Introduction
Alport syndrome (AS) is a hereditary disease
characterized by hematuria, proteinuria and chronic progressive
failure of renal function, as well as potential ocular
abnormalities and sensorineural deafness (1,2). The
basic pathological mechanism of AS is glomerular basement membrane
(GBM) abnormalities, due to pathogenic mutations in the genes
encoding the α3, α4 and α5 chains of type IV collagen (2,3). AS is
generally divided into three inherited types. The most common type
is X-linked dominant hereditary type AS (XLAS), which accounts for
>80% of AS cases, and involves a mutation in the gene encoding
the α5 chain (COL4A5) located at chromosome Xq2. Meanwhile,
autosomal recessive AS (ARAS), which accounts for 15% of AS cases,
and rare autosomal dominant AS (ADAS), which accounts for 5% of AS
cases, are induced by mutations in the collagen type IV α 3 chain
(COL4A3) and collagen type IV α 4 chain (COL4A4)
genes located at autosomal chromosome 2q36.3 (4,5).
AS had been historically diagnosed based on the
clinical manifestations of hematuria, proteinuria and renal
dysfunction; however, the use of renal biopsy with light microscopy
(LM) and electron microscopy (EM), or immunohistochemical staining
has become more popular in the past five decades (4). Ear and eye tests, alongside pedigree
analysis have proven helpful for improving the accuracy of an AS
diagnosis (4,6). However, missed diagnoses are still
common in AS, and early intervention is crucial to delay disease
progression; therefore, more precise diagnostic tools are required
(4). Advances in genetic diagnostic
technologies have largely made up for the shortcomings of
traditional tools and have revealed that genotype defects are not
linearly correlated with the various clinical phenotypes (2,6). In
particular, patients with ADAS with heterozygous mutations are
clinically characterized by slower end-stage renal disease (ESRD)
progression with less typical extra-renal manifestations compared
with ARAS, in which gene mutations usually occur in both alleles
(6). Thus, the incidence of missed
diagnoses for ADAS could potentially be higher than previously
suggested (7), which could be
sufficiently treated by early administration of
angiotensin-converting-enzyme inhibitor (ACEi) (4,6,8).
Therefore, the introduction of whole-exome sequencing (WES)
analysis can markedly improve the diagnostic and prognostic
accuracy for AS (3,6).
In the present study, a proposed AS proband was
identified based on the patients' clinical manifestations and was
further confirmed by pathological examination. WES was recommended
for the proband to determine the exact mutation sites. Two novel
pathogenic COL4A3 mutations (c.2603G>A; p.G868E, and
c.583G>A; p.G195S) were identified. The results of the present
study enrich the pool of mutations associated with ADAS, whilst
showing that WES may be a powerful tool for AS diagnosis and
mutation characterization, as well as for assisting with precise
medical intervention, prognosis prediction and a pre-implantation
genetic diagnosis (6).
Case report
The present study was approved by the Ethics
Committee of Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology (Wuhan, China). Written
informed consent was obtained from each patient, or from the
parents of any patients <18 years old, for participation in the
study. The proband (III.2) was a 13-year-old male patient who was
admitted to the pediatric nephrology department of Tongji Hospital
after presenting with persistent hematuria for >8 years. The
proband (III.2) and 9 members of the family (6 men and 4 women)
clinically suspected of AS were recruited. All patients underwent
blood and urine routine examination, as well as liver and kidney
function evaluations. Eyesight and visual filed inspection, hearing
tests and vestibular function tests were also performed, where
necessary.
The family's clinical history was examined carefully
for each case through the reconstruction of the pedigree for at
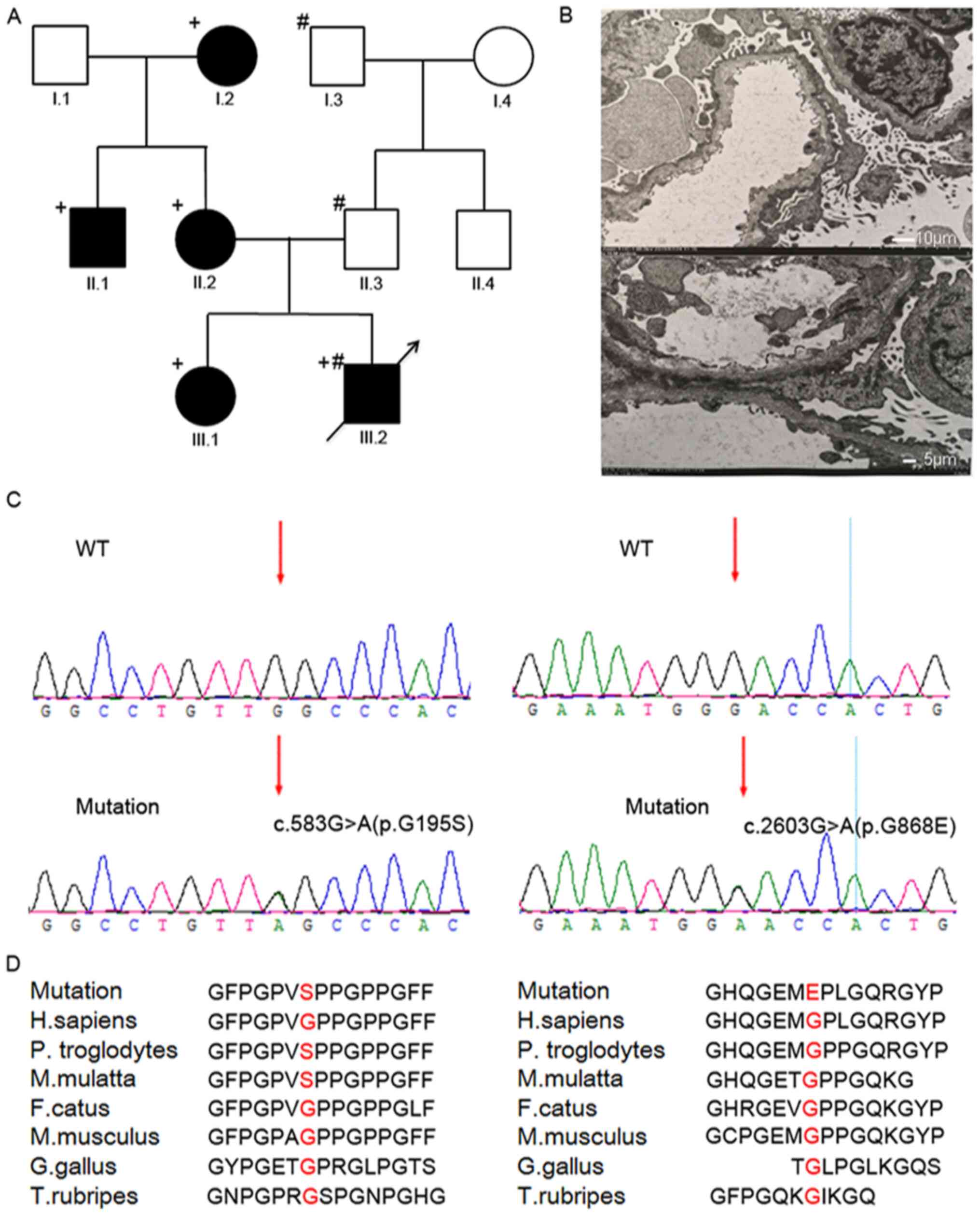
least three generations (Fig. 1A).
The renal biopsy of the proband (III.2) and his older sister
(III.1) were examined in the nephrology laboratory of Tongji
Hospital. The proband fulfilled three of the AS clinical diagnostic
criteria: The family history of renal failure, hematuria and poor
vision (6). The hearing test and
vestibular function test were normal. The eye test revealed retinal
detachment in the left eye. At the age of 13 years, urine analysis
revealed proteinuria (2.8 g/24 h), so a renal biopsy was performed.
The abnormal GBM was identified by EM, which was compatible with
the typical ultrastructural abnormalities characteristic of thin
basement membrane disease. Antiproteinuric treatment was reinforced
with ACEi and tacrolimus.
Materials and methods
Whole blood genomic DNA
extraction
Fresh venous blood (3 ml) was obtained from patients
of the pedigree for genomic DNA extraction, following the
manufacturer's instructions (QIAamp; Qiagen AB).
WES and bioinformatics analysis
WES was performed by Beijing Genomics Institution in
Shenzhen, China. The experimental procedures were performed as
described previously (9). Briefly,
the concentration and purification of the proband's DNA
(OD260/280>1.8) was detected using a NanoDrop™ 2000
spectrophotometer (Thermo Fisher Scientific, Inc.), and a 1 µg
sample was used for DNA library construction. Genomic DNA was
randomly broken into 150-250-bp-long fragments using a Covaris S2
Sonicator (Covaris). End repairing, A-tailing and adapter ligation
of fragments were performed in a single tube (total 110 µl) for 15
min at 22˚C (iGeneTech, Inc.), followed by a purification with 88
µl Agencourt Ampure XP beads (Beckman Coulter, Inc.), and collected
in 20 µl nuclease-free water. After pre-PCR amplification (9˚C for
3 min; followed by 8 cycles of 98˚C for 20 sec, 60˚C for 15 sec and
72˚C for 30 sec; with a final extension step at 72˚C for 10 min;
and held at 4˚C) using a Library Prep kit (iGeneTech, Inc.), the
product was purified using Agencourt Ampure XP beads and quantified
using a Qubit dsDNA BR assay kit (Thermo Fisher Scientific,
Inc.).
The purified PCR products were hybridized with a
human gene chip for 68-72 h using an MGIEasy Exome Capture V4 Probe
Set (MGI Tech Co., Inc.). The target fragments were captured by the
Cap beads (iGeneTech, Inc.), and then underwent further capture
post-PCR (95˚C for 3 min; followed by 8 cycles of 98˚C for 20 sec,
60˚C for 15 sec and 72˚C for 30 sec; with a final extension step at
72˚C for 10 min; and held at 4˚C) using a TargetSeq One kit
(iGeneTech, Inc.). The PCR products were purified using Agencourt
Ampure XP beads, qualified using the Qubit ssDNA kit (Thermo Fisher
Scientific, Inc.) and amplified to make DNA nanoballs (10), which consist of >300 copies of
one molecule. Pair-end 100 bases reads were generated using the
combinatorial Probe-Anchor Synthesis-based BGISEQ-500 platform
(BGI) as described previously (10).
The obtained sequences were compared to those of the
human reference genome (hg19) using the Burrows-Wheeler Aligner
software (version 0.7.15) to acquire valid sequences, and potential
pathogenic variants [by comparing with dbSNP, 1000 Genomes Project,
Online Mendelian Inheritance in Man (OMIM) and ClinVar]. The
function of variants was predicted using SIFT version 6.1
(sift.bii.a-star.edu.sg) and PolyPhen-2
(genetics.bwh.harvard.edu/pph2). The
conservation of amino acid sequences of COL4A3 gene variants
was analyzed using the NCBI database (ncbi.nlm.nih.gov/protein).
Sanger sequencing verification of
COL4A3 gene mutation
According to the gene identification site, fragments
were amplified by PCR with the following thermocycling conditions:
95˚C for 15 min; followed by 30 cycles of 94˚C for 30 sec, 53˚C for
30 sec and 72˚C for 30 sec; with a final extension step of 72˚C for
10 min. The sequences of the primers were: c.583G>A forward,
5'-AGGGAGGCGGAGGGTAC-3' and reverse, 5'-ATGGCCCCTGGTTTCTTAC-3'; and
c.2603G>A forward, 5'-AGGGAGGCGGAGGGTAC-3' and reverse,
5'-ATGGCCCCTGGTTTCTTAC-3'. PCR products were sequenced by Sanger
sequencing (ABI 3730XL sequencer; Thermo Fisher Scientific, Inc.)
and the Sanger sequencing peak map and results were analyzed on
DNASTAR Lasergene version 13.0 (DNASTAR, Inc.).
Results
The clinical investigation of the pedigree suggested
the presence of hereditary ADAS within the family (Fig. 1A). The proband was a 13-year-old
boy, who presented with repeated gross hematuria aggravated by
fever for 8 years, and whose left vision had been progressively
decreasing for 2 years due to retinal detachment. Routine urine
routine revealed the following abnormal results: Red blood cells
(RBC), 3+; white blood cells, ±; urinary albumin, 1+; microscopic
RBC per full field/high power; irregular RBC %, 30.0%; and 24 h
urinary albumin, 2.8 g. The levels of serum creatinine (Scr) and
blood urea nitrogen (BUN) were normal. Renal biopsy showed partial
glomerular sclerosis, mild mesangial cell hyperplasia and slightly
increased mesangial matrix content without segmental necrosis and
thrombosis under LM. No obvious abnormalities were identified by
immunofluorescence. EM revealed that the capillary basement
membrane was segmentally thinned, and the lowest thickness was 160
nm (physiological thickness is ~300 nm), and even some dense layers
were torn and delaminated (Fig.
1B). These results suggested that the proband could be
diagnosed with AS and recommended for genetic testing.
The pedigree investigation revealed hereditary ADAS
(Table I). The proband's
15-year-old sister had abnormal urinalysis findings for 1 year,
including the following: RBC, 3+; urinary albumin, 2+; 24 h urinary
albumin, 500 mg. The levels of Scr (65 µmol/l) and BUN (4.52
mmol/l) were normal. Renal biopsy under LM revealed segmental
mesangial cells and mild stromal hyperplasia with segmental
endothelial proliferation. Her EM results were similar to those of
her younger brother (data not shown). The sister had normal hearing
and vision after examination. Thus, the sister could also be
diagnosed with AS. In the maternal family, the grandmother had
uremia, whereas the mother and uncle had chronic glomerulonephritis
without ocular or auditory abnormalities. No significant abnormal
manifestations were observed in the paternal members.
 | Table IClinical and molecular characteristics
of the recruited patients. |
Table I
Clinical and molecular characteristics
of the recruited patients.
| Patient | Sex | Age | Hematuria | Proteinuria | Renal Function | Mutations |
|---|
| I1 | M | 68 | No | No | Normal | Normal |
| I2 | F | 65 | Yes | >3.5 g/24 h | Chronic renal
failure-dialysis | c.2603G>A;
p.G868E |
| I3 | M | 70 | No | No | Normal | c.583G>A;
p.G195S |
| I4 | F | 67 | No | No | Normal | Normal |
| II1 | M | 35 | Yes | 1.2 g/24 h | Normal | c.2603G>A;
p.G868E |
| II2 | F | 37 | Yes | No | Normal | c.2603G>A;
p.G868E |
| II3 | M | 41 | No | No | Normal | c.583G>A;
p.G195S |
| II4 | M | 43 | No | No | Normal | Normal |
| III1 | F | 15 | Yes | 0.5 g/24 h | Normal | c.2603G>A;
p.G868E |
| III2 | M | 13 | Yes | 2.8 g/24 h | Normal | c.2603G>A;
p.G868E +c.583G>A; p.G195S |
The proband's WES raw data output was 22.71 G, and
the target area length was 58.68 Mb with 99.92% coverage and an
average sequencing depth of 82.93X, which met the experimental
design requirements. WES identified two mutations in the proband.
One located in exon 10 of the COL4A3 gene (NM_000091.4) on
chromosome 2, in which G in the 583rd position was changed to A
(c.583G>A), leading to the GGC→AGC codon change and the amino
acid change from Gly to Ser (p.G195S; Fig. 1C). The other mutation was the change
from G to A in the 2,603rd position in exon 32 of the same gene,
resulting in the GGA→GAA codon change and 868th amino acid changing
from Gly to Glu(p.G868E; Fig. 1D).
These two mutations were extremely rare and had not been detected
before in databases such as dbSNP, 1000 Genomes Project, OMIM,
Human Gene Mutation Database and ClinVar.
All members of the pedigree underwent Sanger
sequencing for the COL4A3 gene. The c.2603G>A mutation
was found in four family members who presented with various AS
symptoms, including the mother, uncle and grandmother of the
maternal family and the proband's older sister (Table I). The c.583G>A mutation was
found in the proband's father and grandfather, who had normal
urinalysis results, whereas the grandfather-in-law, who also had
normal urinalysis readings, did not carry any mutations (Table I). Therefore, this pedigree could be
diagnosed with hereditary ADAS, with the causal mutation site of
the COL4A3 gene being c.2603 G>A (p.G868E).
Protein function prediction using SIFT and
PolyPhen-2 software revealed both p.G195S and p.G868E to be
pathogenic mutations. In addition, conserved sequence analysis
showed the Gly at the 195th and 868th position in the COL4A3
gene was highly conserved in several species (Fig. 1D), suggesting that these positions
could have fundamental influence over the structure of the three
chains of type IV collagen and the biological function of the
organism. According to the ACMG standards (11), the p.G868E variant is classified as
a pathogenic variant and p.G195S is classified as likely
pathogenic.
Discussion
AS is typically characterized by hematuria,
proteinuria and progressive chronic renal failure, and may be
accompanied by ocular and auditory abnormalities in certain
patients, based on their gene mutations and disease progression
(1,2). The pathology of AS involves GBM
abnormalities due to developmental defects in the α3, α4 and α5
chains of type IV collagen (2,4). GBM
abnormalities underlie the various manifestations of patients with
AS, with gene mutations being the primary culprits of the GBM
abnormalities (4). Of note, a
patient's diagnosis and genotypes are not entirely deterministic,
as several parameters are involved in AS progression (4,6).
However, early ACEi treatment is particularly effective in
preventing disease progression to ESRD (2,4). Thus,
early precise diagnosis and intervention are crucial for patient
prognosis, and next-generation sequencing (NGS) testing may be a
useful tool in the selection of a therapeutic strategy for AS
(6,7).
The clinical manifestations of AS in patients
primarily depends on their gene type classification, that is XLAS,
ARAS and ADAS (4,5). Each type has different presentation
and prognosis, since the outcomes of AS are based on various
factors, such as mutation site, sex, age, family history, course,
complications and treatment (2,12).
With regard to ADAS, the manifestations typically emerge during the
childhood or teenage years, and will ultimately deteriorate
gradually into ESRD if no medical interventions are performed;
however, these patients have less auditory or ocular abnormalities
(6). In the present study, the
proband presented with gross hematuria for 8 years, following
laboratory examinations and further confirmation using a kidney
biopsy. The proband's 15-year-old sister was also diagnosed with AS
following laboratory examinations and significant findings in a
pathological biopsy, which deemed pedigree investigation necessary.
As suspected, three maternal members had AS, presenting with
heterogenous symptoms; the middle-aged mother and uncle had chronic
glomerulonephritis, whereas the disease in the grandmother had
progressed into ESRD. The proband had ocular abnormalities, but no
such abnormalities were observed in any of the other family
members. Imafuku et al (13)
reported that only 2 out of the 14 patients with ADAS exhibited
loss of hearing (14.2%) and none had ocular changes. These results
imply that the clinical manifestations of AS are varied, from
isolated hematuria, proteinuria, mild renal insufficiency to
ESRD.
All patients with AS in the maternal family were
found to possess a single p.G868E mutation that had given rise to
various AS manifestations, which largely depended on the age of the
three maternal members. These findings were consistent with those
of a previous report of ADAS, where hematuria first emerged during
childhood, likely as the only symptom, and proteinuria then
gradually became prominent during puberty, rendering the ultimate
progression to ESRD inevitable, given the persistent deterioration
of renal function and lack of effective medical intervention for
decades (14). Therefore, p.G868E
is a pathogenic mutation and the patients carrying it can be
diagnosed with ADAS.
However, the proband's clinical examination results
were not in line with the typical course of ADAS development. The
patient first presented with hematuria at the age of 5 years, which
is not rare amongst pediatric patients with ADAS; however, the
patient gradually presented with proteinuria and the aggravation of
hematuria, which are not common findings amongst teenagers with
ADAS. In particular, the age of onset preceded that of his older
sister (who had abnormal hematuria and proteinuria for only 1 year)
by several years. These findings suggested that other auxiliary
mutations may have been implicated in the proband's disease
course.
The proband had two mutations on the COL4A3
gene, whereas his older sister had only one mutation, which she had
inherited from the mother. The aforementioned p.G868E mutation was
derived from the maternal family, which laid the foundation for
ADAS development. The other p.G195S mutation originated from the
paternal family. The latter was not deterministic for AS onset,
which was demonstrated by the fact that the carriers, including the
father and grandfather, did not exhibit any abnormal AS
manifestations. Thus, it is inferred that p.G195S is a pathogenic
mutation that can potentially contribute to AS acceleration, but is
not sufficient to induce AS on its own. Type IV collagen is the
primary component of GBM, with embryonic α1α1α2 being replaced with
the mature α3α4α5 heterotrimer in childhood to ensure integral
structure and full function (1,15).
The α3 chain encoded by the COL4A3 gene is
composed of 1,670 amino acids, including a 28-amino acid signal
peptide at the amino terminus, a carboxyl terminus containing 225
amino acids, and the important middle collagen area, which is rich
in Gly (Gly-X-Y) triple-helix structure sequences (16). The intact triple-helix structure of
type IV collagen is essential for GBM development and the
maintenance of renal function, while a mutated triple-helix
structure can induce a vicious cycle of abnormal signaling in the
nephron and lead to marked extracellular matrix accumulation and
glomerulosclerosis, causing progressive renal disorder (17). Since the molecular weight of Gly is
the lowest amongst the amino acids, the three Gly residues can
exactly match the inside part of the tightly folded triple-helix
structure to maintain structural integrity (16,18).
Otherwise, if Gly is replaced by any other residue, an abnormally
folded triple-helical collagen molecule would be highly sensitive
to proteases and thus prone to degradation (18-20).
Since the middle Gly (Gly-X-Y) triple-helix structure sequence is
considerably long, gene mutations in different mutation sites would
cause different clinical manifestations, largely depending on their
role in the structural integrity (16,18).
In the present study, the proband was a patient with ADAS
presenting with hematuria at only 5 years of age, at the time of IV
collagen heterotrimer replacement (1), whereas the older sister exhibited
overt symptoms considerably later. Therefore, the p.G868E mutation
was pivotal in this case of ADAS. It was inferred that p.G195S was
a less important conspirator, which did not cause abnormal clinical
findings independently, but could synergistically accelerate
disease progression. In particular, certain Gly substitutions in
the triple-helix structure do not lead to pathological alterations
and overt symptoms, further illustrating that the mutation site is
critical in AS development and classification (19,20).
Abnormal urinalysis and renal dysfunction are the
important clinical manifestations of AS, and genetic screening is
helpful for the diagnosis and prognosis of AS (21), as in the case of the pedigree in the
present study. It was feasible to detect the pathogenic mutations
of AS by targeted NGS or first-generation sequencing. However,
there are three genes associated with AS, namely COL4A3,
COL4A4 and COL4A5, which contain 52, 48 and 51 exons
respectively (22).
First-generation sequencing is very time-consuming and laborious
and the results may be very frustrating, due to the pitfalls of
sequencing large genes and the absence of known mutational hot
spots (21,22). At present, if the causative genes of
inherited diseases are not clear, WES is a favorable alternative
for identifying potentially causative variants with the advantages
of time- and cost-efficiency (22-24).
WES can provide detailed information on the sites of the inherited
mutation to ensure timely medical intervention; if the proband and
their family are diagnosed and administered ACEi earlier, renal
function can be preserved for longer (6).
However, there were some limitations in the present
study. Firstly, immunofluorescence examinations of all the family
members of the pedigree were not performed due to the lack of
resources and the patients' willingness. Secondly, the progress of
the ADAS in the pedigree requires further follow-up to unveil the
clinical characteristics and prognosis caused by these two
mutations in the long-term. Finally, in order to thoroughly prove
the effect of the two mutations' functional disadvantage,
loss-of-function experiments for both in vivo and in
vitro verification should be performed. In the future, the AS
conditions of these siblings' future generations should be
assessed, and any potential disease progression in the carriers of
these two mutations should also be monitored if possible, to assist
in timely medical interventions.
In conclusion, p.G868E is a vital mutation that
leads to ADAS, whereas the p.G195S mutation can synergistically
promote ADAS progression. These two mutations can provide novel
insights into AS pathogenesis, and NGS may prove very effective in
AS diagnosis and treatment.
Acknowledgements
We would like to thank Professor Han Min (Department
of Nephrology, Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology) for assistance with the
electron microscopy analysis and pathological analysis of renal
biopsies.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 8200021897).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article. The novel COL3A4
mutation described in the present study has been submitted to
ClinVar (accession no. VCV001177517.1; variant, NM_000091.5.).
Authors' contributions
DN and CX performed the experiments. DN and ZZ
designed the study and drafted the manuscript. DN, KH and JL
analyzed and interpreted the data. CW and TG collected the
patient's data and followed up the case. All authors have read and
approved the final manuscript. DN, CX and ZZ confirmed the
authenticity of all raw data.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology. Written informed consent for
participation in the study was obtained from each patient, or from
the parents of patients <18 years old.
Patient consent for publication
Written informed consent for publication of this
study was obtained from each patient, or from the parents of
patients <18 years old.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hudson BG, Tryggvason K, Sundaramoorthy M
and Neilson EG: Alport's syndrome, Goodpasture's syndrome, and type
IV collagen. New Engl J Med. 348:2543–2556. 2003.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kashtan C: Alport syndrome: Facts and
opinions. F1000Res. 6(50)2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Naylor RW, Morais M and Lennon R:
Complexities of the glomerular basement membrane. Nat Rev Nephrol.
17:112–127. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kruegel J, Rubel D and Gross O: Alport
syndrome-insights from basic and clinical research. Nat Rev
Nephrol. 9:170–178. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Crockett DK, Pont-Kingdon G, Gedge F,
Sumner K, Seamons R and Lyon E: The Alport syndrome COL4A5 variant
database. Hum Mutat. 31:E1652–E1657. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kashtan CE: Alport syndrome: Achieving
early diagnosis and treatment. Am J Kidney Dis. 77:272–279.
2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Fallerini C, Dosa L, Tita R, Del Prete D,
Feriozzi S, Gai G, Clementi M, Manna AL, Miglietti N, Mancini R, et
al: Unbiased next generation sequencing analysis confirms the
existence of autosomal dominant Alport syndrome in a relevant
fraction of cases. Clin Genet. 86:252–257. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Omachi K and Miner JH: Alport syndrome
therapeutics: Ready for prime-time players. Trends Pharmacol Sci.
40:803–806. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Xu Y, Lin Z, Tang C, Tang Y, Cai Y, Zhong
H, Wang X, Zhang W, Xu C, Wang J, et al: A new massively parallel
nanoball sequencing platform for whole exome research. BMC
Bioinformatics. 20:153–161. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Huang J, Liang X, Xuan Y, Geng C, Li Y, Lu
H, Qu S, Mei X, Chen H, Yu T, et al: A reference human genome
dataset of the BGISEQ-500 sequencer. Gigascience. 6:1–9.
2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Richards CS, Bale S, Bellissimo DB, Das S,
Grody WW, Hegde MR, Lyon E and Ward BE: ACMG recommendations for
standards for interpretation and reporting of sequence variations:
Revisions 2007. Genet Med. 10:294–300. 2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Goka S, Copelovitch L and Levy Erez D:
Long-term outcome among females with Alport syndrome from a single
pediatric center. Pediatr Nephrol. 36:945–951. 2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Imafuku A, Nozu K, Sawa N, Hasegawa E,
Hiramatsu R, Kawada M, Hoshino J, Tanaka K, Ishii Y, Takaichi K, et
al: Autosomal dominant form of type IV collagen nephropathy exists
among patients with hereditary nephritis difficult to diagnose
clinicopathologically. Nephrology (Carlton). 23:940–947.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Marcocci E, Uliana V, Bruttini M, Artuso
R, Silengo MC, Zerial M, Bergesio F, Amoroso A, Savoldi S, Pennesi
M, et al: Autosomal dominant Alport syndrome: Molecular analysis of
the COL4A4 gene and clinical outcome. Nephrol Dial Transplant.
24:1464–1471. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ninomiya Y, Kagawa M, Iyama K, Naito I,
Kishiro Y, Seyer JM, Sugimoto M, Oohashi T and Sado Y: Differential
expression of two basement membrane collagen genes, COL4A6 and
COL4A5, demonstrated by immunofluorescence staining using
peptide-specific monoclonal antibodies. J Cell Biol. 130:1219–1229.
1995.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Heidet L, Arrondel C, Forestier L,
Cohen-Solal L, Mollet G, Gutierrez B, Stavrou C, Gubler MC and
Antignac C: Structure of the human type IV collagen gene COL4A3 and
mutations in autosomal Alport syndrome. J Am Soc Nephrol.
12:97–106. 2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yeo J, Qiu Y, Jung GS, Zhang YW, Buehler
MJ and Kaplan DL: Adverse effects of Alport syndrome-related Gly
missense mutations on collagen type IV: Insights from molecular
simulations and experiments. Biomaterials.
240(119857)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Inoue Y, Nishio H, Shirakawa T, Nakanishi
K, Nakamura H, Sumino K, Nishiyama K, Iijima K and Yoshikawa N:
Detection of mutations in the COL4A5 gene in over 90% of male
patients with X-linked Alport's syndrome by RT-PCR and direct
sequencing. Am J Kidney Dis. 34:854–862. 1999.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Gross O, Netzer KO, Lambrecht R, Seibold S
and Weber M: Meta-analysis of genotype-phenotype correlation in
X-linked Alport syndrome: Impact on clinical counselling. Nephrol
Dial Transplant. 17:1218–1227. 2002.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Savige J, Ariani F, Mari F, Bruttini M,
Renieri A, Gross O, Deltas C, Flinter F, Ding J, Gale DP, et al:
Expert consensus guidelines for the genetic diagnosis of Alport
syndrome. Pediatr Nephrol. 34:1175–1189. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Artuso R, Fallerini C, Dosa L, Scionti F,
Clementi M, Garosi G, Massella L, Epistolato MC, Mancini R, Mari F,
et al: Advances in Alport syndrome diagnosis using next-generation
sequencing. Eur J Hum Genet. 20:50–57. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Morinière V, Dahan K, Hilbert P, Lison M,
Lebbah S, Topa A, Bole-Feysot C, Pruvost S, Nitschke P, Plaisier E,
et al: Improving mutation screening in familial hematuric
nephropathies through next generation sequencing. J Am Soc Nephrol.
25:2740–2751. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tran KT, Le VS, Vu CD and Nguyen LT: A
novel de novo variant of LAMA2 contributes to merosin deficient
congenital muscular dystrophy type 1A: Case report. Biomed Rep.
12:46–50. 2020.PubMed/NCBI View Article : Google Scholar
|