Introduction
There has been increasing attention focused on
multiple chemical sensitivity (MCS) in the last decade, a
multi-system chronic disorder that is currently included in the
broader definition of sensitivity-related illnesses (SRI), and also
central sensitization syndromes, together with fibromyalgia (FM),
chronic fatigue syndrome (CFS) and electromagnetic hypersensitivity
(EHS) (1,2).
MCS develops as a result of loss of tolerance to
chronic exposure to various environmental contaminants
(organophosphates, solvents, heavy metals) at concentrations below
the threshold limit values considered toxic for the general
population (3). The wide variety of
multi-organ symptoms includes chronic muscular fatigue, bronchitis
and asthma, as well as effects on eye-nose-throat,
gastrointestinal, cardiac, psychosomatic, neurological, memory and
mood disorders, post-traumatic distress and autoimmunity. The
presence of these symptoms varies extensively from one individual
to another, based on individual sensitivity (4).
MCS has an estimated prevalence of 0.5-6.5% in
patients that received a medical diagnosis, whilst self-diagnosed
patients show a prevalence of 9.0-11.2% in the general population
(1). The main challenges hindering
a correct clinical framework of MCS are: The absence of a specific
and defined pathogenic mechanism or mechanisms; a range of
potential triggers; an absence of dose-dependent responses to
triggers; and the presence of common comorbidity features with
known autoimmune diseases, such as systemic lupus erythematosus,
rheumatoid arthritis or vitiligo (5,6).
Due to difficulties in finding unique and
differential diagnostic markers, defining MCS has been approached
with genetic, metabolic, immunological, etiological, symptomatic,
therapeutic and epidemiological tools (7). To date, the importance of oxidative
stress as an etiopathogenetic mechanism of this condition is widely
recognized, in addition to the role of redox status alterations in
the development of chronic mild systemic inflammation (5-7).
In previous studies it was demonstrated in the clinical setting how
oxidative status and inflammatory markers were elevated in patients
with MCS compared with healthy subjects (8-12).
Moreover, almost all patients with MCS are characterized by a
genetic background, potentially predisposing them to the
development of oxidative/nitrosative stress, given the presence of
several single nucleotide polymorphisms (SNPs) in genes coding for
enzymes involved in phase I and II detoxification reactions, as
well as antioxidant enzymes (9-11,13-17).
In particular, the rs4880 SNP (C47>T) of the superoxide
dismutase 2 (SOD2) gene has been reported as one of the genetic
determinants of MCS risk (15). The
C to T nucleotide change leads to an alanine to valine amino acid
change at codon 16 (Ala16Val, A16V), leading to a reduction of SOD2
levels in the mitochondria and thus reduced superoxide removal,
resulting in superoxide accumulation and mitochondrial damage
(18). Notably, an impairment in
liver function after exposure to bisphenol A has been reported in
carriers of the SOD2 variant (19).
Despite the reported association of the SOD2 A16V
polymorphism with MCS, the role of SOD2 A16V genetic background in
the modifications of the redox status in patients with MCS has not
yet been investigated.
Starting from biochemical features of patients with
MCS characterized in our previous study (8), the influence of the SOD2 A16V
polymorphism on the alterations of selected biomarkers of systemic
oxidative damage in patients with MCS compared with healthy
subjects were evaluated in the present study.
Materials and methods
Study cohort
The cases examined in this study consisted of 67
Italian patients with MCS (44 female/23 male; median age, 48 years;
inter-quartile range, 12) randomly selected amongst patients that
had been enrolled by medical staff at Department of Medical
Pathophysiology, University of Rome ‘La Sapienza’ - Polyclinic
Hospital ‘Umberto I’ (Rome, Italy), at Istituto Dermopatico
dell'Immacolata (IDI-IRCCS, study protocol n.121/CE/2008) (Rome,
Italy), and at Istituto of Ricerca Medica Ambientale (IRMA,
Acireale, Italy) between 2009 and 2014.
Patients had been selected on the basis of diagnosis
for MCS, established according to Cullen's criteria (20) and the Quick Environment Exposure
Sensitivity Inventory (QEESI) score (21), adapted for local application. QEESI
is a reference validated questionnaire to determine the levels of
sensitization to chemical environmental triggers (airborne or
other), to score the type, localization and severity of symptoms
after exposure, and their life impact. MCS subjects are stratified
based on a QEESI score >20.
Controls examined in this study were 55 Italian
healthy subjects (44 female/11 male, median age, 53 years old;
inter-quartile range, 12) randomly selected amongst volunteers that
had been recruited, between 2011 and 2017, at IDI-IRCCS (Rome,
Italy) (protocol approval no. 52/CE/2010) and Polyclinic Hospital
University ‘G. Martino’ (Messina, Italy) (protocol approvals no.
37/17 and 51/17), according to the following established criteria:
i) Absence of any clinically diagnosed disease, in particular
allergic or immunologic disturbances; ii) no drug or nutraceutical
supplement use in at least the 6 weeks prior to blood sampling; and
iii) whole blood total production of reactive oxygen
species/reactive nitrogen species (ROS/RNS) below 650 cps/µl, as
determined by luminol-dependent chemiluminescent response to
phorbol-myristate-acetate (PMA) (22).
Non-smokers accounted for 76.2% of individuals in
the MCS group and 90.9% of the control group. Alcohol or other drug
users were absent in both groups of participants. None of the
enrolled subject had taken any antioxidant supplementation in the 3
months preceding the recruitment.
All subjects enrolled for the study provided written
informed consent to participate in the study and to blood sampling
as well as anamnestic data collection, as specified in the
Declaration of Helsinki (23).
Genotyping
Genomic DNA was isolated from frozen (-80˚C) blood
samples using the Puregene-DNA purification system (GENTRA, Qiagen
GmbH), according to manufacturer's protocol. The DNA was quantified
by spectrophotometric measurement at 260 nm using a Biophotometer
(Eppendorf). DNA quality was considered acceptable for samples with
a 260/280 ratio ≥1.6. DNA integrity and the presence of contaminant
RNA were assessed by electrophoresis on a 0.8% agarose gel, and
subsequent UV detection of DNA bands using a gel photodocumentation
system (Vilber Lourmat).
Genotyping of patients with MCS and controls for
SOD2 A16V (C47> T, rs4880) SNP were performed using quantitative
PCR-based allelic discrimination using a pre-designed TaqMan SNP
genotyping assay (ID: C_8709053_10), available from Applied
Biosystems (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol.
Genotyping was performed on a 7900HT Fast Real-Time
PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.),
using thermal cycling conditions as previously described (24).
Analysis of redox markers
The whole blood luminol-dependent chemiluminescence
(CL) response to PMA and the plasma levels of nitrites/nitrates
(NO2-/NO3-) reacting
with Griess reagent were measured as described previously by
(22,25).
The levels of reduced glutathione (GSH) in
erythrocytes, as well as the plasma concentrations of ubiquinol
(Ubi-ol), the reduced form of coenzyme Q10
(CoQ10H2), were assessed by High-Performance
Liquid Chromatography equipped with array photodiode and
electrochemical detection, as previously described (7). Enzyme activities of superoxide
dismutase (SOD), catalase (CAT), glutathione-S-transferase (GST)
and glutathione peroxidase (GPx) in erythrocytes were measured
spectrophotometrically, as described previously (26-29).
The antioxidant activity (AOA) in plasma was determined as
described (30).
Analysis of fatty acid (FA) levels in
erythrocyte membranes
The pattern of FA esterified to the phospholipids of
the erythrocyte membranes was analyzed by gas chromatography
coupled with mass spectrometry on a crosslinked-FFAP capillary
column, following purification of lipid fractions by thin layer
chromatography (TLC), as described (31). Results were expressed as a
percentage of the total FA content.
Statistical analysis
Continuous data were expressed as the mean ±
standard deviation, and the categorical variables as the number and
percentage. The difference between cases and controls, in terms of
categorical variables, was assessed using a Fisher's exact test, as
well as the compliance of genotype distributions to the
Hardy-Weinberg equilibrium, using an online tool (ihg.gsf.de/cgi-bin/hw/hwa1.pl). The
distribution of data was assessed using a Kolmogorov-Smirnov test,
and was found to exhibit skewed distribution. Thus, non-parametric
tests were used for subsequent statistical analyses. A Mann-Whitney
U test for independent samples was used to compare comparisons
between cases and controls. Statistical analysis was performed
using SPSS version 22.0 (IBM Corp.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Genotype distributions for SOD2 A16V polymorphism in
patients with MCS and healthy controls are shown in Table I. Genotype frequencies, both in
cases and controls, were in Hardy-Weinberg equilibrium and,
similarly, allele frequencies amongst cases and controls were
within the 95% confidence interval.
 | Table IGenetic background of the patients
with MCS and healthy controls at the SOD2 locus. |
Table I
Genetic background of the patients
with MCS and healthy controls at the SOD2 locus.
| SOD2 genotype
(C47>T, A16V) | CTR (n) | MCS (n) | P-value |
|---|
| CC47 (AA16) | 24% (13) | 10% (7) | 0.0836 |
| CT47 (AV16) | 47% (26) | 51% (34) | 0.7197 |
| TT (VV16) | 29% (16) | 39% (26) | 0.3386 |
| C allele
frequency | 0.472 | 0.358 | |
| T allele
frequency | 0.527 | 0.641 | |
The analysis of the SOD2 A16V (C>T) genotype
distribution showed that the AV heterozygous genotype was the most
represented in both populations, as compared with other SOD2
genotypes, even if it was slightly more frequent in the MCS group
compared with the control group (Table
I).
The AA16 genotype was observed more frequently
amongst healthy subjects vs. patients, whereas the TT16 genotype
was more frequently observed in patients with MCS compared with the
controls. However, no statistically significant differences were
found between cases and controls.
Comparative analysis of metabolic redox biomarkers
and FA profiles was performed, as a whole and also related to the
different SOD2 A16V genetic background of the patients with MCS and
healthy controls.
MCS cases displayed a severely reduced antioxidant
capacity as compared to healthy controls, as evidenced by
significantly lower plasma AOA values (0.42±0.18 vs. 0.61±0.24
mmol/l; P=0.000). A significantly lower AOA level was observed in
patients with MCS with either the AA or AV genotype, compared with
their healthy counterparts (P<0.05, Table II).
| Table IIVariability of non-enzymatic
antioxidant defense biomarkers in MCS patients and healthy controls
having different genetic backgrounds. |
Table II
Variability of non-enzymatic
antioxidant defense biomarkers in MCS patients and healthy controls
having different genetic backgrounds.
| | Erythrocyte content
of reduced glutathione, mg/l | Plasma ubiquinol,
µg/l | Antioxidant
activity, µmol/l |
|---|
| Genotype
SOD2A16V | CTR | MCS | CTR | MCS | CTR | MCS |
|---|
| AA16 | 347.0±45.5 | 307.1±5.6 | 578.0±95.1 | 575.8±45.6 | 0.66±0.30 |
0.44±0.2a |
| AV16 | 342.5±61.2 | 331.4±40.1 | 633.6±75.8 | 599.2±33.0 | 0.60±0.24 |
0.48±0.2a |
| VV16 | 342.9±60.5 | 310.7±31.4 | 655.7±71.8 |
565.7±76.4b | 0.59±0.21 |
0.47±1.6a |
The concentration of Ubi-ol, was also significantly
depleted in patients with MCS vs. healthy subjects (573.9±99.40 vs.
626.92±86.55 µg/l; P=0.007), along with erythrocyte content of GSH
(297.0±68.0 vs. 343.72±53.50 mg/l; P=0.008), accounting for the
impaired antioxidant capacity typical in patients with this
condition. Patients with MCS with the AA genotype showed the lowest
levels of GSH and Ubi-ol, lower than those observed in the
controls, although the differences were not statistically
significant.
PMA-triggered release of ROS/RNS from granulocytes,
as non-specifically determined by PMA-induced-CL (416.3±160.9 vs.
386.6±140.2 cps/µl, MCS vs. control), and the
NO2-/NO3- plasma levels (23.4±130.9 vs.
18.9±6.6 µmol/l, MCS vs. control), an indirect measurement of the
extracellular superoxide anion and nitric oxide production by
circulating leukocytes and endotheliocytes, were not significantly
different between the two groups.
Moreover, the erythrocyte antioxidant GPx activity
was significantly higher in patients with MC compared with the
controls (25.44±8.12 vs. 21.39±7.67 U/mg Hb; P=0.018). The SOD
antioxidant activity was found to be increased in MCS cases
compared with controls (0.27±0.33 vs. 0.25±0.07 U/g prot; P=0.064),
while CAT activity was higher in controls than in patients with MCS
(8.54±3.86 vs. 7.40±5.45 U/g prot; P=0.057). Despite being not
significant, the P-values associated with these differences may
suggest a trend towards statistical significance. GST enzyme
activity was lower in patients with MCS compared with healthy
controls (1.70±0.62 vs. 1.93±0.69 U/mg Hb; P=0.101).
No significant differences were found between
genotype subgroups in MCS cases and control subjects with regard to
antioxidant enzyme activities, except for erythrocyte GPx activity,
which was significantly increased in patients bearing the AV16
genotype compared with their healthy counterparts (Table III).
| Table IIIVariability of antioxidant enzyme
activities in patients with MCS and CTR with different SOD2 A16V
genotypes. |
Table III
Variability of antioxidant enzyme
activities in patients with MCS and CTR with different SOD2 A16V
genotypes.
| | Superoxide
dismutase, U/g prot | Catalase, U/g
prot | Glutathione
peroxidase, U/mg Hb |
Glutathione-S-transferase, U/mg Hb |
|---|
| Genotype SOD2
A16V | CTR | MCS | CTR | MCS | CTR | MCS | CTR | MCS |
|---|
| AA | 0.33±0.1 | 0.27±0.3 | 8.5±3.8 | 6.7±0.3 | 22.8±10 | 25.4±2.3 | 2.0±0.7 | 1.6±0.6 |
| AV | 0.31±0.3 | 0.29±0.1 | 8.6±2.7 | 7.8±1.5 | 19.4±7.0 |
27.0±4.0a | 1.9±0.8 | 1.8±0.2 |
| VV | 0.23±0.1 | 0.28±0.1 | 8.4±3.9 | 8.1±2.1 | 23.4±7.6 | 21.1±4.6 | 1.9±0.5 | 1.8±0.4 |
The FA composition of the phospholipids in the
erythrocyte membrane of patients with MCS and controls, as possible
markers of oxidative damage.
The comparative analysis of FA profiles showed that
the percentage of saturated FA (SFA) was significantly higher in
patients with MCS compared with the controls (38.31±2.74 vs.
37.5±1.02%; P=0.041). Patients with MCS also showed a significantly
higher concentration of monounsaturated fatty acids (UFA) compared
with controls (20.03±2.25 vs. 18.63±1.35%; P=0.035). SFA was
primarily represented by palmitic acid (C16:0) and stearic acid
(C18:0) in patients with MCS (Fig.
1).
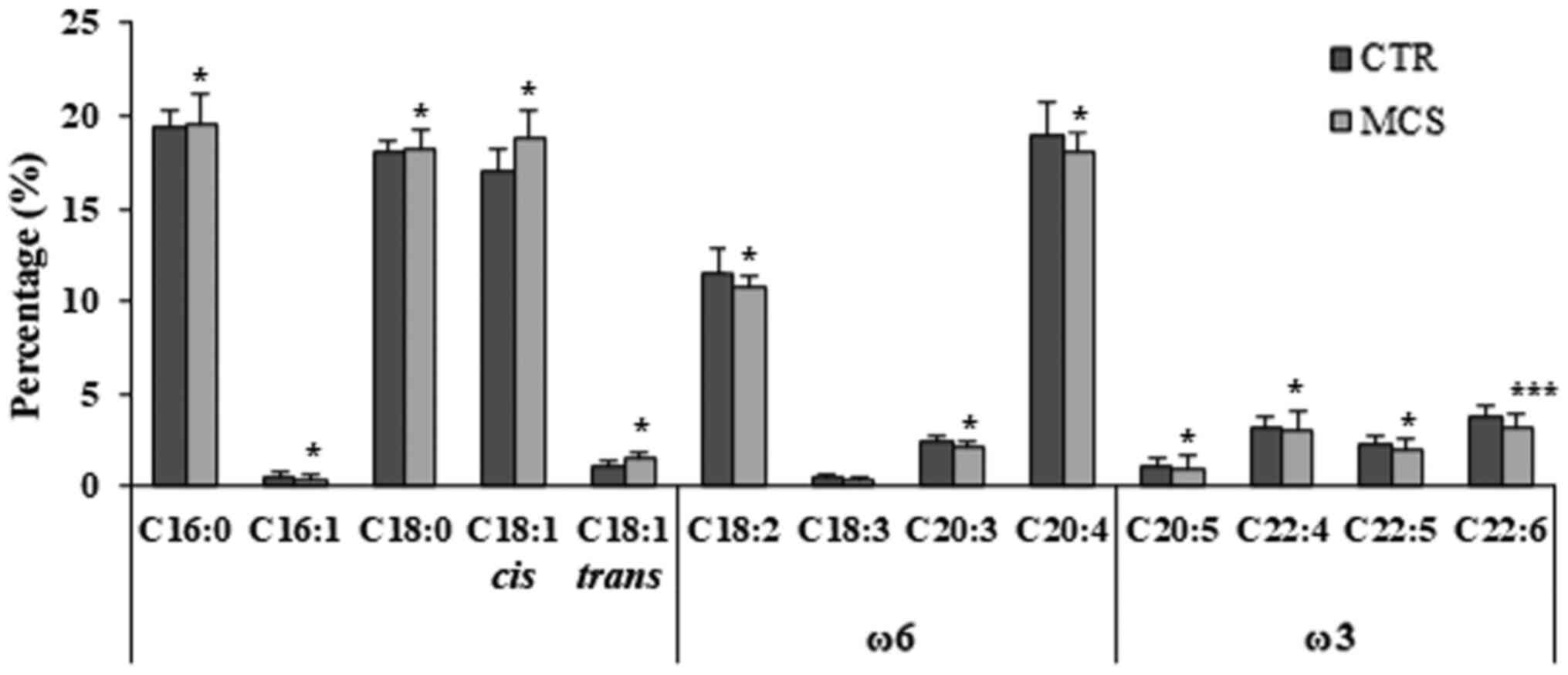 | Figure 1Fatty acids composition of
erythrocyte membranes in MCS cases and healthy controls. Relative
abundance (%) of representative saturated fatty acids (C16:0,
palmitic acid; C18:0, stearic acid), monounsaturated fatty acids
(C16:1, palmitoleic acid; cis C18:1 oleic acid; trans
C18:1, elaidic acid) and polyunsatured fatty acids (ω6 C18:2,
linoleic acid; ω6 C18:3, γ-linolenic acid; ω6 C20:3,
dihomo-γ-linolenic acid; ω6 C20:4, arachidonic acid; ω3 C20:5,
eicosapentaenoic acid; ω3 C22:4, docosatetraenoic acid (DTA); ω3
C22:5, docosapentaenoic acid (DPA); ω3 C22:6, cervonic acid. A
Mann-Whitney U test for independent samples was used to make
comparisons between the cases and controls. *P<0.05,
***P<0.01 vs. respective CTR value. MCS, multiple
chemical sensitivity patients (n=67); CTR, controls (n=55). |
As a key result relevant to the measurement of
oxidative damage, the percentage of the highly oxidable
polyunsaturated fatty acids (PUFA) was significantly lower in
patients with MCS compared with the controls (43.3±1.6 vs.
41.7±1.8%; P=0.021). In particular, the levels of omega-6 PUFA,
linoleic acid (C18:2 ω6), arachidonic acid (C20:4 ω6) and
dihomo-γ-linolenic acid (C20:3 ω6), as well as the omega-3 FA,
eicosapentaenoic acid (C20:5 ω3), docosatetraenoic acid (DTA C22:4
ω3), docosapentaenoic acid (C22:5 ω3) and docosahexaenoic acid
(C22:6 ω3), were significantly reduced in the MCS group (Fig. 1). Finally, the ω6/ω3 ratio was
significantly higher in patients with MCS compared with the
controls (3.68±0.95 vs. 3.36±0.60 %; P=0.038).
The MCS group presented with no overall functionally
relevant differences in the FA profiles between patient and control
groups (Table IV). Minor
significant differences were observed in individuals possessing a
AA genotype, with a significantly increased percentage of the
amount of palmitic acid (C16:0) and, at to a minor extent, stearic
acid (C18:0) (Table IV), and
significantly lower amounts of linoleic acid (18:2) (Table IV), the progenitor of the ω-6
series, in comparison with healthy subjects. Instead, the
monounsaturated elaidic acid (trans C18:1) was significantly
increased in MCS cases possessing either the VV or the AV genotype
compared with controls (Table IV),
while the ω-6 γ-linolenic acid (C18:3) levels were significantly
lower in the same patients (Table
IV). Moreover, oleic acid (cis C18:1) levels were
significantly higher in the patients with MCS with the VV genotype
compared with the controls (Table
IV). The amount of ω6 arachidonic acid was reduced in all
patients, and significant differences were found only in those
possessing an AV genotype compared with controls.
| Table IVVariability of erythrocyte membrane
phospholipid fatty acid composition in patients with MCS and CTR
with a different SOD2 genetic background. |
Table IV
Variability of erythrocyte membrane
phospholipid fatty acid composition in patients with MCS and CTR
with a different SOD2 genetic background.
| | CTR | MCS |
|---|
| Fatty acid | SOD2 AA16 | SOD2 AV16 | SOD2 VV16 | SOD2 AA16 | SOD2 AV16 | SOD2 VV16 |
|---|
| C16:0, % | 19.6±0.9 | 20.0±1.1 | 19.9±1.1 |
20.7±0.9b |
19.3±0.4a | 19.1±1.1 |
| C16:1, % | 0.5±0.3 | 0.5±0.3 | 0.4±0.3 | 0.4±0.1 | 0.3±0.1 | 0.3±0.0 |
| C18:0, % | 18.0±0.6 | 18.4±0.8 | 18.4±0.6 | 18.6±1.2 | 18.0±1.4 | 17.9±0.7 |
| C18:1 cis,
% | 16.9±1.5 | 17.1±1 | 17±1.1 | 18.1±0.7 | 17.5±1.1 |
18.1±1.0a |
| C18:1 trans,
% | 1.2±0.4 | 1.0±0.2 | 1.1±0.1 | 1.3±0.4 |
1.3±0.4b | 1.4±0.5b |
| C18:2, % | 11.7±1.1 | 11.4±1.7 | 11.4±0.9 |
10.4±0.9b | 11.2±1.1 | 11.3±0.9 |
| C18:3, % | 0.42±0.2 | 0.5±0.2 | 0.51±0.7 | 0.38±0.1 |
0.42±0.1a |
0.44±0.7a |
| C20:3, % | 2.37±0.4 | 2.56±0.3 | 2.27±0.3 | 2.3±0.3 | 2.4±1.0 | 2.2±0.3 |
| C20:4, % | 18.7±1.8 | 19.5±7.9 | 19.1±1.4 | 18.2±1.2 |
18.2±1.8a | 19.0±1.2 |
| C20:5, % | 1.28±0.6 | 1.0±0.2 | 0.9±0.4 | 0.85±0.3 | 0.9±0.4 | 1.0±0.3 |
| C22:4, % | 3.2±0.8 | 3.0±0.6 | 3.2±0.5 | 2.9±0.4 | 3.0±0.4 | 3.1±0.4 |
| C22:5, % | 2.3±0.4 | 2.2±0.6 | 2.2±0.5 | 2.0±0.4 | 2.2±0.5 | 2.1±0.5 |
| C22:6, % | 3.8±0.8 | 3.9±0.7 | 3.7±0.8 | 3.2±1.9 | 3.6±0.5 | 3.8±0.5 |
| SFA, % | 38.2±1.4 | 37.4±1.6 | 37.0±1.3 | 39.2±1.9 | 38.4±1.7 | 38.3±2.0 |
| UFA, % | 18.5±1.6 | 18.7±1.2 | 18.5±1.3 | 29.6±4.2 | 19.1±0.6 | 22.0±1.4 |
| PUFA, % | 43.3±1.7 | 43.6±1.8 | 44.0±1.9 | 40.0±1.8 | 41.2±0.9 | 41.9±0.7 |
| ω6/ω3 | 3.1±0.5 | 3.2±0.4 | 3.3±0.5 | 3.7±0.4 | 3.4±0.5 | 3.6±0.7 |
All FAs of the ω-3 series were found to be depleted
in patients with MCS compared with controls, and the lowest levels
were found in patients with SOD2 AA16 genotype (Table IV). However, no significant
differences were observed between cases and controls.
SFA and UFA were increased in patients with MCS
compared with the controls, and the highest percentage amounts were
found in subjects with the AA genotype, whereas in the same
individuals, the lowest levels of PUFA were observed, that were
generally reduced in comparison with healthy subjects. Notably, the
ratio ω6/ω3 was found to be higher in MCS cases compared with the
healthy controls, and the highest values were observed in patients
with the SOD2 AA16 genotype (Table
IV). However, no significant differences were observed between
the two groups, and only a trend to statistical significance was
observed for differences in SFA amounts in patients with MCS with
either the AV genotype (P=0.05) or the VV genotype (P=0.07).
Discussion
In the last two decades several studies have
provided evidence for a correlation between MCS and chemical
defense system alterations, occurring in the presence of gene
polymorphisms of detoxification phase I (CYPs) and phase II enzymes
(GST, NAT and UGT, amongst others), as well as antioxidant enzymes
SOD2 and GPX (5,9-17).
Previous study by De Luca et al (8) on a large group of patients with MCS
highlighted the relevance of oxidative stress in this syndrome. In
particular, a reduction of CAT and GST enzyme activities,
associated with glutathione reduction, increased GPx activity,
PUFA-depleted profiles of erythrocyte membranes, and specifically
altered pro-inflammatory plasma cytokine patterns have been shown
in a MCS cohort as a whole (8,9).
Moreover, in our previous study, it was reported that there was a
significantly higher frequency of polymorphisms in genes coding for
CYP enzymes in patients with MCS compared with controls. These
findings indicated that these genetic variants may increase the
individuals' susceptibility to develop MCS, and oxidative stress
conditions typically associated with this complex syndrome
(14). Notably, Cui et al
(15) also reported that the
missense polymorphism A16V in gene SOD2, coding for the antioxidant
mitochondrial superoxide dismutase, is associated with MCS
development. In silico and in vitro experiments
showed that this polymorphism can significantly affect enzyme
activity, reducing antioxidant defenses (15). However, the relationship between
SOD2 genetic background and redox metabolism features in patients
with MCS have not been investigated thus far, to the best of our
knowledge.
In the present study, the role of SOD2 A16V
polymorphism in the modifications was examined in the previously
established panel of redox biomarkers assessed in a representative
group of 67 patients with MCS as compared with an age-matched group
of 55 healthy controls. The goal was to search for possible
correlations between the inter-individual variations existing in
the blood metabolic parameters and the genetic background.
Taking into consideration the MCS group as a whole,
there was a reduced capacity of the antioxidant system of these
individuals compared with the healthy counterparts, as evidenced by
the decreased values of plasma AOA and Ubi-ol, as well as
erythrocyte GSH. GSH represents the most powerful antioxidant
agent, capable of preventing ROS-induced damage to important
cellular components, and is also largely used for cell and body
detoxification from xenobiotics (5). Ubi-ol is a potent endogenous
antioxidant, and its depletion is viewed as a reliable biomarker of
systemic oxidative stress (7). It
acts as a reducing agent in the mitochondria and in lipid membranes
by either directly scavenging free radicals or in conjunction with
α-tocopherol (5). These results
confirmed the findings of Miyamae et al (32) with fibromyalgic patients, that
showed a drastic reduction of Ubi-ol levels compared with healthy
controls, suggesting an increased formation of ROS in the
circulating blood in patients with juvenile FM.
The measurement of antioxidant enzymes SOD, CAT and
GPx in erythrocytes allows assessment, in a non-invasive manner, of
the circulating antioxidant defense in humans. Cell damage by ROS
is initiated by the production of the superoxide radicals, which is
closely associated with the metabolism of molecular oxygen in
mitochondria and in cellular membranes. Thus, the first-line
antioxidant defense system against oxidative damage is represented
by SOD, the cytosolic Cu-Zn SOD (SOD1, dimeric), the mitochondrial
Mn-SOD (SOD2), and the extracellular Cu-Zn SOD (SOD3, tetrameric),
which convert superoxide anion to hydrogen peroxide. Subsequently,
hydrogen peroxide is scavenged by CAT and GPx (33). Although it was not significant,
there was a trend-to-reduction of CAT and GST activities, and a
trend-to-increase in SOD enzyme activity in patients with MCS,
coupled with an overall significant glutathione depletion. Both CAT
and GST are stress proteins that are upregulated under pathological
conditions, such as that represented by chemical stress. However,
chemical stress can also induce cytotoxicity in blood cells
producing CAT, that in turn may result in a reduction in CAT
activity and accumulation of excessive amounts of hydrogen
peroxide. Hydrogen peroxide excess initiates a chain-reaction of
lipid peroxidation, the major feature of which is the decomposition
of PUFA to aldehydes, with an overall reduction of PUFA content in
erythrocyte membranes. Under these conditions, GPx likely becomes
the major second-line antioxidant enzyme to neutralize hydrogen
peroxide through the oxidation of GSH to oxidized glutathione, and
counteract the accelerated production of lipid hydroperoxides.
These observations highlight a general trend of increased oxidative
stress in patients with MCS. As a likely consequence, patients with
MCS presented with markedly increased GPx activity and GSH
depletion (5). Consistently, a
significant increase in GPx activity has been previously confirmed
in the muscles of patients with CFS, sharing several symptomatic
features with patients with MCS (34). This possible adaptive response of
GPx to excess hydroperoxides is in line with previous findings of
our group in psoriasis, a pathological condition characterized by
chronic inflammation and immune system activation (35). Notably, the genetic background at
the GPx locus does not seem to be altered in patients with MCS as
compared with the healthy population, at least as concerns the
rs1800668 (C/T) variant within the promoter of GPx1 gene, which is
able to affect enzyme activity (11).
The increased lipid peroxidation observed in the MCS
group was also indirectly confirmed by the observed alterations in
the FA profiles of red blood cell membranes. The contents of all
PUFAs and of selected PUFAs relevant to
inflammatory/anti-inflammatory processes, primarily arachidonic
(AA; C20:4, ω6) were much lower-than-normal in the MCS cohort, thus
confirming the occurrence of a sustained lipoperoxidation in the
erythrocyte membranes of patients with MCS.
A key chemical feature of lipid peroxidation is the
ability to decompose PUFA to form a broad array of end-products,
namely aldehydes, such as such as malondialdehyde and
4-hydroxy-2-nonenal (4-HNE), a stable electrophile formed during
the lipid peroxidation of ω-6-PUFA, namely linoleic and arachidonic
acids, which readily react with proteins and DNA to affect enzyme
gene expression, enzymatic activity, as well as the formation of
autoantigens (36).
A similar pattern of increased GPx activity and
depleted erythrocyte membrane PUFA, with increased levels of
end-products of lipoperoxidation, was shown in patients affected
with psoriasis, a chronic inflammatory immune-mediated pathology,
which shares with MCS a severe imbalance of the systemic redox
status coupled with a marked dysregulation of plasmatic
inflammatory cytokines (37).
The condition of impaired redox status and reduced
antioxidant capacity, seen at a general level in the MCS group, was
also found considering the different genotypic structures of SOD2
A16V polymorphism in both cohorts under study. Indeed, all four
major markers found as depleted in the overall MCS group, AOA, GSH,
Ubi-ol and PUFA levels, along with increased GPx activity, showed
the same trend in the MCS subgroup with the SOD2 AA genotype. This
subgroup showed the largest differences between patients with MCS
and controls, and also the most differing prevalence between
patients and controls. Conversely, even healthy controls bearing
this genotype showed oxidative stress levels higher than those
found in other control subgroups with different genotypes. Notably,
the lowest levels of oxidative stress in both study cohorts were
found in individuals bearing the SOD2 VV16 genotype.
These findings suggest that the SOD2 AA16 wild-type
genotype represents a genetic risk factor for increased
susceptibility to oxidative stress, while the mutated homozygous
SOD2 VV16 displays a protective effect. The Ala16-wild-type variant
of SOD2 gene allows a more efficient importation of Mn-SOD into the
mitochondria, resulting in turn in the generation of a more active
enzyme compared with the Val16-variant, that is related to the
induction of oxidative stress. Likely, this is the reason why the
SOD2 Ala16 variant has a much lower prevalence than the Val16 one
in all ethnic groups worldwide (18). Notably, the heterozygous AV16
genotype seems to represent the better conditions for optimal
enzyme activity of mitochondrial Mn-SOD, given the highest
frequency observed in our study cohorts, that is in line with
findings from several studies carried out in Italian and Caucasian
populations (23,37-41).
Most importantly, these same studies, and several others not cited
here, suggested that the SOD2 AA16V (rs4880) polymorphism may have
an impact on acute and chronic oxidative-related damage, and
increase the susceptibility to various oxidative stress-related
pathological conditions, such as pregnancy complications, cardio-
and cerebrovascular disorders, cancer, glaucoma, diabetes (37,38,40-43),
and even accelerates telomere shortening with age (39).
These findings are in line with the report of Cui
et al (15) suggesting that
SOD2 AA16 genotype increase the risk for MCS, even if no
correlation with biochemical features were assessed. Notably, it
has been reported that this genotype increases the susceptibility
to oxidative stress-related cyto-genotoxicity induced by
pyridostigmine bromide, that has been implicated as a causal factor
in Gulf War syndrome, a disorder sharing several features with MCS
(44). Moreover, cytotoxic effects,
at all times of exposure to static magnetic field (SMF), have been
observed in peripheral blood mononuclear cells isolated from
individuals bearing the AA16 genotype, while AV and VV cells
presented mortality only after longer times of exposure to SMF.
These results suggest a toxico-genetic effect of SMF exposure
related to an imbalance in SOD2 activity associated with the AA16
genotype (45). Interestingly,
hypersensitivity to electromagnetic field, also called EHS, is a
common co-morbidity of MCS (9,46) in
which oxidative stress plays a major role (47).
In conclusion, the analysis of the metabolic markers
of antioxidant defense and redox imbalance confirmed the occurrence
in the MCS group of a statistically solid reduction of the main
four blood metabolic markers of oxidative stress, the depletion of
the low-molecular weight antioxidant Ubi-ol, of the total plasmatic
antioxidant activity, of the red blood cell membrane PUFA, with
possible generation of lipid hydroperoxide by-products leading to
an increase of GPx activity. This specific pattern of metabolic
alterations was more striking in MCS carriers of the SOD2AA
genotype than either in other patients or controls.
The results of the present study provide additional
evidence that functional and/or genetic defects of endogenous
enzymes detoxifying H2O2, lipid peroxides, or
stable toxic products of lipid peroxidation may cause chronic
oxidative stress with increased pro-inflammatory cytokine release
and consequent metabolic alterations, characteristic for the
patients with SRI, as originally hypothesized by our team (5). The observed correlation of these
selected metabolic alterations with SOD2 A16V polymorphism may
contribute to an improved understanding of the abnormal
susceptibility to low-level xenobiotic stimuli in these peculiar
clinical settings.
These results await confirmation by larger studies
where the patients with MSC shall be necessarily further stratified
based on the severity of their clinical manifestations and on their
specific patterns of co-morbidities, thus taking into account the
large amount of heterogeneity of MCS pathogenesis and the
persisting lack of consensus on the diagnostic protocols (48). Additional confirmatory studies will
possibly contribute to finally validate the clinical relevance of
the described targeted panel of gene polymorphisms and of
biomarkers of the systemic redox status impairment, as a feasible
laboratory approach for MCS management, for an evidence-based
process of diagnosis, prognosis and treatment follow-up of this
multi organ syndrome.
Acknowledgements
Dr Maria Grazia Bruccheri for helping in the
recruitment of patients with MCS at IRMA (Acireale, CT).
Funding
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LK and DC conceived the study. LK, MC, NF and RI
performed patient/volunteer recruitment. CDL, AC and GA performed
the investigations. AA analyzed the data. AC and GA prepared tables
and figures. AC and CDL wrote the manuscript. DC and LK reviewed
and edited the manuscript. All authors read and approved the final
manuscript. AC, CDL, GA, DC, MC, NF, RI, AA and LK confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The recruitment of patients and controls was
approved by the Ethics Committee of IDI-IRCCS (approval no.
121/CE/2008 and 52/CE/2010) (Rome, Italy) and the Ethics Committee
of Polyclinic Hospital University ‘G. Martino’ (approval nos. 37/17
and 51/17) (Messina, Italy). All subjects enrolled for the study
provided written informed consent to participate in the study and
to blood sampling as well as anamnestic data collection, as
specified in the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Genuis SJ: Sensitivity-related illness:
The escalating pandemic of allergy, food intolerance and chemical
sensitivity. Sci Total Environ. 408:6047–6061. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yunus MB: Central sensitivity syndromes: A
new paradigm and group nosology for fibromyalgia and overlapping
conditions, and the related issue of disease versus illness. Semin
Arthritis Rheum. 37:339–352. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Miller CS: Toxicant-induced loss of
tolerance - an emerging theory of disease? Environ Health Perspect.
105 (Suppl 2):445–453. 1997.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lacour M, Zunder T, Schmidtke K, Vaith P
and Scheidt C: Multiple chemical sensitivity syndrome (MCS) -
suggestions for an extension of the U.S. MCS-case definition. Int J
Hyg Environ Health. 208:141–151. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Korkina L, Scordo MG, Deeva I, Cesareo E
and De Luca C: The chemical defensive system in the pathobiology of
idiopathic environment-associated diseases. Curr Drug Metab.
10:914–931. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Pall ML: Multiple chemical sensitivity:
Toxicological questions and mechanisms. In: General, Applied and
Systems Toxicology. 3rd edition. John Wiley & Sons (eds). John
Wiley & Sons, Ltd. Hoboken, NJ, pp2303-2352, 2009.
|
|
7
|
De Luca C, Raskovic D, Pacifico V, Thai
JCS and Korkina L: The search for reliable biomarkers of disease in
multiple chemical sensitivity and other environmental intolerances.
Int J Environ Res Public Health. 8:2770–2797. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
De Luca C, Scordo MG, Cesareo E, Pastore
S, Mariani S, Maiani G, Stancato A, Loreti B, Valacchi G, Lubrano
C, et al: Biological definition of multiple chemical sensitivity
from redox state and cytokine profiling and not from polymorphisms
of xenobiotic-metabolizing enzymes. Toxicol Appl Pharmacol.
248:285–292. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
De Luca C, Thai JC, Raskovic D, Cesareo E,
Caccamo D, Trukhanov A and Korkina L: Metabolic and genetic
screening of electromagnetic hypersensitive subjects as a feasible
tool for diagnostics and intervention. Mediators Inflamm.
2014(924184)2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
De Luca C, Gugliandolo A, Calabrò C, Currò
M, Ientile R, Raskovic D, Korkina L and Caccamo D: Role of
polymorphisms of inducible nitric oxide synthase and endothelial
nitric oxide synthase in idiopathic environmental intolerances.
Mediators Inflamm. 2015(245308)2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Gugliandolo A, Gangemi C, Calabrò C,
Vecchio M, Di Mauro D, Renis M, Ientile R, Currò M and Caccamo D:
Assessment of glutathione peroxidase-1 polymorphisms, oxidative
stress and DNA damage in sensitivity-related illnesses. Life Sci.
145:27–33. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cannata A, De Luca C, Korkina LG, Ferlazzo
N, Ientile R, Currò M, Andolina G and Caccamo D: The SNP rs2298383
reduces ADORA2A gene transcription and positively associates with
cytokine production by peripheral blood mononuclear cells in
patients with Multiple Chemical Sensitivity. Int J Mol Sci.
21(1858)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Schnakenberg E, Fabig KR, Stanulla M,
Strobl N, Lustig M, Fabig N and Schloot W: A cross-sectional study
of self-reported chemical-related sensitivity is associated with
gene variants of drug-metabolizing enzymes. Environ Health.
6(6)2007.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Caccamo D, Cesareo E, Mariani S, Raskovic
D, Ientile R, Currò M, Korkina L and De Luca C: Xenobiotic sensor-
and metabolism-related gene variants in environmental
sensitivity-related illnesses: A survey on the Italian population.
Oxid Med Cell Longev. 2013(831969)2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cui X, Lu X, Hiura M, Oda M, Miyazaki W
and Katoh T: Evaluation of genetic polymorphisms in patients with
multiple chemical sensitivity. PLoS One. 8(e73708)2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Micarelli A, Cormano A, Caccamo D and
Alessandrini M: Olfactory-Related Quality of Life in Multiple
Chemical Sensitivity: A Genetic-Acquired Factors Model. Int J Mol
Sci. 21(156)2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
D'Attis S, Massari S, Mazzei F, Maio D,
Vergallo I, Mauro S, Minelli M and Bozzetti MP: Assessment of
CYP2C9, CYP2C19, and CYP2D6 Polymorphisms in Allergic Patients with
Chemical Sensitivity. Int Arch Allergy Immunol. 179:173–186.
2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bastaki M, Huen K, Manzanillo P, Chande N,
Chen C, Balmes JR, Tager IB and Holland N: Genotype-activity
relationship for Mn-superoxide dismutase, glutathione peroxidase 1
and catalase in humans. Pharmacogenet Genomics. 16:279–286.
2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kim JH, Lee MR and Hong YC: Modification
of the association of bisphenol A with abnormal liver function by
polymorphisms of oxidative stress-related genes. Environ Res.
147:324–330. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cullen MR: The worker with multiple
chemical sensitivities: An overview. Occup Med. 2:655–661.
1987.PubMed/NCBI
|
|
21
|
Hojo S, Kumano H, Yoshino H, Kakuta K and
Ishikawa S: Application of Quick Environment Exposure Sensitivity
Inventory (QEESI) for Japanese population: Study of reliability and
validity of the questionnaire. Toxicol Ind Health. 19:41–49.
2003.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Brown GE, Silver GM, Reiff J, Allen RC and
Fink MP: Polymorphonuclear neutrophil chemiluminescence in whole
blood from blunt trauma patients with multiple injuries. J Trauma.
46:297–305. 1999.PubMed/NCBI View Article : Google Scholar
|
|
23
|
World Medical Association. World Medical
Association Declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194.
2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Vecchio M, Currò M, Trimarchi F, Naccari
S, Caccamo D, Ientile R, Barreca D and Di Mauro D: The oxidative
stress response in elite water polo players: Effects of genetic
background. BioMed Res Int. 2017(7019694)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Giovannoni G, Land JM, Keir G, Thompson EJ
and Heales SJ: Adaptation of the nitrate reductase and Griess
reaction methods for the measurement of serum nitrate plus nitrite
levels. Ann Clin Biochem. 34:193–198. 1997.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sun Y, Oberley LW and Li Y: A simple
method for clinical assay of superoxide dismutase. Clin Chem.
34:497–500. 1988.PubMed/NCBI
|
|
27
|
Aebi H: Catalase in vitro. Methods
Enzymol. 105:121–126. 1984.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Habig WH, Pabst MJ and Jakoby WB:
Glutathione S-transferases. The first enzymatic step in mercapturic
acid formation. J Biol Chem. 249:7130–7139. 1974.PubMed/NCBI
|
|
29
|
Paglia DE and Valentine WN: Studies on the
quantitative and qualitative characterization of erythrocyte
glutathione peroxidase. J Lab Clin Med. 70:158–169. 1967.PubMed/NCBI
|
|
30
|
Miller NJ, Rice-Evans C and Davies MJ: A
new method for measuring antioxidant activity. Biochem Soc Trans.
21(95S)1993.PubMed/NCBI View Article : Google Scholar
|
|
31
|
De Luca C, Filosa A, Grandinetti M, Maggio
F, Lamba M and Passi S: Blood antioxidant status and urinary levels
of catecholamine metabolites in β-thalassemia. Free Radic Res.
30:453–462. 1999.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Miyamae T, Seki M, Naga T, Uchino S,
Asazuma H, Yoshida T, Iizuka Y, Kikuchi M, Imagawa T, Natsumeda Y,
et al: Increased oxidative stress and coenzyme Q10 deficiency in
juvenile fibromyalgia: Amelioration of hypercholesterolemia and
fatigue by ubiquinol-10 supplementation. Redox Rep. 18:12–19.
2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Matés JM, Pérez-Gómez C and Núñez de
Castro I: Antioxidant enzymes and human diseases. Clin Biochem.
32:595–603. 1999.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Fulle S, Mecocci P, Fanó G, Vecchiet I,
Vecchini A, Racciotti D, Cherubini A, Pizzigallo E, Vecchiet L,
Senin U, et al: Specific oxidative alterations in vastus lateralis
muscle of patients with the diagnosis of chronic fatigue syndrome.
Free Radic Biol Med. 29:1252–1259. 2000.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Pastore S, Mariani V, Lulli D, Gubinelli
E, Raskovic D, Mariani S, Stancato A, de Luca C, Pecorelli A,
Valacchi G, et al: Glutathione peroxidase activity in the blood
cells of psoriatic patients correlates with their responsiveness to
Efalizumab. Free Radic Res. 45:585–599. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Marantos C, Mukaro V, Ferrante J, Hii C
and Ferrante A: Inhibition of the lipopolysaccharide-induced
stimulation of the members of the MAPK family in human
monocytes/macrophages by 4-hydroxynonenal, a product of oxidized
omega-6 fatty acids. Am J Pathol. 173:1057–1066. 2008.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Giusti B, Vestrini A, Poggi C, Magi A,
Pasquini E, Abbate R and Dani C: Genetic polymorphisms of
antioxidant enzymes as risk factors for oxidative stress-associated
complications in preterm infants. Free Radic Res. 46:1130–1139.
2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Palmirotta R, Barbanti P, De Marchis ML,
Egeo G, Aurilia C, Fofi L, Ialongo C, Valente MG, Ferroni P,
Della-Morte D, et al: Is SOD2 Ala16Val polymorphism associated with
migraine with aura phenotype? Antioxid Redox Signal. 22:275–279.
2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Hernando B, Gil-Barrachina M, Tomás-Bort
E, Martinez-Navarro I, Collado-Boira E and Hernando C: The effect
of long-term ultra-endurance exercise and SOD2 genotype on telomere
shortening with age. J Appl Physiol (1985). 129:873–879.
2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Synowiec E, Wigner P, Cichon N, Watala C,
Czarny P, Saluk-Bijak J, Miller E, Sliwinski T, Zielinska-Nowak E
and Bijak M: Single-nucleotide polymorphisms in oxidative
stress-related genes and the risk of a stroke in a Polish
population - a preliminary study. Brain Sci. 11(391)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Atanasovska Velkovska M, Goričar K, Blagus
T, Dolžan V and Cvenkel B: Association of genetic polymorphisms in
oxidative stress and inflammation pathways with glaucoma risk and
phenotype. J Clin Med. 10(1148)2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Crawford A, Fassett RG, Geraghty DP, Kunde
DA, Ball MJ, Robertson IK and Coombes JS: Relationships between
single nucleotide polymorphisms of antioxidant enzymes and disease.
Gene. 501:89–103. 2012.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kang SW: Superoxide dismutase 2 gene and
cancer risk: Evidence from an updated meta-analysis. Int J Clin Exp
Med. 8:14647–14655. 2015.PubMed/NCBI
|
|
44
|
Azzolin VF, Barbisan F, Teixeira CF,
Pillar D, Mastella MH, Duarte T, Turra BO, Ribeiro EE, Duarte MMFM
and da Cruz IBM: The Val16Ala-SOD2 polymorphism affects
cyto-genotoxicity of pyridostigmine bromide on human peripheral
blood mononuclear cells. Toxicol In Vitro. 60:237–244.
2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Dornelles EB, Goncalves BD, Schott KL,
Barbisan F, Unfer TC, Glanzner WG, Machado AK, Cadona FC, Azzolin
VF, Montano MA, et al: Cytotoxic effects of moderate static
magnetic field exposure on human periphery blood mononuclear cells
are influenced by Val16Ala-MnSOD gene polymorphism. Environ Sci
Pollut Res Int. 24:5078–5088. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Belpomme D, Campagnac C and Irigaray P:
Reliable disease biomarkers characterizing and identifying
electrohypersensitivity and multiple chemical sensitivity as two
etiopathogenic aspects of a unique pathological disorder. Rev
Environ Health. 30:251–271. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Irigaray P, Caccamo D and Belpomme D:
Oxidative stress in electrohypersensitivity self reporting
patients: Results of a prospective in vivo investigation
with comprehensive molecular analysis. Int J Mol Med. 42:1885–1898.
2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Rossi S and Pitidis A: Multiple Chemical
Sensitivity: Review of the State of the Art in Epidemiology,
Diagnosis, and Future Perspectives. J Occup Environ Med.
60:138–146. 2018.PubMed/NCBI View Article : Google Scholar
|














