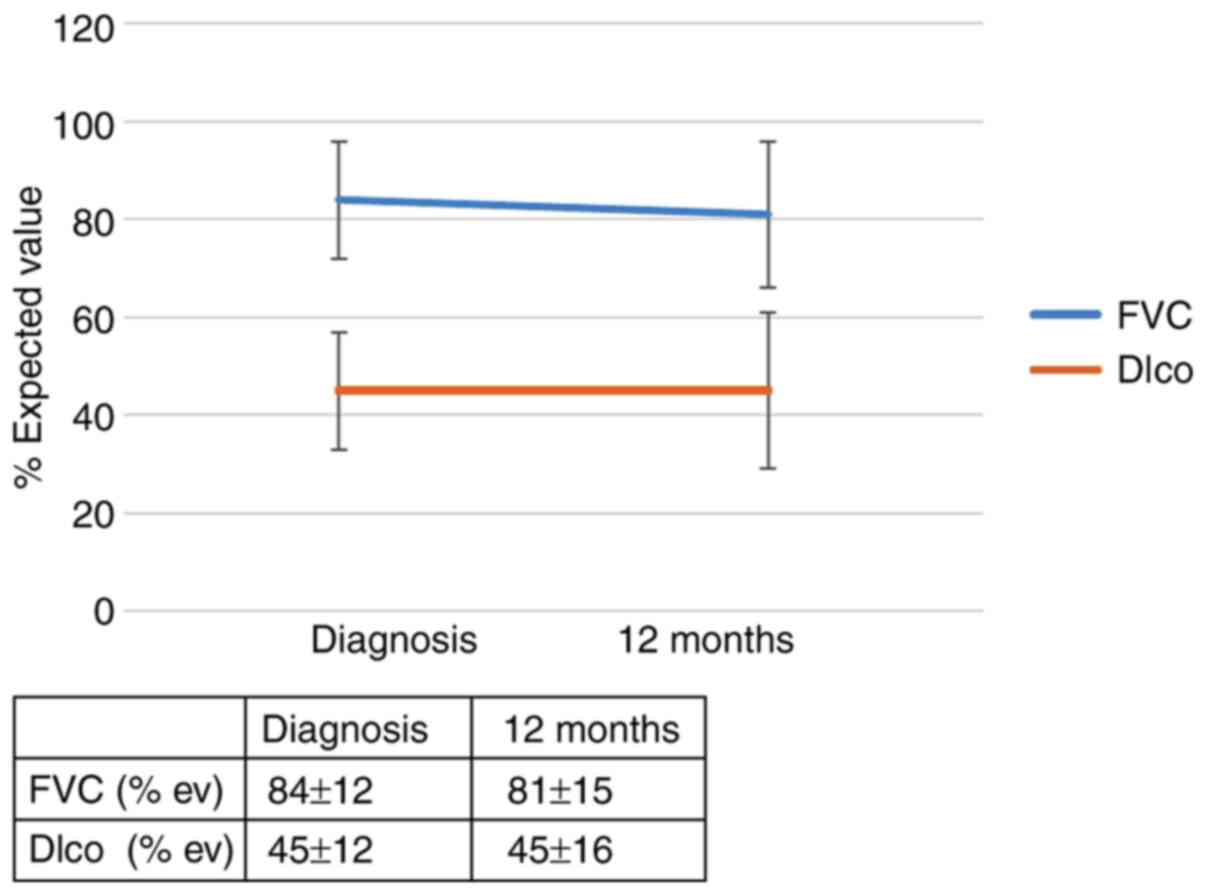
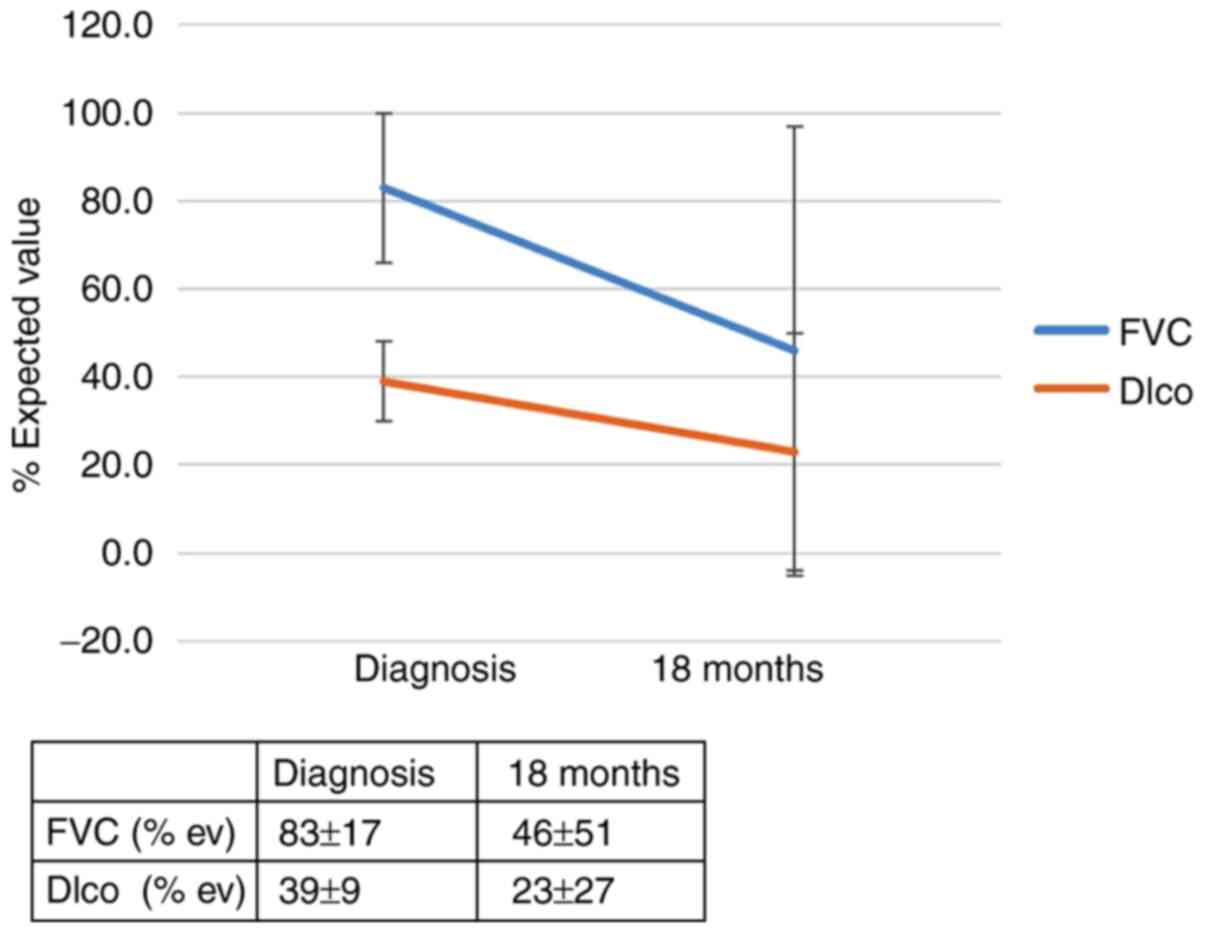
the IL-4 SNP (rs 2070874) on patient outcomes was observed; RR of
progression for the T allele: 5.56 (95% CI, 0.79-39.0; P=0.053).
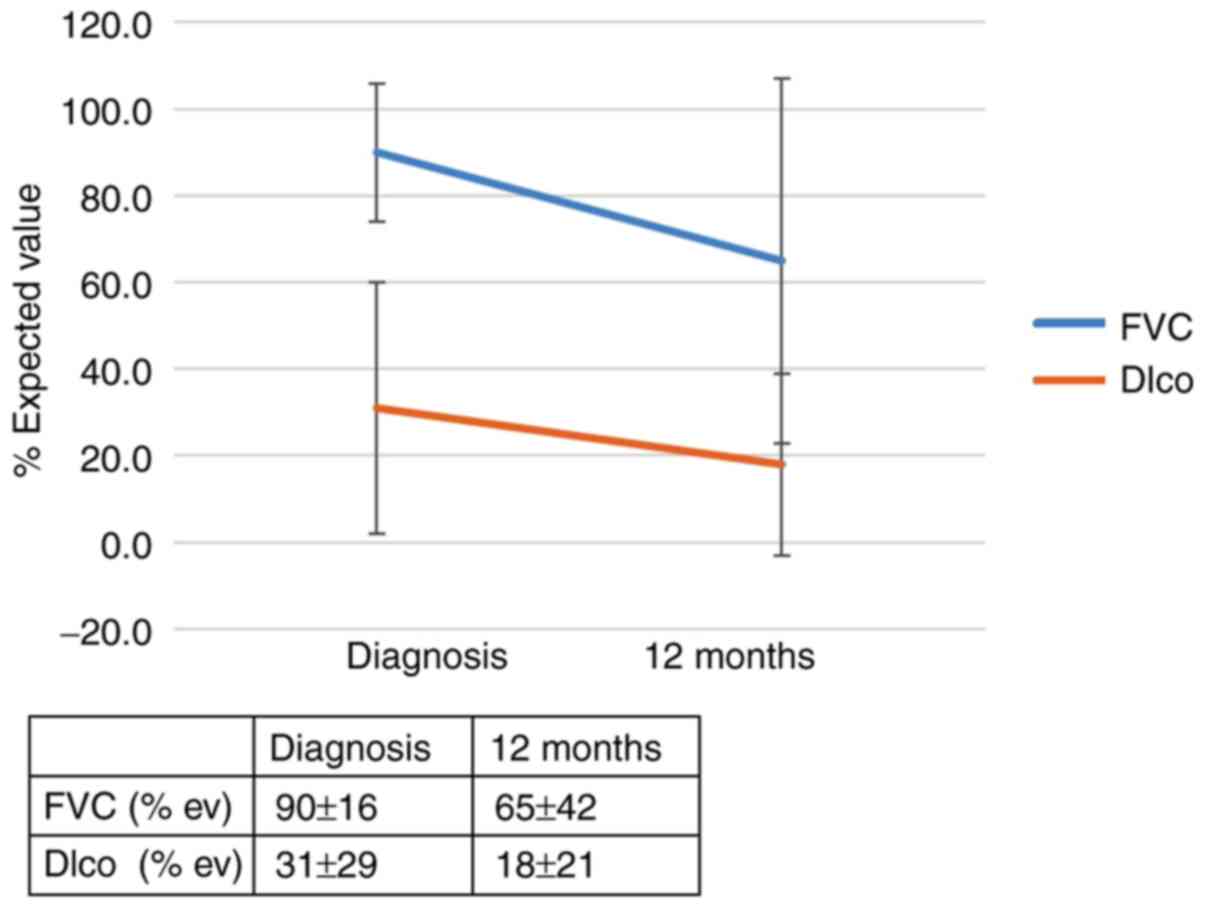
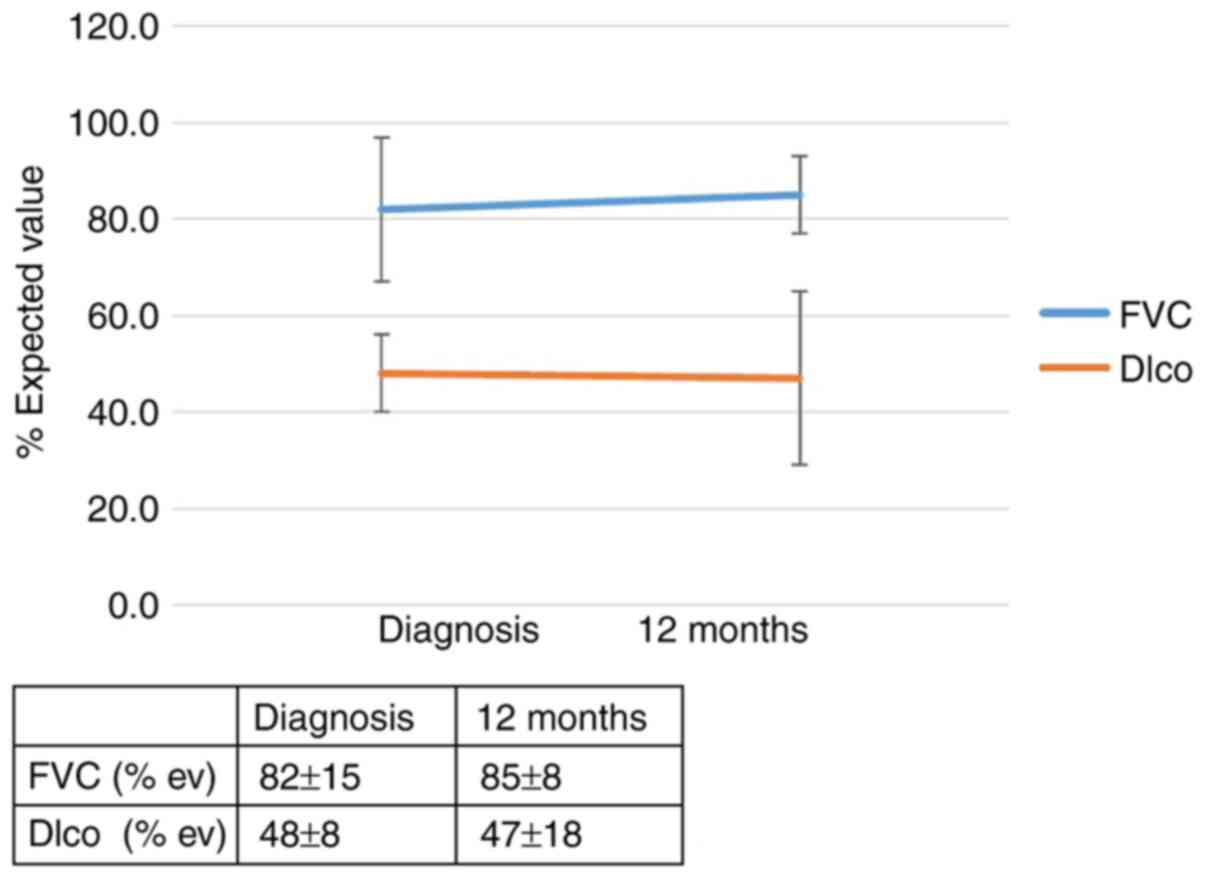
The RR of progression in patients with the IL-4 SNP (rs 2243250)
and the CT and TT genotypes (allele CC was found to be protective)
was 4.3 (95% CI, 1.1-17.5; P=0.046). The effect of the genotype
IL-4 (rs 2070874) on the outcome of patients with IPF 12 months
after diagnosis is summarized in Figs.
1 and 2 and in Table SIIA. The effect of the genotype IL-4
(rs 2243250) on the outcomes of patients with IPF 12 months after
diagnosis is shown in Figs. 3 and
4 and Table SIIB. Call cluster plots showing
distribution of rs 2070874, rs 2243250 and rs 111521887 genotypes
from mass spectrometry are included in Fig. S1A-C, respectively.
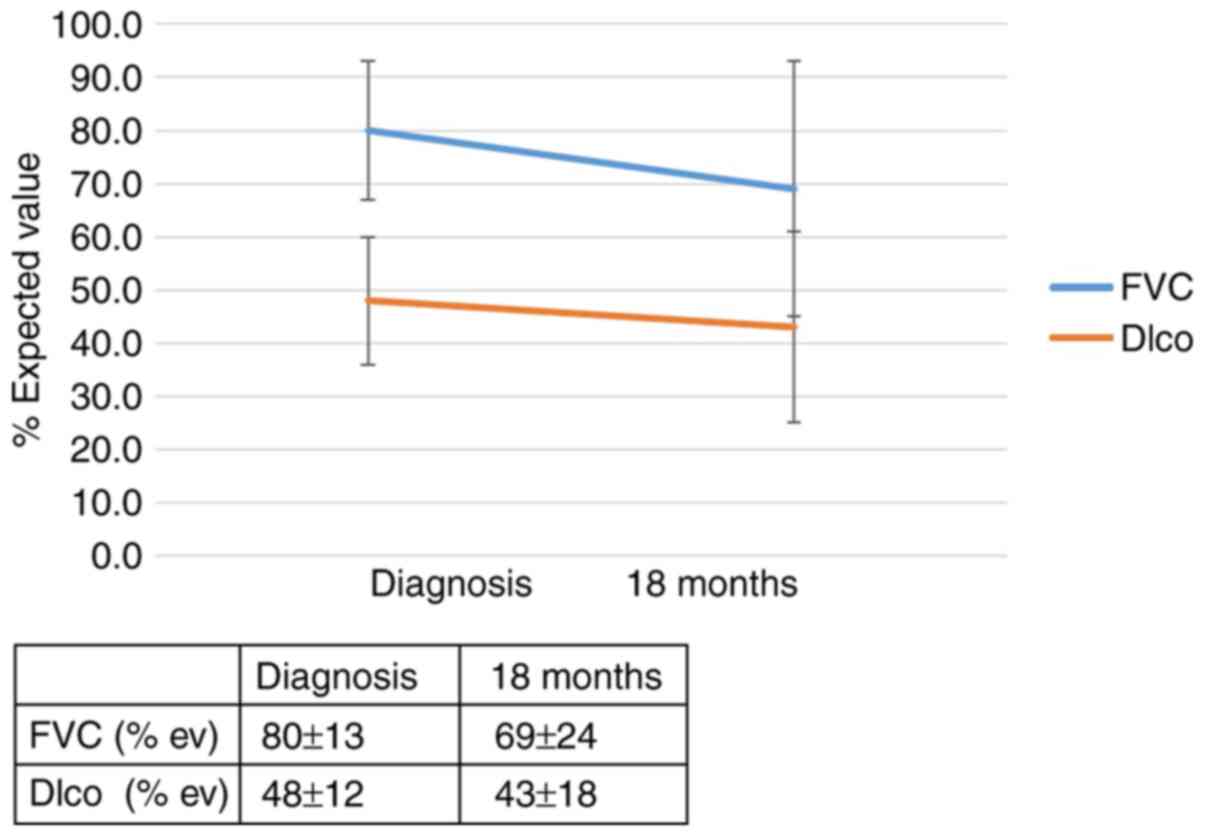
A total of 18 months after the diagnosis of IPF, a
clinically significant effect of the TOLLIP polymorphism was
detected on patient outcome (rs 111521887, risk allele GC; RR, 7.2;
95% CI, 0.97-53.6; P=0.052). The effect of the genotype of TOLLIP
(rs 111521887) on the outcome of patients with IPF 18 months after
diagnosis is shown in Figs. 5 and
6 and in Table SII. No relationship was found
between the patient outcomes and polymorphisms of the MUC5B, TGF-β,
TAP1, TAP2 and SFTPA genes.
Discussion
In the present study, the effects of IL-4, MUC5B,
TGF-β, TOLLIP, TAP1, TAP2 and SFTPA gene polymorphisms on the IPF
course over 18 months were evaluated. The results showed an effect
of the IL-4 (rs 2070874 and rs 2243250) polymorphism on disease
progression 12 months after diagnosis of IPF and an effect of the
TOLLIP gene polymorphism (rs 111521887) on IPF outcomes 18 months
after diagnosis. No relationship was found between the patient
outcomes and polymorphisms of the MUC5B, TGF-β, TAP1, TAP2 and
SFTPA genes in the study group.
The patient data were evaluated at the time of
diagnosis and again 6, 12 and 18 months after diagnosis. No studies
have evaluated the optimal visit interval to the best of our
knowledge, yet most clinical trials evaluate patient outcomes after
12 months. As cases of rapid IPF progression have been described
and longitudinal data were available for most of the enrolled
subjects, data from months 6 and 18 were also included into the
study protocol. IPF is the most aggressive form of progressive
fibrosing interstitial lung disease, and a 6-month interval and 10%
decline in FVC were found to be meaningful in a previous study
(29). A Dlco decline of >15% was
incorporated as a marker of disease progression. The reason for
including Dlco is explained in the study by Nathan et al
(30); an FVC decline during the
first year of follow-up may not be predictive of further
progression of the disease. Despite the Dlco limitations, adding
this parameter to the FVC decline is a reasonable means of defining
the progression of IPF (29).
SFTPA mutations are detected primarily in patients
with familial interstitial pneumonia or those with a concomitant
diagnosis of lung fibrosis and lung cancer (31). However, no patient in the present
study had a history of familial interstitial pneumonia or lung
cancer, possibly explaining why no associations were found between
SFTPA gene polymorphisms and prognosis.
There are several literature reports concerning
various TGF-β gene polymorphisms and either the risk of developing
IPF or the disease outcome. For example, the rs 1800470 variant has
been reported to be a significant risk factor for IPF (32). The TGF-β polymorphism (rs 1800470)
can modify the expression of the protein, and the G allele was
shown to increase TGF-β secretion (33). However, not only the concentration of
TGF-β but also the concentration of TOLLIP orchestrates the process
of fibrosis. TOLLIP antagonizes TGF-β signaling by degrading TGF-β1
receptors through SMAD7-dependent mechanisms, and thus dampens the
profibrotic cascade (34).
Noth et al (13) identified three TOLLIP SNPs associated
with IPF risk; two of these (rs 111521887 and rs 5743890) were
associated with an increased risk of IPF, and the third (rs
5743894) was associated with a lower risk of the disease. However,
if subjects carrying this mutation develop IPF, they have a higher
risk of mortality (34). Both the rs
111521887 and rs 5743894 variants are associated with a 20-50%
reduction in TOLLIP mRNA expression. Despite the low number of
studied subjects, the present study showed that the effects of
different gene polymorphisms on the disease course may be time
dependent, the effect of the TOLLIP rs 111521887 polymorphism on
IPF outcomes were not observed until 18 months after the IPF
diagnosis. Thus, it may not be a driver of the disease, but instead
modify/enhance the fibrotic process. Since TOLLIP and TGF-β
interact with each other, a more complex evaluation of the effects
of multiple gene polymorphisms is required.
Both TAP 1 and TAP 2 polymorphisms may result in
TAP1/2 protein deficiency (reported for rs 1057141, rs 1135216 and
rs 241447), which leads to bare lymphocyte syndrome (35). They were also found to be associated
with chronic infections and impaired antigen clearance, which could
potentially lead to chronic exposure to potentially harmful
substances, and enhance the risk of persistent lung damage
(36). Exposure to inhalation of
antigens, as well as their role in IPF pathogenesis, has been
widely discussed as an important trigger of lung injury, followed
by impaired healing (37). However,
neither persistent viral infection nor chronic exposure to
inhalation of antigens represent major contributors to IPF
pathogenesis. This may explain why no association between TAP gene
polymorphisms and IPF outcomes were found in the present study.
Although certain studies have suggested that the
MUC5B minor allele does not affect IPF survival (38,39),
others have challenged the initial study of Peljto et al
(38), declaring an improved
prognosis in patients with IPF with the minor allele and, after
adjusting for index event bias, proposing a significant association
of MUC5B polymorphism and decreased survival (40,41). The
results of the present study support the prognostic benefit of the
rs35705950 polymorphism. Previous study has suggested an effect of
the MUC5B variant on the bacterial burden in patients with IPF,
possibly explaining its positive effect on the evolution of the
disease (41).
It is difficult to establish the specific effect of
IL-4 gene polymorphisms in the very complex setting of an immune
response. Certain polymorphisms affect the concentration of the
encoded protein; however, as different levels may be evident in
different compartments and other gene-protein interactions or
protein-protein interactions may occur, the effects are not easily
assessed. This situation is further complicated by the detection of
the splice variant IL-4δ2 mRNA, which is hypothesized to regulate
IL-4 production and acts as a factor to abrogate the Th2 response.
However, the regulation of its production is currently unknown, and
the effect of IL-4 gene polymorphisms on alternative splicing and
production of the IL-4δ2 mRNA isoform remains elusive (42,43).
This may lead to the assumption that one patient with IPF may carry
more than one gene mutation, and that multiple genetic factors and
mechanisms might play roles in IPF progression, as reported in the
study by Dressen et al (44)
in which variations of the course of IPF according to the patient's
genetic background was observed. They showed that patients with
telomere complex-associated mutations had a more rapidly
progressive IPF course than those with MUC5B mutations, and
suggested that different pathogenetic events may lead to the same
fibrotic outcome in affected subjects. Interestingly, neither MUC5B
nor most of the identified IPF-associated telomere complex gene
mutations were identified as prognostic factors in a study using a
functional genomic model approach (45).
The major limitation of the present study was the
number of enrolled patients and the relatively short observation
period. Despite finding data associating IL-4 and TOLLIP gene
polymorphisms with the course of IPF, additional studies with
larger patient cohorts are required to further explore the
significance of the findings. The presented results should be
interpreted with caution; however, they may suggest that a broader
approach, involving not only the risk of developing the disease but
also the disease course, should be implemented in future
studies.
In conclusion, the identified IL-4 and TOLLIP gene
polymorphisms may represent disease course-modifying factors, but
not drivers of IPF.
Supplementary Material
Call cluster plots showing the
distributions of the different polymorphisms. Call cluster plots
showing distribution of (A) rs 2070874, (B) rs 2243250 and (C) rs
111521887 genotypes.
List of investigated gene
polymorphisms.
Effect of the SNPs on IPF
outcomes.
Acknowledgements
Not applicable.
Funding
The present study was supported by funding from the Ministry of
Health, Internal Grant Agency, Czech Republic (grant no.
LF_2020_004/2021_014), Palacky University, Olomouc, Czech Republic
(grant no. 61989592), Thomayer Hospital, Prague, Czech Republic
(grant no. TN00064190) and in part from ENOCH (grant no.
(CZ.02.1.01/0.0/0.0/16_019/0000868).
Availability of data and materials
The datasets generated and/or analyzed during the
present study are available in the European Variation Archive
repository (https://www.ebi.ac.uk/ena/data/view/PRJEB44734).
Author's contributions
MS designed the study, collected patient data and
blood samples, and wrote the manuscript. AK and KS performed the
laboratory studies. JS performed the statistical analysis. MV and
MP assisted with study design. All authors have read and approved
the final manuscript. MS, MP and KS confirmed the authenticity of
all the raw data.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
Thomayer Hospital and Institute of Clinical and Experimental
Medicine, Prague, Czech Republic. Written informed consent was
obtained from all enrolled subjects.
Patient consent for publication
Patient consent for publication was obtained as part
of the written informed consent.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Michalski JE and Schwartz DA: Genetic risk
factors for idiopathic pulmonary fibrosis: Insights into
immunopathogenesis. J Inflamm Res. 13:1305–1318. 2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Churg A: Centrilobular fibrosis in
fibrotic (Chronic) hypersensitivity pneumonitis, usual interstitial
pneumonia, and connective tissue disease-associated interstitial
lung disease. Arch Pathol Lab Med. 144:1509–1516. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jain R, Yadav D, Puranik N, Guleria R and
Jin JO: Sarcoidosis: Causes, diagnosis, clinical features, and
treatments. J Clin Med. 9(1081)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Meyer KC: Pulmonary fibrosis, part I:
Epidemiology, pathogenesis, and diagnosis. Expert Rev Respir Med.
11:343–359. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ballester B, Milara J and Cortijo J:
Mucins as a new frontier in pulmonary fibrosis. J Clin Med.
8(1447)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Stock CJW and Renzoni EA: Telomeres in
interstitial lung disease. J Clin Med. 10(1384)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Snijder J, Peraza J, Padilla M, Capaccione
K and Salvatore MM: Pulmonary fibrosis: A disease of alveolar
collapse and collagen deposition. Expert Rev Respir Med.
13:615–619. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Schwartz DA: Idiopathic pulmonary fibrosis
is a genetic disease involving mucus and the peripheral airways.
Ann Am Thorac Soc. 15 (Suppl):S192–S197. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Barros A, Oldham J and Noth I: Genetics of
idiopathic pulmonary fibrosis. Am J Med Sci. 357:379–383.
2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Walters GI: Occupational exposures and
idiopathic pulmonary fibrosis. Curr Opin Allergy Clin Immunol.
20:103–111. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhang L, Ikegami M, Korfhagen TR,
McCormack FX, Yoshida M, Senior RM, Shipley JM, Shapiro SD and
Whitsett JA: Neither SP-A nor NH2-terminal domains of SP-A can
substitute SP-D in regulation of alveolar homeostasis. Am J Physiol
Lung Cell Mol Physiol. 291:L181–L190. 2006.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Froidure A, Marchal-Duval E, Homps-Legrand
M, Ghanem M, Justet A, Crestani B and Milleux C: Chaotic activation
of developmental signalling pathways drives idiopathic pulmonary
fibrosis. Eur Respir Rev. 29(190140)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Noth I, Zhang Y, Ma SF, Flores C, Barber
M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, et al:
Genetic variants associated with idiopathic pulmonary fibrosis
susceptibility and mortality: A genome-wide association study.
Lancet Respir Med. 1:309–317. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Aquino-Galvez A, Camarena A, Montaño M,
Juarez A, Zamora AC, González-Avila G, Checa M, Sandoval-López G,
Vargas-Alarcon G, Granados J, et al: Transporter associated with
antigen processing (TAP) 1 gene polymorphisms in patients with
hypersensitivity pneumonitis. Exp Mol Pathol. 84:173–177.
2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hanalioglu D, Ayvaz DC, Ozgur TT, van der
Burg M, Sanal O and Tezcan I: A novel mutation in TAP1 gene leading
to MHC class I deficiency: Report of two cases and review of the
literature. Clin Immunol. 178:74–78. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
1000 Genome Project: https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes,
visited on 14-Mar-2020.
|
|
17
|
Cho YA and Kim J: Association of IL4,
IL13, and IL4R polymorphisms with gastrointestinal cancer risk: A
meta-analysis. J Epidemiol. 27:215–220. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yoshimoto T: The hunt for the source of
primary interleukin-4: How we discovered that natural killer T
cells and basophils determine T helper type 2 cell differentiation
in vivo. Front Immunol. 9(716)2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gieseck RL III, Wilson MS and Wynn TA:
Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol.
18:62–76. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Raghu G, Remy-Jardin M, Myers JL, Richeldi
L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SV, Morell F,
et al: Diagnosis of idiopathic pulmonary fibrosis. An official
ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care
Med. 198:e44–e68. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kocova E, Vanasek J, Koblizek V, Novosad
J, Elias P, Bartos V and Sterclova M: Scoring of the radiological
picture of idiopathic interstitial pneumonia: A study to verify the
reliability of the method. Acta Radiol Open.
4(2058460115605865)2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Macintyre N, Crapo RO, Viegi G, Johnson
DC, van der Grinten CP, Brusasco V, Burgos F, Casaburi R, Coates A,
Enright P, et al: Standardisation of the single-breath
determination of carbon monoxide uptake in the lung. Eur Respir J.
26:720–735. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Pellegrino R, Viegi G, Brusasco V, Crapo
RO, Burgos F, Casaburi R, Coates A, van der Grinten CP, Gustafsson
P, Hankinson J, et al: Interpretative strategies for lung function
tests. Eur Respir J. 26:948–968. 2005.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wuyts WA, Wijsenbeek M, Bondue B, Bouros
D, Bresser P, Robalo Cordeiro C, Hilberg O, Magnusson J, Manali ED,
Morais A, et al: Idiopathic pulmonary fibrosis: Best practice in
monitoring and managing a relentless fibrotic disease. Respiration.
99:73–82. 2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Vasakova M, Sterclova M, Matej R, Olejar
T, Kolesar L, Skibova J and Striz I: IL-4 polymorphisms, HRCT score
and lung tissue markers in idiopathic pulmonary fibrosis. Hum
Immunol. 74:1346–1351. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kishore A, Žižková V, Kocourková L and
Petřek M: A dataset of 26 candidate gene and pro-inflammatory
cytokine variants for association studies in idiopathic pulmonary
fibrosis: Frequency distribution in normal Czech population. Front
Immunol. 6(476)2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Sikorova K, Kishore A, Rapti A, Adam K,
Kocourkova L, Zizkova V, Charikiopoulou M, Kalianos A, Bouros E,
Bouros D and Petrek M: Association of TGF-β3 and ANXA11 with
pulmonary sarcoidosis in Greek population. Expert Rev Respir Med.
14:1065–1069. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Collard HR, Ryerson CJ, Corte TJ, Jenkins
G, Kondoh Y, Lederer DJ, Lee JS, Maher TM, Wells AU, Antoniou KM,
et al: Acute exacerbation of idiopathic pulmonary fibrosis. An
international working group report. Am J Respir Crit Care Med.
194:265–75. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Taha N, D'Amato D, Hosein K, Ranalli T,
Sergiacomi G, Zompatori M and Mura M: Longitudinal functional
changes with clinically significant radiographic progression in
idiopathic pulmonary fibrosis: Are we following the right
parameters? Respir Res. 21(119)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Nathan N, Giraud V, Picard C, Nunes H,
Dastot-Le Moal F, Copin B, Galeron L, De Ligniville A, Kuziner N,
Reynaud-Gaubert M, et al: Germline SFTPA1 mutation in familial
idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet.
25:1457–1467. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Deng Y, Li Z, Liu J, Wang Z, Cao Y, Mou Y,
Fu B, Mo B, Wei J, Cheng Z, et al: Targeted resequencing reveals
genetic risks in patients with sporadic idiopathic pulmonary
fibrosis. Hum Mutat. 39:1238–1245. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Juarez I, Gutierrez A, Vaquero-Yuste C,
Molanes-López EM, López A, Lasa I, Gómez R and Martin-Villa JM:
TGFB1 polymorphisms and TGF-β1 plasma levels identify gastric
adenocarcinoma patients with lower survival rate and disseminated
disease. J Cell Mol Med. 25:774–783. 2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhu L, Wang L, Luo X, Zhang Y, Ding Q,
Jiang X, Wang X, Pan Y and Chen Y: Tollip, an intracellular
trafficking protein, is a novel modulator of the transforming
growth factor-β signaling pathway. J Biol Chem. 287:39653–3963.
2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kaur A, Mathai SM and Schwartz DA:
Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis,
and treatment. Front Med (Lausanne). 4(154)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Gadola SD, Moins-Teisserenc HT, Trowsdale
J, Gross WL and Cerundolo V: TAP deficiency syndrome. Clin Exp
Immunol. 121:173–178. 2000.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Qiu B, Huang B, Wang X, Liang J, Feng J,
Chang Y and Li D: Association of TAP1 and TAP2 polymorphisms with
the outcome of persistent HBV infection in a northeast Han Chinese
population. Scand J Gastroenterol. 47:1368–1374. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Biondini D, Cocconcelli E, Bernardinello
N, Lorenzoni G, Rigobello C, Lococo S, Castelli G, Baraldo S, Cosio
MG, Gregori D, et al: Prognostic role of MUC5B rs35705950 genotype
in patients with idiopathic pulmonary fibrosis (IPF) on
antifibrotic treatment. Respir Res. 22(98)2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Peljto AL, Zhang Y, Fingerlin TE, Ma SF,
Garcia JG, Richards TJ, Silveira LJ, Lindell KO, Steele MP, Loyd
JE, et al: Association between the MUC5B promoter polymorphism and
survival in patients with idiopathic pulmonary fibrosis. JAMA.
309:2232–2239. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Seibold MA, Wise AL, Speer MC, Steele MP,
Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong
SD, et al: A common MUC5B promoter polymorphism and pulmonary
fibrosis. N Engl J Med. 364:1503–1512. 2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Jiang H, Hu Y, Shang L, Li Y, Yang L and
Chen Y: Association between MUC5B polymorphism and susceptibility
and severity of idiopathic pulmonary fibrosis. Int J Clin Exp
Pathol. 8:14953–14958. 2015.PubMed/NCBI
|
|
41
|
Molyneaux PL, Cox MJ, Willis-Owen SA,
Mallia P, Russell KE, Russell AM, Murphy E, Johnston SL, Schwartz
DA, Wells AU, et al: The role of bacteria in the pathogenesis and
progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care
Med. 190:906–913. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Luzina IG, Lockatell V, Todd NW, Highsmith
K, Keegan AD, Hasday JD and Atamas SP: Alternatively spliced
variants of interleukin-4 promote inflammation differentially. J
Leukoc Biol. 89:763–770. 2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Luzina IG, Keegan AD, Heller NM, Rook GA,
Shea-Donohue T and Atamas SP: Regulation of inflammation by
interleukin-4: A review of ‘alternatives’. J Leukoc Biol.
92:753–764. 2012.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Dressen A, Abbas AR, Cabanski C, Reeder J,
Ramalingam TR, Neighbors M, Bhangale TR, Brauer MJ, Hunkapiller J,
Reeder J, et al: Analysis of protein-altering variants in
telomerase genes and their association with MUC5B common variant
status in patients with idiopathic pulmonary fibrosis: A candidate
gene sequencing study. Lancet Respir Med. 6:603–614.
2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Huang Y, Ma SF, Vij R, Oldham JM,
Herazo-Maya J, Broderick SM, Strek ME, White RW, Hogarth DK, Sandbo
NK, et al: A functional genomic model for predicting prognosis in
idiopathic pulmonary fibrosis. BMC Pulm Med. 15(147)2015.PubMed/NCBI View Article : Google Scholar
|