Introduction
Wolfram syndrome 1 (WS1) is a rare autosomal recessive neurodegenerative disease that is characterized by the presence of diabetes insipidus (DI), diabetes mellitus (DM), optic atrophy (OA) and deafness (1). The prevalence of WS1 in the general population is ~1/770,000 in the United Kingdom, 1/710,000 in Japan and 1/100,000 in North America (2). Other clinical features may include renal abnormalities, intellectual impairment and psychiatric illness, such as cerebellar ataxia, dysarthria and areflexia. Non-insulin dependent, non-autoimmune DM of juvenile onset and OA are the minimal diagnostic criteria for WS1, and these symptoms may present in childhood, adolescence or early adult life (3). However, phenotypic heterogeneity can occur in patients, especially in different ethnic populations. Recently, Rigoli et al (4) reported an atypical case of late-onset WS1 without DI in an Italian family. The clinical diagnosis of WS1 is therefore challenging, especially in children.
The genetic basis of WS1 is mutation in the wolframin endoplasmic reticulum ER transmembrane glycoprotein (WFS1) gene, which is located on chromosome 4p16.1. This gene encodes the transmembrane protein Wolframin, which plays a key role in the pathway of ER stress and in regulating calcium levels in cells. Wolframin is located primarily in the ER and is ubiquitously expressed at the highest levels in the brain, pancreas, heart and insulinoma β cells. To date, hundreds of variants of the WFS1 gene have been deposited in ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), including those that lead to missense, frameshift, nonsense and splice mutations (5-7). Similar to other genetic diseases, WS1 heterogeneity is also reported, as variants in another gene, WFS2, are also responsible for this syndrome (8). Mitochondrial variants may also be involved in WS1. Rötig et al (9) found a 7.6-kb pair heteroplasmic deletion of the mitochondrial DNA in the tissues of a WS case, which supported the view that a respiratory chain defect could present with WS1. Thus, genetic analysis of WS1 is necessary in order to make a definite diagnosis.
With the progress in next-generation sequencing technology, whole exome sequencing (WES) has been clinically applied to analyses of suspected WS1 cases, and more pathogenic mutations causing WS1 are being discovered. The present study reports two homologous variants of WFS1 found by WES, which may be the molecular basis of the disease.
Case report
Patient
A 3.5-year-old Chinese Han boy was admitted to the Department of Nephrology, Guiyang Maternal and Child Health Care Hospital (Guiyang, China) in August 2021 due to abnormal renal function and a persistent fever for 3 days, with a single convulsion. The highest temperature reached was 38.8˚C. Approximately 1 year before, the patient had suffered from nyctalopia, and 4 months before the current admittance, retinitis pigmentosa (RP) was diagnosed in another hospital (Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China). The patient was the first baby born after a full-term pregnancy in a non-consanguineous family. For further treatment, the patient was referred to Guiyang Maternal and Child Health Care Hospital in August 2021.
The patient exhibited a pale and weak appearance. Several palpable lymph nodes in the neck were detected by physical examination, with throat congestion and tonsil enlargement. The blood pressure was abnormal (140/80 mmHg), and a decreased hemoglobin level (68 g/l) and red blood cell count (2.35x1012/l) were detected on blood tests (Table I). Serum chemistry showed elevated serum creatinine (639 µM) and fasting plasma glucose (6.43 mM) levels (Table I), but other serum chemistry and immunological tests were normal. In addition, other abnormal data, including lower complement C3 and calcium levels, and higher urea, BNP and Troponin T, were also revealed by serum chemistry (Table I). Urine tests showed proteinuria (2+) and positive urine glucose (+), but hematuria was ruled out by microscopy. Abdominal ultrasound examination with a Philips EPIQ5 (Probe model C5-1; Philips Medical Systems, Inc.; Table SI) revealed that both kidneys were 53x23 mm, with hyperechogenicity and an unclear boundary between the cortex and medulla. According to the guidance of a nephrologist, this patient was diagnosed with stage 5 chronic kidney disease (Kidney Disease: Improving Global Outcomes guidelines), which was a contraindication for a renal biopsy. Increased liver echogenicity, intrahepatic bile duct dilation and splenomegaly were also detected. Cardiac ultrasound showed congenital heart disease, with left ventricular enlargement and mild mitral insufficiency. In addition, visional impairment had been noticed in the previous admission to Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine. Therefore, a fundus camera was now used to collect high-resolution images of the fundus, which can be accurately obtained in a short time without mydriasis. Bilateral RP was confirmed, but OA was not evident (Fig. 1). Based on these findings, a diagnosis of renal failure, moderate ammonia and congenital heart disease was considered. Simultaneously, a diagnosis of WS1 was also suspected, as an abnormal plasma glucose level and RP had been found.
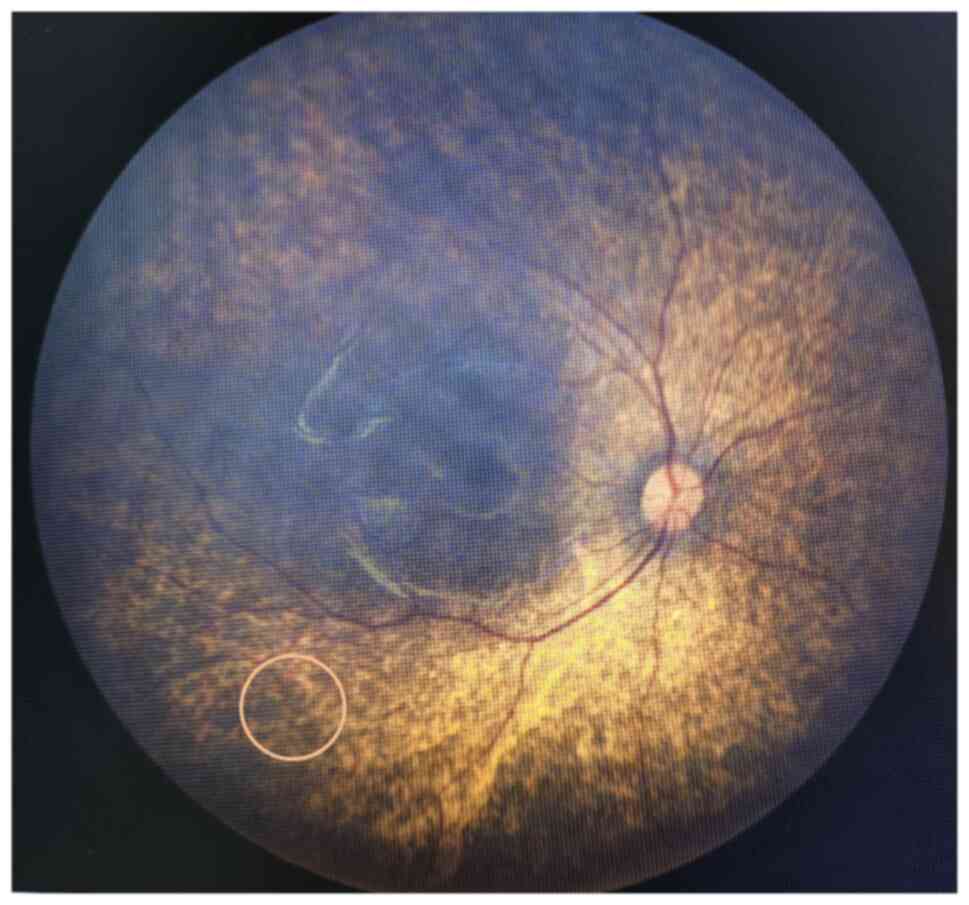 |
Figure 1
Bilateral retinitis pigmentosa was detected by fundus photography. The circle indicates osteocytoid pigmentation.
|
 |
Table I
Abnormal laboratory data at presentation.
|
Table I
Abnormal laboratory data at presentation.
| Parameters |
Patient |
Reference range |
| Blood pressure, mmHg |
140/80 |
75-125/44-90 |
| Blood routine tests |
|
|
| HGB, g/l |
68 |
105-140 |
| RBC, 1012/l |
2.35 |
4.50-6.50 |
| Urine routine tests |
|
|
| Urine protein |
2+ |
Negative |
| Urine glucose |
1+ |
Negative |
| Serum chemistry |
|
|
| Fasting plasma glucose, mM |
6.4 |
3.9-6.1 |
| Albumin, g/l |
39.3 |
40.0-55.0 |
| Globulin, g/l |
19.8 |
20.0-40.0 |
| Serum creatinine, µmol/l |
639 |
57-97 |
| Urea, mmol/l |
26.4 |
2.7-7.0 |
| Complement 3, g/l |
0.72 |
0.77-1.95 |
| Calcium, mmol/l |
1.51 |
2.10-2.80 |
| BNP, pg/ml |
5729.00 |
0.00-125.00 |
| Troponin T, pg/ml |
35.24 |
0.00-14.00 |
| Liver function |
|
|
| AST, U/l |
45 |
15-40 |
| Coagulation function |
|
|
| D-Dimer, mg/l |
1.00 |
0.00-0.55 |
| Folate, ng/ml |
>23.90 |
2.3- |
| Lymphocyte sub population |
|
|
| NK cells, % |
29.14 |
4.00-26.00 |
| CD3+/CD19, % |
9.86 |
10.00-31.00 |
| CD4/CD8 |
0.92 |
0.98-1.94 |
The patient was treated by blood purification to remove uremic toxins, and the standard peritoneal dialysis protocol included Tenckhoff catheter placement by laparoscopy and post-Tenckhoff catheter insertion. In-out exchanges were immediately performed post-catheter insertion for the first five exchanges, and hourly exchanges were performed for 24-48 h until the effluent was clear of gross blood. The patient was also administered erythropoietin (1,100 IU every 5 days) to promote erythropoiesis. Alfacalcidol (0.25 µg every other day) was also administered orally to inhibit the production of parathyroid hormone for 2 weeks. Blood glucose level and blood pressure were dynamically monitored. There is currently no effective therapy for patients with WS1. The patient currently has stage 5 chronic kidney disease and is being treated by automated peritoneal dialysis. The medical prognosis is the requirement for lifetime peritoneal dialysis, eventual hemodialysis or a kidney transplant.
Genetic analysis
Considering the early-onset syndromic phenotype and the suspected diagnosis of WS1, the patient was recommended to undergo genetic testing. DNA samples were isolated from the peripheral blood cells of the proband and their parents with a QIAamp DNA Blood MiniKit (Qiagen GmbH), according to the manufacturer's instructions, and WES was performed by Chigene (Beijing) Translational Medical Research Center, Co., Ltd. In brief, genomic DNA samples were fragmented to 150-200 bp and subjected to DNA library preparation [final loading concentration of ≥5 ng/µl measured by measured by Qubit 2.0 (Thermo Fisher Scientific, Inc.)]. Protein-coding exome enrichment was performed using xGen Exome Research Panel v1.0 (Integrated DNA Technologies, Inc.), and adaptor-ligated libraries were amplified via PCR. Paired-end sequencing was carried out on a DNBSEQ-T7 sequencing platform [Chigene (Beijing) Translational Medical Research Center, Co., Ltd.] with a PE150 sequencing kit (FCL PE150 V2.0; cat. no. 1000028454) to obtain a read length of 2x150 bp. Primary data were in fastq format and filtered to generate ‘clean reads’ by removing adapters and low-quality reads. All clean reads were aligned to the Human Reference genome (HG19) using the Burrows-Wheeler Aligner Multi-Vision software package (version no. 0.7.17-r1188; https://github.com/lh3/bwa). Single nucleotide variants and indels were identified by using the SOAPsnp software and Samtools Indel Genotyper (version no. 1.13; https://github.com/samtools/samtools). The variants were annotated and were analyzed using the ATAV software (https://atavdb.org/) (10). Two parallel reactions were performed for each sample. Finally, sequence analysis revealed two homozygous variants of the WFS1 gene, NM_006005 c.1820C>T and c.2603G>A, which changed an amino-acid residue of the Wolframin protein. Both parents of the proband were heterogeneous for the variants. Considering the homologous variants in the proband, both variants of the WFS1 gene were most likely in a cis pattern in the parents. The variants were included in the Human ClinVar Database, with a classification of uncertain significance. Sanger sequencing was then performed to validate the identified variation (Fig. 2). According to the American College of Medical Genetics and Genomics standards and guidelines (11), both variants were classified as likely pathogenic (PM1+PM2+PP3+PM3-Supporting). Based on the genetic analysis, the patient was finally diagnosed with WS1.
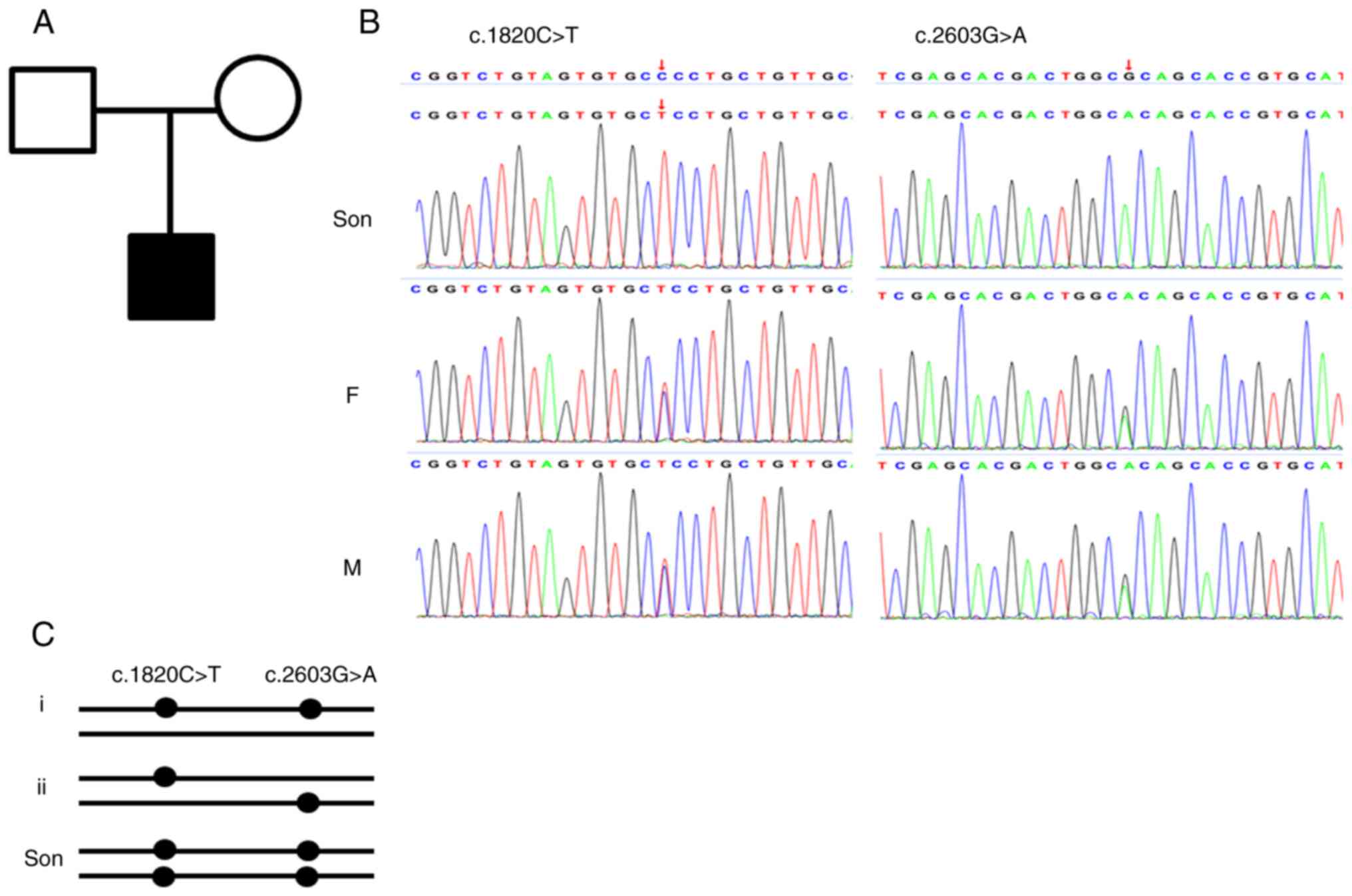 |
Figure 2
Identification of the variants in the family. (A) Pedigree of the family. Affected family members are denoted in black. Squares indicate males and circles indicate females. (B) Direct Sanger sequencing confirmed the variants in the wolframin endoplasmic reticulum transmembrane glycoprotein gene. (C) The genotype of the parents and the proband. The genotype of the parents might be i or ii, but the genotype of the proband must be homologous at both loci. Son, the proband; F, father; M, mother.
|
Discussion
As an autosomal recessive rare disease, WS1 is poorly recognized by physicians, especially in early onset patients. DM of juvenile onset and OA are the minimal diagnostic criteria. WFS1 mutations cause WS1, and the spectrum of WFS1 mutations causing WS1 is broad. Due to the development of sequencing technology, WES has been used clinically for the analysis of suspected gene mutations (12,13).
Patients with WS1 present with DM at an average age of 6 years, while OA manifests at the age of 11 years (14). In the present case, RP was diagnosed first, and then an abnormal plasma glucose level was detected in the Department of Nephrology, Guiyang Maternal and Child Health Care Hospital. Although the diagnosis of DM could not be established due to the inability to perform a glucose tolerance test and the lack of diabetic manifestations, the patient was classified as prediabetic when taking age into consideration. Thus, the diagnosis of WS1 was suspected. In addition, the detected renal abnormalities and heart malformations matched with those found in other reported cases (15).
The WFS1 gene encodes a transmembrane protein that is located primarily in the ER and is ubiquitously expressed in various tissues. Hundreds of variants of the WFS1 gene have been deposited in the ClinVar database, and 180 variants have been classified as pathogenic or likely pathogenic. For the point mutation or indel variants, most of these variants are located in exon 8 of WFS1 (16). In the present case, both variants were located on exon 8 and categorized as missense mutations. Based on the genotype of the proband, the variants of the parents were most likely in the cis pattern, which means that both variants were located on the same chromosome (Fig. 2C). Under this condition, the offspring of the compound heterozygous parents have a probability of 1/4 to be homologous at both loci. Although both variants are classified as of uncertain significance in Clinvar Database, they can now be classified as likely pathogenic based on the present findings, including co-segregation of the genotype and phenotype in the core family, and the variant frequency in the population. Both variants were located in the hot spot of the pathogenic mutations. However, it is uncertain whether the phenotypes are caused just by one variant or by both variants, as both variants are likely pathogenic.
The present case showed the early clinical features of WS1. Two homozygous variants in the WFS1 gene were the genetic basis of this case of WS1. The genetic diagnosis of this patient was of benefit for deciding any further treatment and for the genetic counseling of the family. This report helps to identify similar cases in clinical practice.
Supplementary Material
Parameters for abdominal ultrasound Philips EPIQ 5 machinea.
Acknowledgements
Not applicable.
Funding
Funding: Not applicable.
Availability of data and materials
According to the Regulation of the People's Republic of China on the Administration of Human Genetic Resources (https://www.gov.cn/zhengce/content/2019-06/10/content_5398829.htm), the raw WES data are not available; however, the VCF file is available from the corresponding author on reasonable request.
Authors' contributions
YT, XS, BY, JQ, SZ and YLiu obtained medical images. YT, XZ and YLi analyzed patient data. All authors contributed to the article and approved the submitted version. All authors have read and approved the final manuscript. YT and YLi confirm the authenticity of all the raw data.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Guiyang Maternal and Child Health Care Hospital (Guiyang, China; approval no. 2021-65).
Patient consent for publication
Written informed consent for publication of identifying images and other personal and clinical details was obtained from the patient's parents.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Fischer TT and Ehrlich BE: Wolfram Syndrome: A Monogenic model to study diabetes mellitus and neurodegeneration. Curr Opin Physiol. 17:115–123. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lima Ferreira J, Carvalho V, Espada F, Massano J, Marques AP and Príncipe RM: Wolfram syndrome: Phenotypic heterogeneity and novel genetic variants in the WFS1 gene. Endocrinol Diabetes Nutr (Engl Ed). 69:153–154. 2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pallotta MT, Tascini G, Crispoldi R, Orabona C, Mondanelli G, Grohmann U and Esposito S: Wolfram syndrome, a rare neurodegenerative disease: From pathogenesis to future treatment perspectives. J Transl Med. 17(238)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Rigoli L, Caruso V, Aloi C, Salina A, Maghnie M, d'Annunzio G, Lamacchia O, Salzano G, Lombardo F and Picca G: An atypical case of late-onset wolfram Syndrome 1 without diabetes insipidus. Int J Environ Res Public Health. 19(2473)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li Y, Gong S, Li M, Cai X, Liu W, Zhang S, Ma Y, Luo Y, Zhou L, Zhang X, et al: The genetic and clinical characteristics of WFS1 related diabetes in Chinese early onset type 2 diabetes. Sci Rep. 13(9127)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chen Y, Zhang M, Zhou Y and Li P: Case Report: A novel mutation in WFS1 gene (c.1756G>A p.A586T) is responsible for early clinical features of cognitive impairment and recurrent ischemic stroke. Front Genet. 14(1072978)2023.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang X, Xie Y, Xu K, Chang H, Zhang X and Li Y: Comprehensive genetic analysis unraveled the 7 missing heritability in a Chinese COHORT WITH WOLFRAM Syndrome 1: Clinical and genetic findings. Invest Ophthalmol Vis Sci. 63(9)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Li L, Venkataraman L, Chen S and Fu H: Function of WFS1 and WFS2 in the central nervous system: Implications for wolfram Syndrome and Alzheimer's disease. Neurosci Biobehav Rev. 118:775–783. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Rötig A, Cormier V, Chatelain P, Francois R, Saudubray JM, Rustin P and Munnich A: Deletion of mitochondrial DNA in a case of early-onset diabetes mellitus, optic atrophy, and deafness (Wolfram syndrome, MIM 222300). J Clin Invest. 91:1095–1098. 1993.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ren Z, Povysil G, Hostyk JA, Cui H, Bhardwaj N and Goldstein DB: ATAV: A comprehensive platform for population-scale genomic analyses. BMC Bioinformatics. 22(149)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Jezkova J, Shaw S, Taverner NV and Williams HJ: Rapid genome sequencing for pediatrics. Hum Mutat. 43:1507–1518. 2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Petersen BS, Fredrich B, Hoeppner MP, Ellinghaus D and Franke A: Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 18(14)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Collier DA, Barrett TG, Curtis D, Macleod A, Arranz MJ, Maassen JA and Bundey S: Linkage of Wolfram Syndrome to chromosome 4p16.1 and evidence for heterogeneity. Am J Hum Genet. 59:855–863. 1996.PubMed/NCBI
|
|
15
|
Urano F: Wolfram Syndrome: Diagnosis, management, and treatment. Curr Diab Rep. 16(6)2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Delvecchio M, Iacoviello M, Pantaleo A and Resta N: Clinical spectrum associated with Wolfram Syndrome type 1 and type 2: A review on genotype-phenotype correlations. Int J Environ Res Public Health. 18(4796)2021.PubMed/NCBI View Article : Google Scholar
|














