Introduction
Psoriatic arthritis (PsA) is a heterogeneous chronic
immune-mediated disease characterized by musculoskeletal
inflammation. Numerous patients develop PsA on the background of
psoriasis, a skin condition of scaly erythematous plaques that
commonly affects the extensor surfaces of the elbows and knees, and
other parts of the body (1-3).
The onset of PsA often occurs between the age of 30 and 50 years
but may arise at any point throughout a patient's lifetime. The
clinical manifestations of PsA vary greatly between patients and
range from relatively mild to severe disease, and disease flares
can alternate with periods of remission (4). Due to the shared similarities in the
clinical presentation of PsA and other arthritic diseases such as
rheumatoid arthritis (RA) and osteoarthritis (OA), PsA is
frequently undiagnosed and/or misdiagnosed (5). However, six clinical domains are
involved in PsA including peripheral arthritis, enthesitis,
dactylitis, psoriasis, psoriatic nail disease and axial disease
(6).
Although the aetiology of PsA is not fully
understood, genetics, epigenetics and environmental factors
contribute to abnormal immune responses and disease expression
(7). At the genetics level, both
human leukocyte antigen (HLA) and non-HLA genes have been
associated with PsA (7). Moreover,
33-50% of patients with PsA have at least one first-degree
relatives who are also affected by psoriasis or PsA (8). Previous studies have shown that
mitochondrial dysfunction contributes significantly to the
pathogenesis of PsA by modulating innate immunity via
redox-sensitive inflammatory pathways (9,10).
Oxidative stress can disrupt redox signalling and cause molecular
damage, which impacts angiogenesis, inflammation and immune cell
function (11). Mitochondria
produce most of the cellular energy through the process of
oxidative phosphorylation (OXPHOS) and are also the major site of
reactive oxygen species (ROS). Besides their central role in
cellular metabolism, mitochondria also participate in other
important cellular processes such as innate immune, inflammatory
and stress responses (12).
Mitochondria have their own genome called mitochondrial DNA
(mtDNA), which is a double-stranded molecule encoding 37 genes. In
total, 13 of the mtDNA genes are involved in the OXPHOS and the
remaining genes are essential in assembling amino acids into
functional proteins (13). mtDNA
presents in multiple copies (1,000-10,000 copies) per cell,
resulting in both homoplasmic and heteroplasmic mtDNA variants.
Moreover, the mtDNA copy number (mtDNAcn) is regulated in a
tissue-specific manner (14) and
correlates positively with the number of mitochondria, and thus is
considered an indicator of mitochondrial function (15). Numerous factors make mtDNA
particularly vulnerable to ROS and oxidative damage, including its
proximity to the site of the electron transport chain (ETC),
absence of protective histones and inadequate DNA repair capacity
(16). Oxidative stress is an
important source of mitochondrial genomic instability and can
induce mtDNA variations and copy number changes, which may lead to
abnormalities in mitochondrial function (17,18).
Both mtDNA variants and copy number alterations have been
implicated in human aging and various pathological conditions
including mitochondrial disorders, cancer, and neurodegenerative
diseases (19,20).
Harty et al (21) evaluated the total mtDNA mutational
load in PsA/RA using a mitochondrial random capture assay, which
revealed a significant increase in the frequency of mtDNA variants
in synovial tissue from patients with RA and PsA compared with
controls. However, mitochondrial random capture assay has
limitations such as low sensitivity and inability to detect
heteroplasmy, which is an important characteristic of numerous
mtDNA-related diseases. Previously, next-generation sequencing
(NGS) has emerged as a robust technique for screening the
mitochondrial genome. It enables comprehensive analysis of the
entire mitochondrial genome for the detection of common and rare
mtDNA variants, mtDNA disease-associated variants, and accurate
measurement of heteroplasmy (22).
Since no previous studies have analysed the entire mitochondrial
genome or evaluated changes in mtDNAcn and oxidative stress in PsA,
the present study aimed to investigate mtDNA variants related to
PsA and/or associated with the risk of PsA via NGS. The present
study also aimed to examine changes in mtDNAcn as well as evaluate
mtDNA oxidative damage in patients with PsA and healthy
controls.
Materials and methods
Study subjects
A total of 43 subjects including 23 patients with
PsA and 20 healthy controls were enrolled in the present study.
Patients with PsA were recruited from the out-patient clinic at the
Department of Rheumatology of Mubarak Hospital, (City of Kuwait,
State of Kuwait). The patients fulfilled the classification
criteria for PsA (CASPAR). Patients with other inflammatory or
autoimmune diseases were excluded from study (23). Clinicopathological characteristics
of patients (including sex and age distribution) are presented in
Table I.
 | Table IDemographic and clinical data of
patients with psoriatic arthritis and controls. |
Table I
Demographic and clinical data of
patients with psoriatic arthritis and controls.
|
Characteristics | PsA | Controls | P-value |
|---|
| Number of
subjects | 23 | 20 | |
| Sex | | | 0.6 |
|
Male, n
(%) | 11(48) | 8(50) | |
|
Female, n
(%) | 12(52) | 12(50) | |
| Age, years (mean ±
SD) | 39±3 | 30±1.3 | 0.01 |
| C-Reactive protein,
mg/dl | 5±0.9 | 0.1±0.05 | 0.02 |
| Rheumatoid factor,
U/ml | 21±2 | 0.4±0.02 | <0.001 |
| Erythrocyte
sedimentation | 28±9 | 2.6±1 | 0.003 |
| rate, mm/h | | | |
| Medications | | | |
|
Topical
treatment | 4 | | |
|
Systemic
treatment | 19 | | |
|
Methotrexate | 4 | | |
|
Adalimumab | 8 | | |
|
Etanercept | 3 | | |
|
Secukinumab | 2 | | |
|
Ixekizumab | 2 | | |
Healthy control individuals without inflammatory
dermatoses or autoimmune diseases were recruited from the Central
Blood Bank, State of Kuwait. Basic clinical characteristics and
laboratory data were obtained from the medical and electronic
records of patients and controls. Written informed consent was
obtained from all participants under protocols approved by Kuwait
University and Ministry of Health (City of Kuwait, State of Kuwait)
(approval no. 2018/496).
Extraction of genomic DNA
Blood samples (5 ml) were collected in EDTA tubes
from the participants and centrifuged at 4˚Cn 1,000 x g for 15 min
to separate the buffy coat which was subjected to genomic DNA
extraction using QIA amp DNA Mini kit (cat. no. 51304; Qiagen GmbΗ)
according to the manufacturer's instructions as previously
described (24). Briefly, a mixture
of 200 µl buffy coat, 20 µl protease and 200 µl lysis buffer was
incubated at 56˚C for 10 min and then centrifuged at 20,000 x g for
1 min at 4˚C. Next, absolute ethanol (200 µl) was added and
centrifuged at 6,000 x g for 1 min followed by washing with 500 µl
washing buffer. The mixture was then centrifuged at room
temperature at 6,000 x g for 1 min and then at 20,000 x g for 3
min. Genomic DNA was eluted with 200 µl elution buffer after
incubation at room temperature for 1 min and centrifugation at
6,000 x g for 1 min at room temperature. The DNA samples were
quantified and assessed for purity using a NanoDrop ND-1000
ultraviolet-visible light spectrophotometer (Thermo Fisher
Scientific, Inc.).
Mitochondrial genome sequencing
The entire mitochondrial genome was sequenced using
the S5™XL NGS system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol, as previously
described (25). After library
preparation and purification, the raw data were automatically
transferred from the Ion Torrent S5 XL sequencer to the Torrent
Suite software version 5.0 (Applied Biosystems; Thermo Fisher
Scientific, Inc.), which allowed the conversion of the raw voltage
semiconductor sequencing data into DNA base calls. For
identification of variants, the Ion Torrent Variant Caller plug-in,
Ion Reporter software version 5.2. and Torrent Variant Caller
version 5.2 (Applied Biosystems; Thermo Fisher Scientific, Inc.)
were used. For alignment, the Revised Cambridge Reference Sequence
of the Human mtDNA (NC_012920.1) was applied as a reference
mitochondrial sequence (26). The
average throughput of the Ion 520 chip was 3.5 Mb. The sequence
data sets were registered in the Sequence Read Archive repository
(reference no. PRJNA 928743).
Bioinformatics analysis
The impact of nonsynonymous mtDNA variants on
protein function and structure was determined using three
in-silico prediction tools used: i) Combined Annotation
Dependent Depletion (CADD): Incorporates multiple annotations
including conservation and functional information into one tool and
categorizes variants as benign or deleterious using a machine
learning approach. Variants with scores ≥20 were predicted to be
deleterious (27); ii) CONsensus
DELeteriousness (Condell): Integrates the output of five algorithms
including Pfam E-value (Logre), MAPP, Mutation Assessor,
Polyphen2, and SIFT to assess the outcome of nonsynonymous single
nucleotide variants (SNVs) on protein function. Variants with
scores ≥0.5 were predicted to be deleterious (28); and iii) Protein Variation Effect
Analyzer (PROVEAN) predicts the impact of an amino acid
substitution or indel on protein function. PROVEAN performance is
comparable to SIFT or PolyPhen-2 and it can process a large number
of protein variants. Variants with scores ≤-2.5 were predicted to
be deleterious, while those with scores >-2.5 were considered
neutral (29).
Analysis of protein stability
Analysis of the impact of nonsynonymous mtDNA
variants on protein stability was conducted using Site-Directed
Mutator (SDM), which is a statistical potential energy function
that uses environment-specific amino acid substitution frequencies
within the family of homologous proteins of known (3-D) structures
to calculate a stability score, which is analogous to the free
energy difference between the wild-type and mutant protein
(30). A change in the Gibbs free
energy for protein stability is expressed as ΔΔG (30).
Determination of relative mtDNAcn
Quantitative polymerase chain reaction (qPCR) was
used to determine the mtDNAcn relative to nuclear DNA (nDNA).
Mitochondrial NADH dehydrogenase subunit 2 (ND2) was used as
a target gene for the amplification of mtDNA with the following
primers: forward, 5'-CAC AGA AGC TGC CAT CAA GTA-3' and reverse,
5'-CCG GAG AGT ATA TTG TTG AAG AG-3'; while nuclear
b2-microglobulin (β2M) was used as a reference gene for the
amplification of nDNA with the following primers: forward, 5'-CCA
GCA GAG AAT GGA AAG TCA A-3' and reverse, 5'-TCT CTC TCC ATT CTT
CAG TAA GTC AAC T-3'. The PCR mixture contained 10 ng genomic DNA,
1X SYBR1 Green PCR Master Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.), forward and reverse primers (50 nM each), and
nuclease-free water to a final volume of 10 µl. PCR was performed
in a 7900HT real-time PCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using the following thermocycling conditions:
Initial denaturation at 95˚C for 10 min, followed by 40 cycles of
95˚C for 10 sec, 60˚C for 30 sec and 72˚C for 30 sec. The
experiments were performed in duplicate and non-template control
was included in each run. Relative quantitation of mtDNAcn was
performed using the 2-ΔΔCq method (31) by obtaining the Cq values of the
ND2 and β2M genes, and then the ΔCq (Cq ND2-Cq
β2M) values for cases and controls were calculated.
Determination of mtDNA oxidative
damage
8-Hydroxyl 2'-deoxyguanosine (8-OHdG) is one of the
most common markers of oxidative DNA lesions and is widely applied
for the measurement of oxidative DNA damage (32,33).
In the detection of oxidative damage with 8-OHdG, the marker does
not deform the DNA structure but causes inhibition of Taq
DNA polymerase during PCR. Therefore, the presence of 8-OHdG in a
certain region of mtDNA can be digested by formamidopyrimidine
[fapy]-DNA glycosylase (FPG) which breaks the DNA fragment at the
lesion site and reduces further amplification of this particular
region. In the present study, 8-OHdG assay was conducted to measure
mtDNA oxidative damage by qPCR. A total of 100 ng DNA was incubated
for 1 h at 37˚C in a 10 µl of reaction mixture containing 1 U FPG
enzyme (New England Bio Labs, Inc), 1X NEB buffer and 0.1 mg/ml
bovine serum albumin (New England Bio Labs, Inc). Next, the
digested DNA was amplified by PCR under the same aforementioned
cycling protocol and conditions. DNA damage was measured as ΔCq
(ΔCq=Cq treated-Cq untreated). The presence of oxidative damage in
DNA after treatment of FPG reduces the PCR efficiency and increases
the Ct value.
Statistical analysis
Statistical analysis of data was performed using
SPSS software (version 23; IBM Corp.). The normal distribution of
data was first evaluated using the Kolmogorov-Smirnov test.
Accordingly, the comparisons of variables between patients and
controls were conducted using the χ2 test for
categorical variables and the Mann-Whitney test for normally
distributed variables. The Fisher's exact test was used to assess
differences in the frequency of variants between patients and
controls and the odds ratio (OR) and 95% confidence interval (CI)
were reported. P≤0.05 was considered to indicate a statistically
significant difference.
Results
Characteristics of the study
subjects
The demographic and clinical data of patients with
PsA (n=23) and healthy control individuals (n=20) are presented in
Table I. The sex ratio (male:
female) was 11:12 for patients and 8:12 for controls. There was no
significant difference in sex distribution between PsA and controls
(P=0.6), but there was a significant difference in mean age between
the two groups (P=0.01). The clinical inflammatory marker
C-reactive protein (CRP) (0-0.8 mg/dl), rheumatoid factor (RF)
(0-20 U/ml), and erythrocyte sedimentation rate (ESR) (0-20 mm/hr)
were all higher than the normal range in patients with PsA. The
mean value of CRP, RF and ESR were significantly higher in PsA
patients compared with controls (P<0.05). Patients were under
the following medications: Topical treatment with corticosteroid
cream (n=4), or systemic treatment (n=19) including Methotrexate
(n=4), Adalimumab (n=8), Etanercept (n=3), Secukinumab (n=2) and
Ixekizumab (n=2).
mtDNA sequence variants
identification
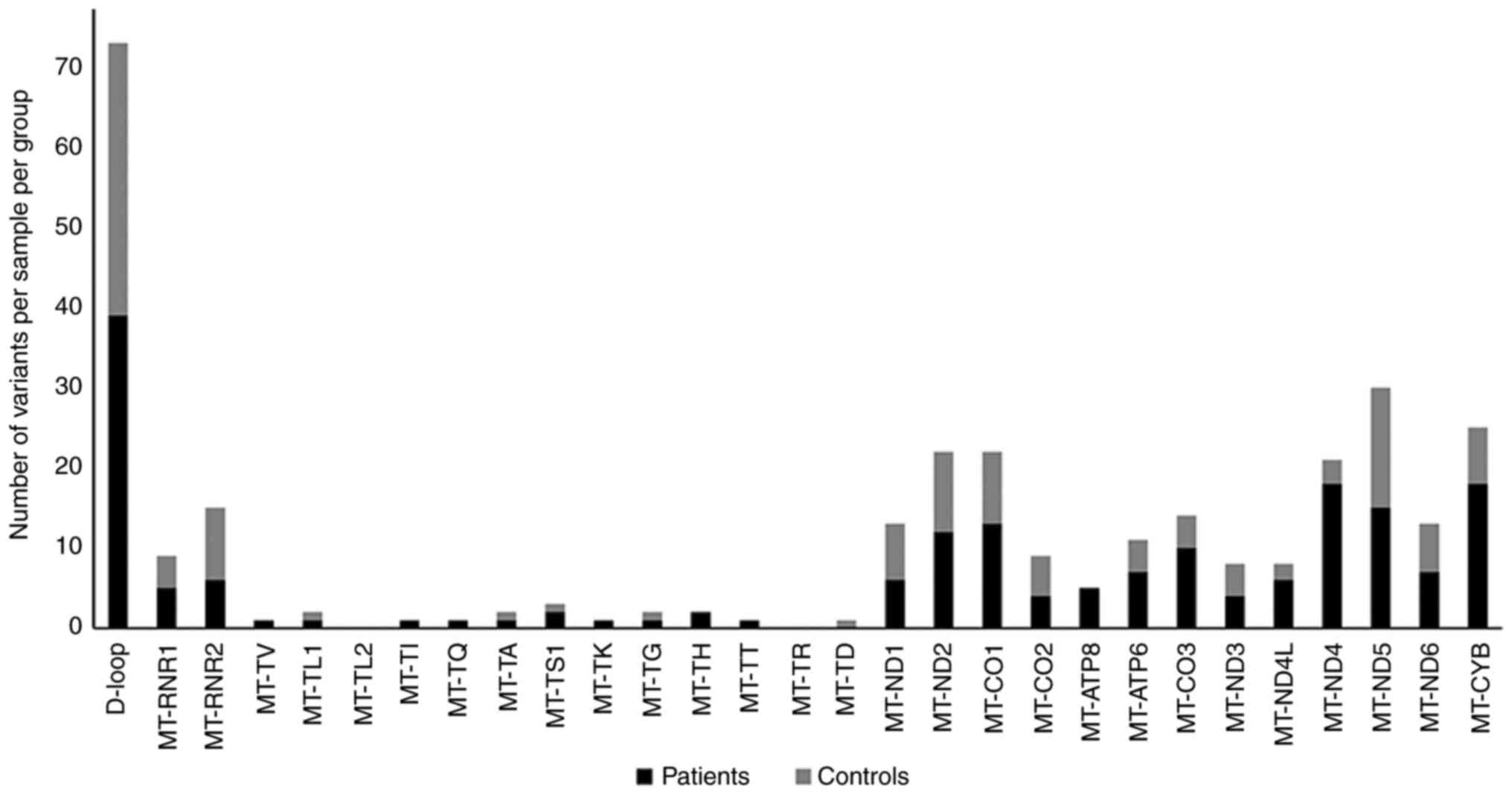
A total of 435 mtDNA sequence variants were
identified in 43 samples. Among them, 187 (43%) variants were
exclusive for patients with PsA, and 122 (28%) variants were found
in control individuals only (Fig.
1). In both patients and controls, the highest number of
variants was observed in the D-loop region, while the lowest number
of variants was observed in the tRNAs genes. In protein-coding
genes of patients with PsA, there were 152 variants including 33
nonsynonymous and 92 synonymous silent variants. By contrast, in
protein-coding genes of control individuals, there were 76
variants, including 18 non-synonymous and 58 synonymous silent
variants.
Particularly, a higher number of variants were
observed in the MT-ND4, MT-ND5 and MT-CYB genes. A higher number of
variants in the MT-ND4, and MT-CYB genes was also observed in
patients than in controls. Moreover, the majority of mtDNA variants
in patients and controls were homoplasmy accounting for 183 and
118, respectively, compared with the small number of heteroplasmy
variants (n=4) in each group. Details of the identified variants in
patients or controls as well as their localization in different
mtDNA regions are shown in Table
II. Notably, of non-synonymous variants in controls only,
4640C>A in the MT-ND2 gene was reported as a pathogenic variant
causing Leber optic atrophy according to the Mito Map (https://www.mitomap.org/MITOMAP) and ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/) databases.
| Table IIDetails of mtDNA variants exclusive
for patients with psoriatic arthritis or controls. |
Table II
Details of mtDNA variants exclusive
for patients with psoriatic arthritis or controls.
| Non-coding
region |
|---|
| | Patients | Controls | |
|---|
| Locus | Variant | Type of
variant | Amino acid
change | rs ID |
Homoplasmy/Heteroplasmy | Variant | Variant type | Affected amino
acid | rs ID |
Homoplasmy/Heteroplasmy |
|---|
| D-loop | m.57T>TC | Insertion | | rs21245905 | Heteroplasmy | m.143G>A | Substitution | | rs375589100 | Homoplasmy |
| D-loop | m.72A>G | Substitution | | rs869183622 | Heteroplasmy | m.178A>G | Substitution | | Not provided | Homoplasmy |
| D-loop | m.93A>G | Substitution | | rs369034419 | Homoplasmy | m.182C>T | Substitution | | rs41473347 | Homoplasmy |
| D-loop | m.153A>G | Substitution | | rs370716192 | Homoplasmy | m.185 G>T | Substitution | | rs879015046 | Homoplasmy |
| D-loop | m.189A>G | Substitution | | rs371543232 | Homoplasmy | m.199T>C | Substitution | | rs72619362 | Homoplasmy |
| D-loop | m.200A>G | Substitution | | rs372099630 | Homoplasmy | m.250 T>C | Substitution | | Not provided | Homoplasmy |
| D-loop | m.207G>A | Substitution | | rs369669319 | Homoplasmy | m.285C>T | Substitution | | rs201801609 | Homoplasmy |
| D-loop | m.215A>G | Substitution | | rs879219259 | Homoplasmy | m.357A>G | Substitution | | rs28678375 | Homoplasmy |
| D-loop | m.217T>C | Substitution | | rs41531144 | Homoplasmy | m.385A>G | Substitution | | rs201801609 | Homoplasmy |
| D-loop | m.235A>G | Substitution | | rs3937037 | Homoplasmy | m.497C>T | Substitution | | rs28660704 | Homoplasmy |
| D-loop | m.236T>C | Substitution | | rs375896687 | Homoplasmy | m.16093T>C | Substitution | | rs2853511 | Heteroplasmy |
| D-loop | m.340C>T | Substitution | | rs117394573 | Homoplasmy | m.16150C>T | Substitution | | rs879004379 | Homoplasmy |
| D-loop | m.508A/G | Substitution | | rs113683159 | Homoplasmy | m.16163 A>G | Substitution | | rs41479950 | Homoplasmy |
| D-loop | m.524 C>CAC | Insertion | | Not provided | Heteroplasmy | m.16185C>T | Substitution | | rs1556424787 | Homoplasmy |
| D-loop | m.16041A>G | Substitution | | rs369904200 | Homoplasmy | m.16186C>T | Substitution | | rs879166752 | Homoplasmy |
| D-loop | m.16051A>G | Substitution | | rs117565943 | Homoplasmy | m.16224T>C | Substitution | | rs386420031 | Homoplasmy |
| D-loop | m.16067C>T | Substitution | | rs1556424732 | Homoplasmy | m.16232C>A | Substitution | | rs1603225749 | Homoplasmy |
| D-loop | m.16111C>T | Substitution | | rs35315169 | Homoplasmy | m.16249T>C | Substitution | | rs372301309 | Homoplasmy |
| D-loop | m.16124T>C | Substitution | | rs386829272 | Homoplasmy | m.16264C>G | Substitution | | rs878922147 | Homoplasmy |
| D-loop | m.16148C>T | Substitution | | rs201893071 | Homoplasmy | m.16288T>C | Substitution | | rs386829301 | Homoplasmy |
| D-loop | m.16178T>C | Substitution | | rs1603225705 | Homoplasmy | m.16293A>G | Substitution | | rs878890610 | Homoplasmy |
| D-loop |
m.16179CA-AAA>CAA§ | Deletion | | rs371240719 | Heteroplasmy | m.16296C>T | Substitution | | rs879138789 | Homoplasmy |
| D-loop | m.16183A>T | Substitution | | rs28671493 | Homoplasmy | m.16318A>T | Substitution | | rs879067317 | Homoplasmy |
| D-loop | m.16192C>T | Substitution | | rs879025248 | Homoplasmy | m.16319G>A | Substitution | | rs35105996 | Homoplasmy |
| D-loop | m.16201C>T | Substitution | | Not provided | Homoplasmy | m.16343A>G | Substitution | | rs374065731 | Homoplasmy |
| D-loop | m.16214C>T | Substitution | | rs368055283 | Homoplasmy | m.16380C>T | Substitution | | rs878952395 | Homoplasmy |
| D-loop | m.16217T>C | Substitution | | rs35134837 | Homoplasmy | m.16381T>C | Substitution | | rs1556424876 | Homoplasmy |
| D-loop | m.16219A>G | Substitution | | rs878960666 | Homoplasmy | m.16526G>A | Substitution | | rs386829315 | Homoplasmy |
| D-loop | m.16230A>G | Substitution | | rs2853514 | Homoplasmy | | | | | |
| D-loop | m.16234C>T | Substitution | | rs368259300 | Homoplasmy | | | | | |
| D-loop | m.16289A>G | Substitution | | rs1603225781 | Homoplasmy | | | | | |
| D-loop | m.16290C>T | Substitution | | rs386828866 | Homoplasmy | | | | | |
| D-loop | m.16295C>T | Substitution | | rs878874012 | Homoplasmy | | | | | |
| D-loop | m.16300A>G | Substitution | | rs879082592 | Homoplasmy | | | | | |
| D-loop | m.16301C>T | Substitution | | rs879194775 | Homoplasmy | | | | | |
| D-loop | m.16304T>C | Substitution | | rs386829305 | Homoplasmy | | | | | |
| D-loop | m.16320C>T | Substitution | | rs62581338 | Homoplasmy | | | | | |
| D-loop | m.16324T>C | Substitution | | rs1556424863 | Homoplasmy | | | | | |
| D-loop | m.16399A>G | Substitution | | rs139001869 | Homoplasmy | | | | | |
| rRNA genes |
| MT-RNR1
(12sRNA) | m.961T>C | Substitution | | rs3888511 | Homoplasmy | m.954C>T | Substitution | | Not provided | Homoplasmy |
| MT-RNR1
(12sRNA) | m.1008A>G | Substitution | | rs727504505 | Homoplasmy | m.980T>C | Substitution | | rs397515731 | Homoplasmy |
| MT-RNR1
(12sRNA) | m.1048C>T | Substitution | | rs2000974 | Homoplasmy | m.1018G>A | Substitution | | rs2856982 | Homoplasmy |
| MT-RNR1
(12sRNA) | m.1442G>A | Substitution | | rs28358573 | Homoplasmy | m.1189T>C | Substitution | | rs28358571 | Homoplasmy |
| MT-RNR1
(12sRNA) | m.1598G>A | Substitution | | rs3135027 | Homoplasmy | | | | | |
| MT-RNR2
(16sRNA) | m.2028G>A | Substitution | | rs2124591154 | Homoplasmy | m.1733C>T | Substitution | | rs878868960 | Homoplasmy |
| MT-RNR2
(16sRNA) | m.2245A>G | Substitution | | rs3020600 | Homoplasmy | m.1738T>C | Substitution | | rs28358574 | Homoplasmy |
| MT-RNR2
(16sRNA) | m.2259C>T | Substitution | | rs201336470 | Homoplasmy | m.2218C>T | Substitution | | rs200813159 | Homoplasmy |
| MT-RNR2
(16sRNA) | m.2283C>T | Substitution | | rs200131896 | Homoplasmy | m.2380C>T | Substitution | | rs1556422622 | Homoplasmy |
| MT-RNR2
(16sRNA) | m.2484C>T | Substitution | | rs2124591301 | Homoplasmy | m.2768A>G | Substitution | | rs3895615 | Homoplasmy |
| MT-RNR2
(16sRNA) | m.2626T>C | Substitution | | rs879158835 | Homoplasmy | m.2772C>T | Substitution | | rs200221487 | Homoplasmy |
| MT-RNR2
(16sRNA) | m.3221A>G | Substitution | | rs1556422691 | Homoplasmy | m.2833A>G | Substitution | | rs3928312 | Homoplasmy |
| MT-RNR2
(16sRNA) | | | | | | m.3158A>AT | Insertion | | rs1556422679 | Homoplasmy |
| MT-RNR2
(16sRNA) | | | | | | m.3221A>G | Substitution | | rs1556422691 | Homoplasmy |
| tRNA genes |
| MT-TV (tRNA) | m.1676A>G | Substitution | | rs1603218612 | Homoplasmy | | | | | |
| MT-TL1 (tRNA) | m.3387T>C | Substitution | | rs1569483877 | Homoplasmy | m.3277G>A | Substitution | | rs386828902 | Homoplasmy |
| MT-TQ (tRNA) | m.4454T>C | Substitution | | rs11510098 | Homoplasmy | | | | | |
| MT-TA (tRNA) | m.5603C>T | Substitution | | rs369496446 | Homoplasmy | m.5655T>C | Substitution | | rs1556423019 | Homoplasmy |
| MT-TS1 (tRNA) | m.7476C>T | Substitution | | rs201950015 | Homoplasmy | m.7474G>C | Substitution | | rs2068703713 | Heteroplasmy |
| MT-TS1 (tRNA) | m.7570A>G | Substitution | | rs1556423311 | Homoplasmy | | | | | |
| MT-TD (tRNA) | | | | | | m.7581T>C | Substitution | | rs201582552 | Homoplasmy |
| MT-TK (tRNA) | m.8292G>A | Substitution | | rs1556423422 | Homoplasmy | | | | | |
| MT-TG (tRNA) | m.10042A>G | Substitution | | rs1603222643 | Homoplasmy | m.10034T>C | Substitution | | rs41347846 | Homoplasmy |
| MT-TH (tRNA) | m.12171A>G | Substitution | | rs1603223589 | Homoplasmy | | | | | |
| MT-TH(tRNA) | m.12175T>C | Substitution | | rs1057520099 | Homoplasmy | | | | | |
| MT-TT (tRNA) | m.15907A>G | Substitution | | rs41383248 | Homoplasmy | | | | | |
| MT-ND1 | m.3516C>A | Silent | p.leu70= | rs2854132 | Homoplasmy | m.3438G>A | Silent | p.Gly44= | rs377699338 | Homoplasmy |
| MT-ND1 | m.3720A>G | Silent | p.Gln138= | rs41355750 | Homoplasmy | m.3546C>A | Silent | p.Thr80= | rs1556422747 | Homoplasmy |
| MT-ND1 | m.3834G>A | Silent | p.Leu176= | rs372080842 | Homoplasmy | m.3591G>A | Silent | p.Leu95= | rs1556422757 | Homoplasmy |
| MT-ND1 | m.3873A>G | Silent | p.Thr189= | rs386828925 | Homoplasmy | m.3666G>A | Silent | p.Gly120= | rs28357968 | Homoplasmy |
| MT-ND1 | m.4104A>G | Silent | p.Leu266= | rs1117205 | Homoplasmy | m.3693G>A | Silent | p.Leu129= | rs193303027 | Homoplasmy |
| MT-ND1 | m.4188A>G | Silent | p.Leu294= | Not provided | Homoplasmy | m.3741C>T | Silent | p.Thr145= | rs878907222 | Homoplasmy |
| MT-ND2 | m.4688T>C | Silent | p.Ala73= | rs878853056 | Homoplasmy | m.4640C>A | Missense | p.Ile57Met | rs387906426 | Homoplasmy |
| MT-ND2 | m.4695T>C | Missense | p.Phe76Leu | rs1556422885 | Homoplasmy | m.4774T>TG | Insertion | | Not provided | Heteroplasmy |
| MT-ND2 | m.4703T>C | Silent | p.Asn78= | rs386828949 | Homoplasmy | m.4991G>A | Silent | p.Gln174= | rs386828958 | Homoplasmy |
| MT-ND2 | m.4742T>C | Silent | p,Asn91= | rs1553139137 | Homoplasmy | m.5036A>G | Silent | p.Trp189= | rs28357982 | Homoplasmy |
| MT-ND2 | m.5075T>C | Silent | p,IIe202= | rs1603219767 | Homoplasmy | m.5046G>A | Missense | p.Val193Ile | rs878927053 | Homoplasmy |
| MT-ND2 | m.5090T>C | Silent | p.IIe207= | Not provided | Homoplasmy | m.5120A>G | Silent | p.Leu217= | rs1603219790 | Homoplasmy |
| MT-ND2 | m.5165C>T | Silent | p.Arg232 | rs1556422959 | Homoplasmy | m.5333T>C | Silent | p.Leu288= | rs1603219906 | Homoplasmy |
| MT-ND2 | m.5300C>T | Silent | p.IIe277= | rs376259646 | Homoplasmy | m.5360C>T | Silent | p.IIe297= | rs879217723 | Homoplasmy |
| MT-ND2 | m.5390A>G | Silent | p. Met307= | rs41333444 | Homoplasmy | m.5393T>C | Silent | p.Ser308= | rs28357987 | Homoplasmy |
| MT-ND2 | m.5442T>C | Missense | p.Phe325Leu | rs3020601 | Homoplasmy | m.5480A>G | Silent | p.Leu337= | rs1603219977 | Homoplasmy |
| MT-ND2 | m.5492T>C | Silent | p.Pro341= | rs377109345 | Homoplasmy | | | | | |
| MT-ND2 | m.5493T>C | Missense | p.Phe342Leu | rs1603219983 | Homoplasmy | | | | | |
| MT-CO1 | m.5981T>C | Silent | p.Ala26= | rs1603220211 | Homoplasmy | m.6026G>A | Silent | p.Leu41= | rs879112886 | Homoplasmy |
| MT-CO1 | m.6045C>T | Silent | p.Leu48= | rs879061193 | Homoplasmy | m.6216T>C | Silent | p.Leu105= | rs367837524 | Homoplasmy |
| MT-CO1 | m.6179G>A | Silent | p.Met92= | rs374303341 | Homoplasmy | m.6261G>A | Missense | p.Ala120Thr | rs201262114 | Homoplasmy |
| MT-CO1 | m.6185T>C | Silent | p.Phe94= | rs1029272 | Homoplasmy | m.6446G>A | Silent | p.Thr181= | rs386420010 | Homoplasmy |
| MT-CO1 | m.6257G>A | Silent | p.Val118= | rs2856983 | Homoplasmy | m.6521C>T | Silent | p.IIe 206= | Not provided | Homoplasmy |
| MT-CO1 | m.6366G>A | Missense | p.Val155Ile | rs370673798 | Homoplasmy | m.6548C>T | Silent | p.Leu215= | rs28358870 | Homoplasmy |
| MT-CO1 | m.6497T>C | Silent | p.Ser198= | rs1556423143 | Homoplasmy | m.6680T>C | Silent | p.Thr259= | rs41352249 | Homoplasmy |
| MT-CO1 | m.6515T>C | Silent | p.Ala204= | rs878998677 | Homoplasmy | m.6989A>G | Silent | p.Ser362= | rs1978001 | Homoplasmy |
| MT-CO1 | m.6546C>T | Missense | p.Leu215Phe | rs1603220531 | Homoplasmy | m.7325A>G | Silent | p.Glu474= | rs1556423269 | Homoplasmy |
| MT-CO1 | m.6599A>G | Silent | p.Gln232 | rs879012660 | Homoplasmy | | | | | |
| MT-CO1 | m.6962G>A | Silent | p.Leu353= | rs1970771 | Homoplasmy | | | | | |
| MT-CO1 | m.7028C>T | Silent | p.Ala375= | rs2015062 | Homoplasmy | | | | | |
| MT-CO1 | m.7193T>C | Silent | p.Phe430= | rs1603220829 | Homoplasmy | | | | | |
| MT-CO2 | m.7861T>C | Silent | p.Asp92= | rs368623956 | Homoplasmy | m.7673A>G | Missense | p.Ile30Val | rs1569484167 | Homoplasmy |
| MT-CO2 | m.8014A>T | Silent | p.Val143= | rs879223416 | Homoplasmy | m.7711T>C | Silent | p.Leu42= | rs372012410 | Homoplasmy |
| MT-CO2 | m.8053A>G | Silent | p.Ser156= | rs56041322 | Homoplasmy | m.7867C>T | Silent | p.Ser94= | rs9783079 | Homoplasmy |
| MT-CO2 | m.8179A>G | Silent | p.Glu198= | rs1603221317 | Homoplasmy | m.8137C>T | Silent | p.Phe148= | rs879043235 | Homoplasmy |
| MT-CO2 | | | | | | m.8155G>A | Silent | p.Gly190= | rs374052533 | Homoplasmy |
| MT-ATP8 | m.8386C>T | Silent | p.Thr7= | rs1603221443 | Homoplasmy | | | | | |
| MT-ATP8 | m.8428C>T | Silent | p.Phe21= | rs1116905 | Homoplasmy | | | | | |
| MT-ATP8 | m.8460A>G | Missense | p.Asn32Ser | rs1116906 | Homoplasmy | | | | | |
| MT-ATP8 | m.8472T>C | Silent | p.Pro36= | rs386829037 | Homoplasmy | | | | | |
| MT-ATP8 | m.8554A>G | Missense | p.Ile10Val | rs1603221583 | Homoplasmy | | | | | |
| MT-ATP6 | m.8618T>C | Missense | p.Ile31Thr | rs28358885 | Homoplasmy | m.8655C>T | Silent | p.IIe43= | rs2853822 | Homoplasmy |
| MT-ATP6 | m.8705T>C | Missense | :p.Met60Thr | rs878959404 | Homoplasmy | m.8684C>T | Missense | p.Thr53Ile | rs201336180 | Homoplasmy |
| MT-ATP6 | m.8860A>G | Missense | p.Thr112Ala | rs2001031 | Homoplasmy | m.8978T>C | Missense | p.Ile151Thr | rs1603221954 | Homoplasmy |
| MT-ATP6 | m.8958C>T | Silent | p.IIe144= | rs1603221942 | Homoplasmy | m.9157G>A | Missense | p.Ala211Thr | rs1556423625 | Homoplasmy |
| MT-ATP6 | m.9007A>G | Missense | p.Thr161Ala | rs1603221973 | Homoplasmy | | | | | |
| MT-ATP6 | m.9042C>T | Silent | p.His172= | rs3020605 | Homoplasmy | | | | | |
| MT-ATP6 | m.9103T>C | Missense | p.Thr161Ala | rs1603222077 | Homoplasmy | | | | | |
| MT-CO3 | m.9336A>G | Missense | p.Met44Leu | rs28474779 | Homoplasmy | m.9302C>T | Silent | p.Ala32= | rs878986141 | Homoplasmy |
| MT-CO3 | m.9347A>G | Silent | p.Leu47= | rs2853824 | Homoplasmy | m.9656T>C | Silent | p.Ser150= | rs1556423706 | Homoplasmy |
| MT-CO3 | m.9494A>G | Silent | p.Gly96= | rs1556423680 | Homoplasmy | m.9899T>C | Silent | p.His231= | rs41345446 | Homoplasmy |
| MT-CO3 | m.9509T>C | Silent | p.Phe101= | rs375478739 | Homoplasmy | m.9956A>G | Silent | p.Leu250= | rs1603222594 | Homoplasmy |
| MT-CO3 | m.9545A>G | Silent | p.Gly113= | rs878853022 | Homoplasmy | | | | | |
| MT-CO3 | m.9575G>A | Silent | p.Pro123= | rs372078920 | Homoplasmy | | | | | |
| MT-CO3 | m.9755G>A | Silent | p.Glu183= | rs2856985 | Homoplasmy | | | | | |
| MT-CO3 | m.9776C>T | Silent | p.Asp190= | Not provided | Homoplasmy | | | | | |
| MT-CO3 | m.9818C>T | Silent | p.His204 | rs2854139 | Homoplasmy | | | | | |
| MT-CO3 | m.9948G>A | Missense | p.Val248Ile | rs1556423747 | Homoplasmy | | | | | |
| MT-ND3 | m.10101T>C | Silent | p.Leu15= | rs1603222669 | Homoplasmy | m.10084T>C | Missense | p.Ile9Thr | rs41487950 | Homoplasmy |
| MT-ND3 | m.10143G>A | Missense | p.Gly29Ser | rs202131419 | Homoplasmy | m.10142C>T | Silent | p.Asn28= | rs878969753 | Homoplasmy |
| MT-ND3 | m.10184C>T | Silent | p.Asp42= | Not provided | Homoplasmy | m.10238T>C | Silent | p.IIe60= | rs193302927 | Homoplasmy |
| MT-ND3 | m.10275T>C | Silent | p.Leu73= | rs373277477 | Homoplasmy | m.10289A>G | Silent | p.Trp77= | rs1556423796 | Homoplasmy |
| MT-ND4L | m.10499A>G | Silent | p.Leu10= | rs1057520074 | Homoplasmy | m.10550A>G | Silent | p.Met27= | rs28358280 | Homoplasmy |
| MT-ND4L | m.10556C>T | Silent | p.Ser29= | rs1603222890 | Homoplasmy | m.10586G>A | Silent | p.Ser39= | rs28358281 | Homoplasmy |
| MT-ND4L | m.10589G>A | Silent | p.Leu40= | rs2853487 | Homoplasmy | | | | | |
| MT-ND4L | m.10628C>T | Silent | p. Ser53= | Not provided | Homoplasmy | | | | | |
| MT-ND4L | m.10632T>C | Silent | p.Leu55= | rs878888873 | Homoplasmy | | | | | |
| MT-ND4L | m.10664C>T | Silent | p.Val65= | rs193302933 | Homoplasmy | | | | | |
| MT-ND4 | m.10819A>G | Silent | p.Lys20= | rs28358283 | Homoplasmy | m.10822C>T | Silent | p.His21= | rs879041592 | Homoplasmy |
| MT-ND4 | m.10876A>G | Silent | p.Leu39= | rs879036391 | Homoplasmy | m.11299T>C | Silent | p.Thr180= | rs28358285 | Homoplasmy |
| MT-ND4 | m.10915T>C | Missense | p.Cys52Trp | rs2857285 | Homoplasmy | m.11476C>T | Silent | p.Gly239= | rs386829131 | Homoplasmy |
| MT-ND4 | m.11002A>G | Silent | p.Gln81= | rs386829114 | Homoplasmy | | | | | |
| MT-ND4 | m.11016G>A | Missense | p.Ser86Thr | rs28594904 | Homoplasmy | | | | | |
| MT-ND4 | m.11050T>C | Silent | p.Ser97= | rs1603223077 | Homoplasmy | | | | | |
| MT-ND4 | m.11143C>T | Silent | p.Pro128= | rs1556423898 | Homoplasmy | | | | | |
| MT-ND4 | m.11172A>G | Missense | p.Asn138Ser | rs2853489 | Homoplasmy | | | | | |
| MT-ND4 | m.11176G>A | Silent | p.Gln139= | rs2853490 | Homoplasmy | | | | | |
| MT-ND4 | m.11287T>C | Silent | p.IIe176= | rs386829125 | Homoplasmy | | | | | |
| MT-ND4 | m.11377G>A | Silent | p.Lys206= | rs193302938 | Homoplasmy | | | | | |
| MT-ND4 | m.11440G>A | Silent | p.Gly227= | rs386829130 | Homoplasmy | | | | | |
| MT-ND4 | m.11590A>G | Silent | p.Leu277= | rs370318850 | Homoplasmy | | | | | |
| MT-ND4 | m.11641A>G | Silent | p.Met294= | rs2853494 | Homoplasmy | | | | | |
| MT-ND4 | m.11776T>C | Silent | p.Ser339= | rs28396842 | Homoplasmy | | | | | |
| MT-ND4 | m.11935T>C | Silent | p.Thr392= | rs1603223480 | Homoplasmy | | | | | |
| MT-ND4 | m.12007G>A | Silent | p.Trp416= | rs2853497 | Homoplasmy | | | | | |
| MT-ND4 | m.12061C>T | Silent | p.Asn434= | rs1556424043 | Homoplasmy | | | | | |
| MT-ND5 | m.12570A>G | Silent | p.Leu78= | rs1603223816 | Homoplasmy | m.12403C>T | Missense | p.Leu23Phe | rs879096684 | Homoplasmy |
| MT-ND5 | m.12720A>G | Silent | p.Met128= | rs2853500 | Homoplasmy | m.12501G>A | Silent | p.Met55= | rs28397767 | Homoplasmy |
| MT-ND5 | m.12771G>A | Silent | p.Glu145= | rs878865822 | Homoplasmy | m.12633C>A | Silent | p.Ser99= | rs3926883 | Homoplasmy |
| MT-ND5 | m.12876C>T | Silent | p.IIe180= | rs1603223952 | Homoplasmy | m.12879T>C | Silent | p.Gly181= | rs1556424182 | Homoplasmy |
| MT-ND5 | m.13020T>C | Silent | p.Gly228= | rs75577869 | Homoplasmy | m.12950A>C | Missense | p.Asn205Thr | rs201361958 | Homoplasmy |
| MT-ND5 | m.13116C>T | Silent | p.Leu260= | rs1603224049 | Homoplasmy | m.13104A>G | Silent | p.Gly256= | rs878871104 | Homoplasmy |
| MT-ND5 | m.13158A>G | Silent | p.Gln274= | rs1556424229 | Homoplasmy | m.13422A>G | Silent | p.Leu362= | rs386829180 | Homoplasmy |
| MT-ND5 | m.13276A>G | Missense | p.Met314Leu | rs2853502 | Homoplasmy | m.13500T>C | Silent | p.Gly388= | rs879066842 | Homoplasmy |
| MT-ND5 | m.13419A>G | Silent | p.Gly361= | rs1603224182 | Homoplasmy | m.13530C>T | Silent | p.Thr398= | rs2068736572 | Homoplasmy |
| MT-ND5 | m.13734T>C | Silent | p.Phe466= | rs41421644 | Homoplasmy | m.13780A>G | Missense | p.Ile482Val | rs41358152 | Homoplasmy |
| MT-ND5 | m.13759G>A | Missense | p.Ala475Thr | rs386420024 | Homoplasmy | m.13789T>C | Missense | p.Tyr485His | rs28359179 | Homoplasmy |
| MT-ND5 | m.13813G>A | Missense | p.Val493Ile | rs1556424332 | Homoplasmy | m.13880C>A | Missense | p.Ser515Tyr | rs28359181 | Homoplasmy |
| MT-ND5 | m.13886T>C | Missense | p.Leu517Pro | rs28359182 | Homoplasmy | m.14070A>G | Silent | p.Ser578= | rs879201732 | Homoplasmy |
| MT-ND5 | m.13967C>T | Missense | p.Thr544Met | rs386829197 | Homoplasmy | m.14110T>C | Missense | p.Phe592Leu | rs371451099 | Homoplasmy |
| MT-ND5 | m.14053A>G | Missense | p.Thr573Ala | rs200134839 | Homoplasmy | m.14139A>G | Silent | p.Leu601= | rs878918283 | Homoplasmy |
| MT-ND6 | m.14212T>C | Silent | p.Val154= | rs28357672 | Homoplasmy | m.14178T>C | Missense | p.Ile166Val | rs28357671 | Homoplasmy |
| MT-ND6 | m.14284C>T | Silent | p.Glu130= | rs28357673 | Homoplasmy | m.14203A>G | Silent | p.Gly157= | rs1569484633 | Homoplasmy |
| MT-ND6 | m.14308T>C | Silent | p.Gly122= | rs28357674 | Homoplasmy | m.14287T>C | Silent | p.Gly129= | rs1603224652 | Homoplasmy |
| MT-ND6 | m.14494T>C | Silent | p.Leu60= | rs879250748 | Homoplasmy | m.14364G>A | Silent | p Leu104= | rs879086798 | Homoplasmy |
| MT-ND6 | m.14562C>T | Missense | p.Val38Ile | rs1603224791 | Homoplasmy | m.14497A>G | Silent | p.Tyr59= | rs1556424454 | Homoplasmy |
| MT-ND6 | m.14587A>G | Silent | p.Gly29= | rs1556424469 | Homoplasmy | m.14560G>A | Silent | p.Val38= | rs28357676 | Homoplasmy |
| MT-ND6 | m.14634T>C | Missense | p.Met14Val | rs1603224816 | Homoplasmy | | | | | |
| MT-CYB | m.14755A>G | Silent | p.Pro3= | rs1603224856 | Homoplasmy | m.14769A>G | Missense | p.Asn8Ser | rs28357679 | Homoplasmy |
| MT-CYB | m.14839A>G | Silent | p.Trp31= | rs1603224921 | Homoplasmy | m.14774C>T | Silent | p.Leu10= | rs1556424490 | Homoplasmy |
| MT-CYB | m.14862C>T | Missense | p.Ala39Val | rs1603224933 | Homoplasmy | m.15077G>A | Missense | p.Glu111Lys | rs201943501 | Homoplasmy |
| MT-CYB | m.14872C>T | Silent | p.IIe42= | rs878879194 | Homoplasmy | m.15115T>C | Silent | p.Thr123= | rs879035822 | Homoplasmy |
| MT-CYB | m.15136C>T | Silent | p.Gly130= | rs2854124 | Homoplasmy | m.15217G>A | Silent | p.Gly157= | rs193302989 | Homoplasmy |
| MT-CYB | m.15172G>T | Silent | p.Gly142= | rs367572771 | Homoplasmy | m.15454T>C | Silent | p.Leu236= | rs879015290 | Homoplasmy |
| MT-CYB | m.15218A>G | Missense | p.Thr158Ala | rs2853506 | Homoplasmy | m.15865A>G | Silent | p.Glu373= | rs879154157 | Homoplasmy |
| MT-CYB | m.15257G>A | Missense | p.Asp171Asn | rs41518645 | Homoplasmy | | | | | |
| MT-CYB | m.15314G>A | Missense | p.Ala190Thr | rs527236176 | Homoplasmy | | | | | |
| MT-CYB | m.15403C>T | Silent | p.Thr219= | rs1603225258 | Homoplasmy | | | | | |
| MT-CYB | m.15431G>A | Missense | p.Ala229Thr | rs193302993 | Homoplasmy | | | | | |
| MT-CYB | m.15468C>T | Missense | p.Thr241Met | rs1603225301 | Homoplasmy | | | | | |
| MT-CYB | m.15490C>T | Silent | p.Asp248= | rs1603225311 | Homoplasmy | | | | | |
| MT-CYB | m.15530T>C | Silent | p.Leu262= | rs1556424600 | Homoplasmy | | | | | |
| MT-CYB | m.15646C>T | Silent | p.IIe300= | rs879113411 | Homoplasmy | | | | | |
| MT-CYB | m.15679A>G | Silent | p.Lys311= | rs1603225420 | Homoplasmy | | | | | |
| MT-CYB | m.15735C>T | Missense | p.Ala330Val | rs1603225446 | Homoplasmy | | | | | |
| MT-CYB | m.15799A>G | Silent | p.Gly351= | rs1603225506 | Homoplasmy | | | | | |
In addition, 126 (28.9%) mtDNA variants were common
for both patients with PsA and controls (Appendix I). The frequency of two common
variants differed significantly between the two groups. The
substitution variant m.152T>C in the D-loop region was
found in 26% of patients and 55% of controls (OR=0.3, 95%
CI=0.1-0.5, P=0.02), whereas the silent variant m.15301G>A in
the MT-CYB gene was found in 30% of patients and 10% of
controls (OR=3.8, 95% CI=1-8, P=0.04). The remaining variants
showed no significant differences in their prevalence among
patients or controls (P>0.05).
Variants with amino acid substitutions
in patients with PsA and their impact on protein function and
protein stability
Out of 187 (43%) mtDNA variants detected only in
patients with PsA, 33 missense variants in mtDNA-encoding genes of
complexes I, III, IV, and V resulted in amino acid
substitutions.
Bioinformatics analysis using CADD, Condel, and
PROVEAN was conducted to determine the potential impact of 33
missense variants that detected in PsA patients, on protein
function and structure. The analysis predicted 25 variants to be
deleterious by at least one bioinformatics tool (Table III). The highest impact on protein
function and structure (by all three in-silico algorithms)
was predicted for 2 variants, namely the m.11172A>G variant
(referred to as rs2853489 polymorphism) in the MT-ND4 gene
and the m.15257G>A variant (referred to as rs41518645
polymorphism) in the MT-CYB gene. The allelic frequencies of
these variants in the gnome AD database were 0.0007. and 0.01,
respectively.
| Table IIImtDNA variants with amino acid
substitutions in patients with psoriatic arthritis and their impact
on protein function and structure. |
Table III
mtDNA variants with amino acid
substitutions in patients with psoriatic arthritis and their impact
on protein function and structure.
| | Prediction |
|---|
| Gene | Variant | Variant type | Amino acid
change | rs ID | MAF | CADD/score | Condel/score | PROVEAN/score |
|---|
| MT-ND2 | m.4695T>C | Missense | p.Phe76Leu | rs1556422885 | 0.0003 | Neutral/5.35 | Neutral/0.07 | Neutral/1,2 |
| MT-ND2 | m.5442T>C | Missense | p.Phe325Leu | rs3020601 | 0.00478 | Neutral/0.03 | Neutral/0.28 | Neutral/0.7 |
| MT-ND2 | m.5493T>C | Missense | p.Phe342Leu | rs1603219983 | 0.007 | Neutral/12.34 | Neutral/0.31 | Neutral/0.52 |
| MT-CO1 | m.6366G>A | Missense | p.Val155Ile | rs370673798 | 0.0018 | Neutral/0.01 | Deleterious/1 | Neutral/-1.1 |
| MT-CO1 | m.6546C>T | Missense | p.Leu215Phe | rs1603220531 | 0.0002 | Neutral/6.11 |
Deleterious/0.84 | Neutral/-0.45 |
| MT-ATP8 | m.8460A>G | Missense | p.Asn32Ser | rs1116906 | 0.00094 | Neutral/12 | Neutral/0.27 | Neutral/-1.6 |
| MT-ATP8 | m.8554A>G | Missense | p.Ile10Val | rs1603221583 | 0.0001 | Neutral/7.68 |
Deleterious/0.54 | Neutral/0.18 |
| MT-ATP6 | m.8618T>C | Missense | p.Ile31Thr | rs28358885 | 0.009 | Neutral/5.55 |
Deleterious/0.5 | Neutral/2 |
| MT-ATP6 | m.8705T>C | Missense | p.Met60Thr | rs878959404 | 0.0043 | Neutral/0.5 |
Deleterious/0.75 | Neutral/0.32 |
| MT-ATP6 | m.8860A>G | Missense | p.Thr112Ala | rs2001031 | 0.99 | Neutral/6.13 |
Deleterious/0.81 |
Deleterious/-3.9 |
| MT-ATP6 | m.9007A>G | Missense | p.Thr161Ala | rs1603221973 | 0.0058 | Deleterious/23 | Neutral/0.06 |
Deleterious/-3.5 |
| MT-ATP6 | m.9103T>C | Missense | p.Thr161Ala | rs1603222077 | 0.00045 | Neutral/9.74 | Deleterious/1 |
Deleterious/-2.5 |
| MT-CO3 | m.9336A>G | Missense | p.Met44Leu | rs28474779 | 0.0004 | Neutral/0.01 |
Deleterious/0.68 | Neutral/-0.32 |
| MT-CO3 | m.9948G>A | Missense | p.Val248Ile | rs1556423747 | 0.0012 | Neutral/9.11 |
Deleterious/0.65 | Neutral/-0.84 |
| MT-ND3 | m.10143G>A | Missense | p.Gly29Ser | rs202131419 | 0.0015 | Neutral/5.51 |
Deleterious/0.96 | Neutral/1.9 |
| MT-ND4 | m.10915T>C | Missense | p.Cys52Trp | rs2857285 | 0.02 | Neutral/16.8 | Neutral/0.36 | Neutral/0.37 |
| MT-ND4 | m.11016G>A | Missense | p.Ser86Thr | rs28594904 | 0.004 | Neutral/0.11 |
Deleterious/0.68 | Neutral/0.21 |
| MT-ND4 | m.11172A>G | Missense | p.Asn138Ser | rs2853489 | 0.0007 | Deleterious/20 |
Deleterious/0.69 |
Deleterious/-3.5 |
| MT-ND5 | m.13276A>G | Missense | p.Met314Leu | rs2853502 | 0.00169 | Neutral/14 | Neutral/0.12 | Neutral/0.2 |
| MT-ND5 | m.13759G>A | Missense | p.Ala475Thr | rs386420024 | 0.0143 | Neutral/0.69 |
Deleterious/0.73 | Neutral /1.6 |
| MT-ND5 | m.13813G>A | Missense | p.Val493Ile | rs1556424332 | 0.0013 | Neutral/6.86 |
Deleterious/0.74 | Neutral/-0.37 |
| MT-ND5 | m.13886T>C | Missense | p.Leu517Pro | rs28359182 | 0.0018 | Neutral/9.6 |
Deleterious/0.6 | Neutral/0.23 |
| MT-ND5 | m.13967 C>T | Missense | p.Thr544Met | rs386829197 | 0.0011 | Neutral/1.23 |
Deleterious/0.61 | Neutral/0.85 |
| MT-ND5 | m.14053 A>G | Missense | p.Thr573Ala | rs200134839 | 0.002 | Neutral/0.17 |
Deleterious/0.76 | Neutral/1.8 |
| MT-ND6 | m.14562 C>T | Missense | p.Val38Ile | rs1603224791 | 0.0002 | Neutral/1.7 |
Deleterious/0.8 | Neutral/0.06 |
| MT-ND6 | m.14634 T>C | Missense | p.Met14Val | rs1603224816 | 0.0011 | Neutral/12 | Neutral/0.17 | Neutral/0.001 |
| MT-CYB | m.14862 C>T | Missense | p. Ala39Val | rs1603224933 | 0.0007 | Neutral/17.5 |
Deleterious/0.65 | Neutral/1.4 |
| MT-CYB | m.15218 A>G | Missense | p.Thr158Ala | rs2853506 | 0.03 | Neutral/13.3 | Neutral/0.29 | Neutral/-1.6 |
| MT-CYB | m.15257 G>A | Missense | p.Asp171Asn | rs41518645 | 0.01 |
Deleterious/23.5 |
Deleterious/0.67 |
Deleterious/-3.5 |
| MT-CYB | m.15314 G>A | Missense | p.Ala190Thr | rs527236176 | 0.002 | Neutral/16.4 |
Deleterious/0.61 | Neutral/-1.6 |
| MT-CYB | m.15431 G>A | Missense | p.Ala229Thr | rs193302993 | 0.002 |
Deleterious/24.4 |
Deleterious/0.61 | Neutral/-0.22 |
| MT-CYB | m.15468 C>T | Missense | p.Thr241Met | rs1603225301 | 0.0004 |
Deleterious/20.8 | Neutral/0.41 | Neutral/-0.81 |
| MT-CYB | m.15735 C>T | Missense | p.Ala330Val | rs1603225446 | 0.0001 | Deleterious/22 |
Deleterious/0.78 | Neutral/-1.7 |
In total, 5 other variants were predicted to be
deleterious by two in-silico algorithms tools: 3 variants in
the MT-ATP6 gene namely m.8860A>G (referred to as
rs2001031 polymorphism), m.9007A>G (referred to as rs1603221973
polymorphism) and m.9103T>C (referred as rs1603222077
polymorphism); and 2 variants in the MT-CYB gene namely
m.15431G>A (referred to as rs193302993 polymorphism) and
m.15735C>T (referred to as rs1603225446 polymorphism). With the
exception of the MT-ATP6 variant m.8860A>G with an
allelic frequency of 0.99, all other variants had allelic
frequencies of <0.01 in the gnome AD database (https://gnomad.broadinstitute.org/).
In addition, 18 other variants were predicted to be
deleterious by one of the in-silico algorithms tools. These
included 2 variants in the MT-CO1 gene, 1 variant in the
MT-ATP8 gene, 2 variants in the MT-ATP6 gene, 2
variants in the MT-CO3, 1 variant in the MT-ND3 gene,
1 variant in the MT-ND4 gene, 5 variants in the
MT-ND5 gene, 1 variant in the MT-ND6 gene and 3
variants in the MT-CYB gene. The majority of these variants
also had low allelic frequencies (<0.01) in the gnome AD
database.
In subsequent analysis, the deleterious mtDNA
variants were further evaluated for their effect on protein
stability using SDM. In SMD, a stability score is calculated using
environment-specific amino acid substitution frequencies within the
family of homologous proteins of known 3-D structures (30). All variants in mtDNA-encoded genes
of complexes I, III and IV were examined at the level of protein
stability. However, the effect of variants in the MT-ATP6
and MT-ATP8 genes of complex V could not be demonstrated as
the human 3-D structures were not available for complex V
proteins.
As shown in Table
IV, SDM predicted 19 destabilizing variants in mtDNA-encoded
genes of complexes I, III and IV. Of them, 13 variants were
predicted to decrease the stability of encoded proteins, including
1 variant in the MT-CO1 gene (m.6546C>T), 2 variants in
the MT-CO3 gene (m.9336A>G and m.9948G>A), 1 variant
in the MT-ND3 gene (m.10143G>A), 2 variants in the
MT-ND4 gene (m.11016G>A and m.11172A>G), 3 variants in
the MT-ND5 gene (m.13759G>A, m.13813G>A and
m.13886T>C), and 4 variants in the MT-CYB gene
(m.14862C>T, m.15314G>A, m.15431G>A, and m.15735C>T).
Furthermore, 6 variants were found to increase the stability of
encoded proteins: 1 variant in the MT-CO1 gene
(m.6366G>A), 2 variants in the MT-ND5 gene (m.13967C>T
and m.14053A>G), 1 variant in the MT-ND6 gene
(m.14562C>T) and 2 variants in the MT-CYB gene
(m.15257G>A and m.15468C>T).
| Table IVDeleterious mtDNA variants in
patients with psoriatic arthritis and their impact on protein
stability. |
Table IV
Deleterious mtDNA variants in
patients with psoriatic arthritis and their impact on protein
stability.
| | SDM |
|---|
| Gene | Variant | Variant type | Amino acid
change | rs ID | DDG | Stability
outcome |
|---|
| MT-CO1 | m.6366G>A | Missense | p.Val155Ile | rs370673798 | 0.68 | Increased |
| MT-CO1 | m.6546C>T | Missense | p.Leu215Phe | rs1603220531 | -0.09 | Decreased |
| MT-ATP8 | m.8554A>G | Missense | p.Ile10Val | rs1603221583 | NP | NP |
| MT-ATP6 | m.8618T>C | Missense | p.Ile31Thr | rs28358885 | NP | NP |
| MT-ATP6 | m.8705T>C | Missense | p.Met60Thr | rs878959404 | NP | NP |
| MT-ATP6 | m.8860A>G | Missense | p.Thr112Ala | rs2001031 | NP | NP |
| MT-ATP6 | m.9007A>G | Missense | p.Thr161Ala | rs1603221973 | NP | NP |
| MT-ATP6 | m.9103T>C | Missense | p.Thr161Ala | rs1603222077 | NP | NP |
| MT-CO3 | m.9336A>G | Missense | p.Met44Leu | rs28474779 | -0.44 | Decreased |
| MT-CO3 | m.9948G>A | Missense | p.Val248Ile | rs1556423747 | -0.94 | Decreased |
| MT-ND3 | m.10143G>A | Missense | p.Gly29Ser | rs202131419 | -0.41 | Decreased |
| MT-ND4 | m.11016G>A | Missense | p.Ser86Thr | rs28594904 | -0.69 | Decreased |
| MT-ND4 | m.11172A>G | Missense | p.Asn138Ser | rs2853489 | -0.64 | Decreased |
| MT-ND5 | m.13759G>A | Missense | p.Ala475Thr | rs386420024 | -0.66 | Decreased |
| MT-ND5 | m.13813G>A | Missense | p.Val493Ile | rs1556424332 | -0.38 | Decreased |
| MT-ND5 | m.13886T>C | Missense | p.Leu517Pro | rs28359182 | -1.5 | Decreased |
| MT-ND5 | m.13967C>T | Missense | p.Thr544Met | rs386829197 | 0.014 | Increased |
| MT-ND5 | m.14053A>G | Missense | p.Thr573Ala | rs200134839 | 1.53 | Increased |
| MT-ND6 | m.14562C>T | Missense | p.Val38Ile | rs1603224791 | 0.35 | Increased |
| MT-CYB | m.14862C>T | Missense | p.Ala39Val | rs1603224933 | -0.409 | Decreased |
| MT-CYB | m.15257G>A | Missense | p.Asp171Asn | rs41518645 | 0.39 | Increased |
| MT-CYB | m.15314G>A | Missense | p.Ala190Thr | rs527236176 | -1.53 | Decreased |
| MT-CYB | m.15431G>A | Missense | p.Ala229Thr | rs193302993 | -2.05 | Decreased |
| MT-CYB | m.15468C>T | Missense | p.Thr241Met | rs1603225301 | 1.19 | Increased |
| MT-CYB | m.15735C>T | Missense | p.Ala330Val | rs1603225446 | -0.84 | Decreased |
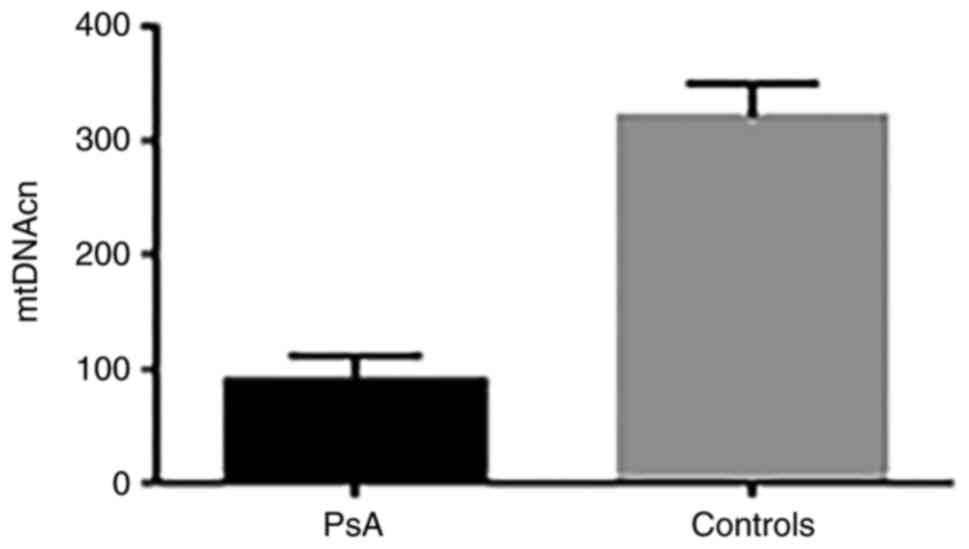
Relative leukocyte mtDNAcn
Using qPCR, the relative mtDNAcn was determined in
the leukocytes of patients with PsA (n=23) and healthy controls
(n=20). The results showed a 3.44-fold reduction in mtDNAcn in
patients with PsA compared with controls. The mean ± SD mtDNAcn was
93.3±10 in patients vs. 321±29 in controls (P=0.0001) (Fig. 2).
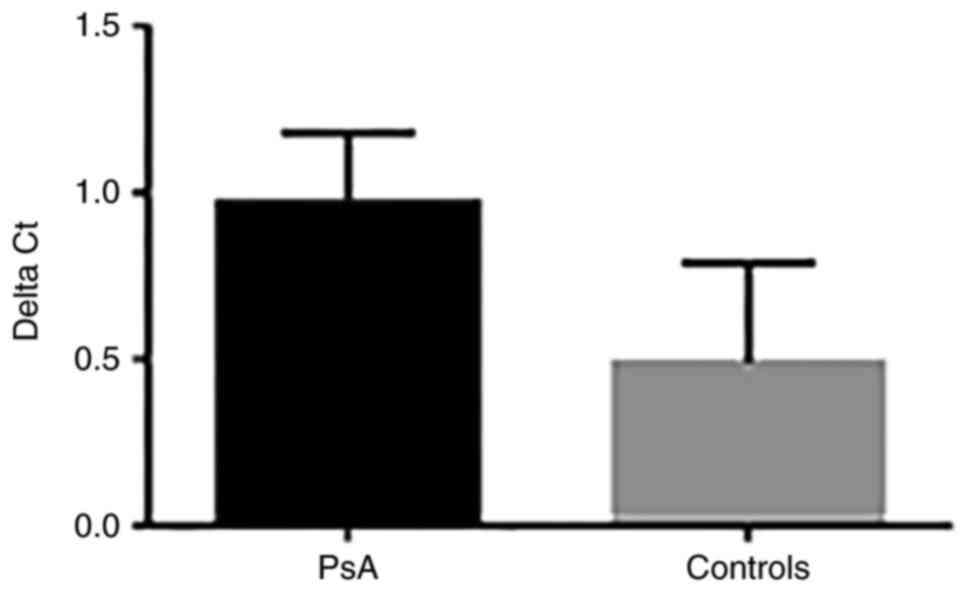
mtDNA oxidative damage
mtDNA oxidative damage was assessed using qPCR by
measuring the 8-OHdG content in patients with PsA and controls. The
ΔCq value of the difference between the Cq value of DNA samples
treated with FPG and the Cq value of DNA samples without FPG
treatment was calculated. The results showed a 2-fold increase in
the level of mtDNA oxidative damage in patients with PsA compared
with controls. The ΔCq value was 0.98±0.29 in patients vs. 0.49±0.3
in controls (P=0.03) (Fig. 3).
Discussion
PsA is a chronic inflammatory disease, which
presents in a significant number of individuals with psoriasis
(1-3).
PsA is widely regarded as a multi-factorial disease with underlying
autoimmune mechanisms including infiltration of plasma cells and
mononuclear cells that are observed in both psoriatic plaque and
PsA articular space (7).
Mitochondrial dysfunction plays an important role in the
pathogenesis of PsA by modulating innate immunity via
redox-sensitive inflammatory pathways or directly through
activation of the inflammatory response (9,10). An
imbalance in oxidant-antioxidant mitochondrial system results in
oxidative stress, which can induce mtDNA variations and copy number
changes leading to mitochondrial functional impairment (17,18).
Currently, there is limited knowledge on whether abnormalities in
mtDNA a possible factor could be involved in PsA.
The present study sequenced the entire mitochondrial
genome using NGS, a high-throughput and sensitive method with the
ability to detect any variants (22), investigated changes in mtDNAcn and
evaluated mtDNA oxidative damage in patients with PsA and healthy
controls.
Analysis of the entire mitochondrial genome revealed
a total of 435 variants, distributed across all regions of the
mtDNA. These included 187 (43%) variants exclusively found in
patients with PsA, and 122 (28%) only present in control
individuals. A higher number of variants were observed in the
D-loop region in both patients and controls, whereas a higher
number of variants in mtDNA-coding genes was found in patients
compared with controls, particularly in the MT-ND4, MT-ND5, and
MT-CYB genes. In addition, common mtDNA variants (126, 28.9%) were
identified among patients and controls. The frequency of two
specific variants differed significantly (P<0.05) between the
two groups and may be linked with the susceptibility to PsA.
Namely, the D-loop m.152T>C variant occurred in 26% of
patients and 55% of controls (OR=0.3, 95% CI=0.1-0.5, P=0.02) and
may confer a protective role against PsA. The D-loop (non-coding or
control region) contains essential regulatory sequences for
replication and transcription (34). Although the majority of harmful
variants are removed by natural selection, some of these variants
are introduced in certain populations and may influence the risk of
developing certain disorders (35),
whereas other variants such as m.152T>C may be
selectively beneficial on some genetic backgrounds. Additionally,
the silent m.15301G>A variant in the MT-CYB gene occurred in 30%
of patients and 10% of controls (OR=3.8, 95% CI=1-8, P=0.04) and
may be associated with the risk of developing PsA. Synonymous
variants in protein-coding genes are generally considered to be
silent with no effect on protein function. However, previous
studies have shown that silent variants can significantly alter
gene expression by affecting the stability and folding of mRNA
(36,37), and can also influence the rate of
translation and posttranslational modification of proteins
(38).
mtDNA variants that cause changes in amino acid
sequences of protein-coding genes have been implicated in the
pathogenicity of numerous diseases such as rheumatoid arthritis
(RA) (39). In the present study,
analysis of mtDNA in patients with PsA only revealed 33 missense
variants in mtDNA-encoded genes of the ETC. In total, 25 of these
variants were predicted to have deleterious effects on encoded
proteins by 1-3 bioinformatics tools (CADD, Condel and
PROVEAN).
Specifically, 2 missense transition variants in
MT-ND4 gene and MT-CYB gene were predicted to have
the highest impact on protein function and structure by all three
in silico algorithms tools. Moreover, 5 mtDNA variants were
predicted to be deleterious by two in silico algorithms
tools, including 3 variants in the MT-ATP6 gene and 2
variants in the MT-CYB gene. A total of 18 other variants in
all mtDNA-encoded complexes of the ETC were predicted to be
deleterious by one of the in silico algorithms tools. The
majority of these variants are considered rare with low allelic
frequencies <0.01 in the gnome AD database.
Protein function and stability are closely related
to protein structure. Variants that cause amino acid alterations
can markedly change protein structure (30,40).
Particularly, the unique amino acid sequence of a protein is
reflected in its folded structure, which is important to perform
proper biological function. Changes in the hydration status of a
protein also affect protein folding (41). Hydrophobic interactions are
important for protein folding, and changes in hydrophobicity can
lead to a collapse of the protein chain in an aqueous environment
(42). The polarity of amino acids
also promotes appropriate folding by interacting with the water
solvent, thus affecting protein stability (41). All the identified deleterious
missense variants in the current study produce amino acids that are
different from the usual amino acids at that position and alter the
function of proteins. For instance, the MT-ND4 m.11172A>G
variant causes changes in amino acid from Asparagine to Serine
(p.Asn138Ser), while the MT-CYB m.15257G>A variant causes
changes in amino acid from Asparagine to Aspartic acid
(p.Asp171Asn). Asparagine is a hydrophilic uncharged polar amino
acid, whereas serine is a neutral uncharged polar amino acid, and
aspartic acid is a hydrophilic negatively charged polar amino
acid.
Previous studies have demonstrated that protein
flexibility is a key factor for its catalytic activity (43), and increased stability of
thermophilic proteins has been shown to be associated with loss of
protein flexibility and reduced enzymatic activity at low
temperatures (44,45). Furthermore, high stability of
proteins leads to increased proteolytic resistance, which make them
difficult to regulate, particularly during cell signalling
(46). It has been also shown that
variants causing increased protein stability can lead to protein
malfunction in human diseases, such as the stabilizing homozygous
variant S37A in patients with parathyroid adenomas (47) and the Parkinson disease-associated
A30P stabilizing variant in human neuroblastoma cells (48).
Analysis of the impact of deleterious variants on
protein stability in present study revealed 19 destabilizing
variants in mtDNA-encoded genes of complexes I, III and IV. Of
them, 13 variants in different mtDNA-encoded complexes of the ETC
were predicted to decrease protein stability. Moreover, 6 variants
were found to increase the stability of encoded proteins. These
variants may affect the stabilizing interaction within folded
proteins, leading to protein instability and malfunction.
The mitochondrial OXPHOS system comprises four
multi-enzymatic respiratory complexes (namely I-IV) and ATP
synthase and is embedded in the inner mitochondrial membrane. A
total of 4 of these complexes (I, III, IV and V) are encoded by
both nDNA and mtDNA genes. Complex I (NADH: ubiquinone
oxidoreductase) is one of the main contributors to ROS production
within the mitochondrial matrix, which is a major cause of cellular
oxidative stress and is associated with neuromuscular diseases and
aging (49-52).
mtDNA-encoded genes of complex I are hotspots for pathological
variants (19). Such variants
affect complex I assembly and activity leading to complex I
deficiency with increased ROS production (51-55).
Particularly, variants in mitochondrial complex I genes have been
previously reported to be associated with severe erosive arthritis
and to be implicated in the pathogenesis of RA (39). Complex III (bc1 complex; ubiquinol
cytochrome c reductase) has been also identified as a main
producer of superoxide within the mitochondrial respiratory chain
(56,57). Previous studies have revealed that
deleterious variants in the MT-CYB gene caused isolated complex III
deficiency, leading to a variety of human diseases such as
cardiomyopathy, encephalomyopathy, and Leber hereditary optic
neuropathy (58-61).
Complex IV or cytochrome c oxidase (COX) is one of the major
regulation sites for OXPHOS and deficiency in the activity of COX
has been linked to a variety of diseases (62). ATP6 and ATP8 are mtDNA-encoded
subunits of the ATP synthase of complex V, which utilizes the
energy provided by the proton electrochemical gradient across the
inner membrane during OXPHOS and synthesizes ATP from ADP (63). Variants in MT-ATP8 gene can disturb
the stability and function of complex V, affecting ATP production
(64). In a previous study by Du
et al (39), a higher rate
of mtDNA variants in complex V was found in patients with RA
compared with controls, suggesting that these variants may be
associated with susceptibility to RA. Variants in complex V were
also linked to ROS production and apoptosis pathways, and
associated with RA progression (39).
Integrity of mtDNA which encodes essential proteins
of the ETC subunits is mandatory for normal mitochondrial function
(13). Improper function of ETC can
enhance ROS production, which is implicated in psoriatic
inflammation and PsA (9,65). Improper function of ETC can enhance
ROS production, which is implicated in psoriatic inflammation and
PsA (9,65). The deleterious mtDNA variants in
patients with PsA identified in the present study were not detected
in control individuals; were located in functionally/structurally
important sites of mtDNA-encoded subunits of the ETC; and resulted
in protein instability. Thus, these variants may be disease-related
and could play a role in the pathogenicity of PsA.
There are several copies per cell of mtDNA,
resulting in both homoplasmy (identical mtDNA) and heteroplasmy
(mixture of mutated and wild-type mtDNA). In the present study, a
higher rate of homoplasmic variants was detected compared with
heteroplasmic variants. While most pathogenic mtDNA variants are
heteroplasmic, the clinical expression of diseases is determined by
the level of heteroplasmy. In this context, mitochondrial
dysfunction becomes clinically apparent when the percentage of
mutant mtDNA exceeds a certain threshold level. Instead, some mtDNA
diseases such as Leber hereditary optic neuropathy are caused by
homoplasmic variants (66).
Pathogenic homoplasmic variants have been also reported in diseases
such as Leigh syndrome (67) and
multiple sclerosis (68), and may
increase the risk of type 2 diabetes (69) and neurodegenerative diseases
(70). Importantly, the phenotypic
expression of homoplasmic variants can be tissue-specific,
suggesting that incomplete penetrance and unidentified nuclear
genetic and/or environmental factors are likely to contribute to
the disease phenotype (66).
The present results also revealed a significant
reduction in mtDNAcn in leukocytes of patients with PsA compared
with healthy controls. mtDNAcn is an important indicator of
mitochondrial biogenesis and function (15). Consequently, changes in mtDNA
content could contribute to various pathological conditions
(20). Low mtDNAcn was previously
reported in numerous diseases such as OA (71). In our previous study, decreased
mtDNAcn in patients with psoriasis was found compared with controls
(24). Decreased mtDNAcn is
associated with oxidative stress-induced mtDNA damage and poor
oxidative capacity which can lead to a reduction in mitochondrial
function and subsequent disruption of cellular functions which
could affect several tissues (24,72).
The present study also found a higher level of mtDNA
oxidative damage in patients with PsA compared with controls. It is
well established that mtDNA is more susceptible to oxidative damage
and has a higher mutational rate than nDNA (16). Oxidative damage to mtDNA can impair
mitochondrial bioenergetics and causes defective mitochondrial ATP
generation, which leads to further mitochondrial dysfunction and
increased ROS production. Therefore, decreased mtDNAcn in patients
with PsA may be a consequence of mtDNA oxidative damage.
In conclusion, the present study identified a number
of unique variants in patients with PsA only or healthy controls
only, as well as common variants in patients and controls, two of
which may be associated with the susceptibility to PsA, and also
identified various missense variants that were present only in
patients with PsA and were predicted to be deleterious with
important effect on the function and stability of encoded proteins.
In addition, lower mtDNAcn and higher levels of mtDNA oxidative
damage were found in patients with PsA compared with controls.
Taken together, the present findings suggested that impaired
mitochondrial function due to deleterious mtDNA variants, low
mtDNAcn, and oxidative damage may be contributory factors in the
pathogenesis of PsA. To the best of our knowledge, the present
study is the first comprehensive analysis of mtDNA in PsA. However,
the present study is limited by the small number of subjects
enrolled; thus, additional large-scale studies are warranted to
further elucidate the role of mtDNA defects in PsA. Moreover, the
possible effect of nuclear genes and environmental factors on
mitochondrial genetics in PsA should be considered in future
studies. In addition, to provide an improved estimate of mtDNA
damage, several regions within the mtDNA genome including the
D-loop region should be targeted in future studies.
Supplementary Material
Common mtDNA variants among PsA
patients and controls.
Acknowledgements
The authors would like to express their gratitude to
Dr Betsy Sheena at research sector of Faculty of Sciences, Kuwait
University (State of Kuwait) for the next generation sequencing
data analysis and also the authors would like to acknowledge the
next generation sequencing facility under the project GS01/02.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets generated and/or analysed during the
current study are available in the Sequence Read Archive (SRA)
repository with reference PRJNA 928743 (accession no. https://www.ncbi.nlm.nih.gov/sra/PRJNA928743).
Authors' contributions
MSA was the project administrator, was responsible
for the conceptualization, methodology, investigation, acquisition
of resources/funding and the data curation of the present study.
MSA, GAK and MA implemented formal analysis. MSA and GAK confirm
the authenticity of all raw data. MSA and GAK wrote the original
draft, reviewed and edited the manuscript. All authors have read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was conducted according to the
guidelines of the Declaration of Helsinki and approved (approval
no. 2018/496) by the Health Science Center Ethics Committee at
Kuwait University and Health and Medical Research Committee in the
Ministry of Health (City of Kuwait, State of Kuwait). Informed
consent was obtained from all subjects involved in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Alinaghi F, Calov M, Kristensen LE,
Gladman DD, Coates LC, Jullien D, Gottlieb AB, Gisondi P, Wu JJ,
Thyssen JP and Egeberg A: Prevalence of psoriatic arthritis in
patients with psoriasis: A systematic review and meta-analysis of
observational and clinical studies. J Am Acad Dermatol. 80:251–265
e219. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ocampo DV and Gladman D: Psoriatic
arthritis. F1000Res. 8(F1000)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Karmacharya P, Chakradhar R and Ogdie A:
The epidemiology of psoriatic arthritis: A literature review. Best
Pract Res Clin Rheumatol. 35(101692)2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sukhov A, Adamopoulos IE and Maverakis E:
Interactions of the immune system with skin and bone tissue in
psoriatic arthritis: A comprehensive review. Clin Rev Allergy
Immunol. 51:87–99. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Saalfeld W, Mixon AM, Zelie J and Lydon
EJ: Differentiating psoriatic arthritis from osteoarthritis and
rheumatoid arthritis: A narrative review and guide for advanced
practice providers. Rheumatol Ther. 8:1493–1517. 2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gladman DD, Mease PJ, Healy P, Helliwell
PS, Fitzgerald O, Cauli A, Lubrano E, Krueger GG, van der Heijde D,
Veale DJ, et al: Outcome measures in psoriatic arthritis. J
Rheumatol. 34:1159–1166. 2007.PubMed/NCBI
|
|
7
|
Carvalho AL and Hedrich CM: The molecular
pathophysiology of psoriatic arthritis-the complex interplay
between genetic predisposition, epigenetics factors, and the
microbiome. Front Mol Biosci. 8(662047)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Eder L, Haddad A, Rosen CF, Lee KA,
Chandran V, Cook R and Gladman DD: The incidence and risk factors
for psoriatic arthritis in patients with psoriasis: A prospective
cohort study. Arthritis Rheumatol. 68:915–923. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Mizuguchi S, Gotoh K, Nakashima Y,
Setoyama D, Takata Y, Ohga S and Kang D: Mitochondrial reactive
oxygen species are essential for the development of psoriatic
inflammation. Front Immunol. 12(714897)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Therianou A, Vasiadi M, Delivanis DA,
Petrakopoulou T, Katsarou-Katsari A, Antoniou C, Stratigos A,
Tsilioni I, Katsambas A, Rigopoulos D and Theoharides TC:
Mitochondrial dysfunction in affected skin and increased
mitochondrial DNA in serum from patients with psoriasis. Exp
Dermatol. 28:72–75. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lin X and Huang T: Oxidative stress in
psoriasis and potential therapeutic use of antioxidants. Free Radic
Res. 50:585–595. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Osellame LD, Blacker TS and Duchen MR:
Cellular and molecular mechanisms of mitochondrial function. Best
Pract Res Clin Endocrinol Metab. 26:711–723. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Garcia I, Jones E, Ramos M,
Innis-Whitehouse W and Gilkerson R: The little big genome: The
organization of mitochondrial DNA. Front Biosci (Landmark Ed).
22:710–721. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Burr SP, Pezet M and Chinnery PF:
Mitochondrial DNA heteroplasmy and purifying selection in the
mammalian female germ line. Dev Growth Differ. 60:21–32.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Montier LL, Deng JJ and Bai Y: Number
matters: Control of mammalian mitochondrial DNA copy number. J
Genet Genomics. 36:125–131. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bohr VA, Stevnsner T and de Souza-Pinto
NC: Mitochondrial DNA repair of oxidative damage in mammalian
cells. Gene. 286:127–134. 2002.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pagano G, Talamanca AA, Castello G,
Cordero MD, d'Ischia M, Gadaleta MN, Pallardó FV, Petrović S, Tiano
L and Zatterale A: Oxidative stress and mitochondrial dysfunction
across broad-ranging pathologies: Toward mitochondria-targeted
clinical strategies. Oxid Med Cell Longev.
2014(541230)2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Liu CS, Tsai CS, Kuo CL, Chen HW, Lii CK,
Ma YS and Wei YH: Oxidative stress-related alteration of the copy
number of mitochondrial DNA in human leukocytes. Free Radic Res.
37:1307–1317. 2003.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Taylor RW and Turnbull DM: Mitochondrial
DNA mutations in human disease. Nat Rev Genet. 6:389–402.
2005.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Filograna R, Mennuni M, Alsina D and
Larsson NG: Mitochondrial DNA copy number in human disease: The
more the better? FEBS Lett. 595:976–1002. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Harty LC, Biniecka M, O'Sullivan J, Fox E,
Mulhall K, Veale DJ and Fearon U: Mitochondrial mutagenesis
correlates with the local inflammatory environment in arthritis.
Ann Rheum Dis. 71:582–588. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang W, Cui H and Wong LJ: Comprehensive
one-step molecular analyses of mitochondrial genome by massively
parallel sequencing. Clin Chem. 58:1322–1331. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Taylor W, Gladman D, Helliwell P,
Marchesoni A, Mease P and Mielants H: CASPAR Study Group.
Classification criteria for psoriatic arthritis: Development of new
criteria from a large international study. Arthritis Rheum.
54:2665–2673. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Alwehaidah MS, AlFadhli S and Al-Kafaji G:
Leukocyte mitochondrial DNA copy number is a potential non-invasive
biomarker for psoriasis. PLoS One. 17(e0270714)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Alwehaidah MS, Al-Kafaji G, Bakhiet M and
Alfadhli S: Next-generation sequencing of the whole mitochondrial
genome identifies novel and common variants in patients with
psoriasis, type 2 diabetes mellitus and psoriasis with comorbid
type 2 diabetes mellitus. Biomed Rep. 14(41)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bandelt HJ, Kloss-Brandstatter A, Richards
MB, Yao YG and Logan I: The case for the continuing use of the
revised Cambridge reference sequence (rCRS) and the standardization
of notation in human mitochondrial DNA studies. J Hum Genet.
59:66–77. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle
E, Wang K and Liu X: Comparison and integration of deleteriousness
prediction methods for nonsynonymous SNVs in whole exome sequencing
studies. Hum Mol Genet. 24:2125–2137. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gonzalez-Perez A and Lopez-Bigas N:
Improving the assessment of the outcome of nonsynonymous SNVs with
a consensus deleteriousness score, Condel. Am J Hum Genet.
88:440–449. 2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Choi Y and Chan AP: PROVEAN web server: A
tool to predict the functional effect of amino acid substitutions
and indels. Bioinformatics. 31:2745–2747. 2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Pandurangan AP, Ochoa-Montano B, Ascher DB
and Blundell TL: SDM: A server for predicting effects of mutations
on protein stability. Nucleic Acids Res. 45:W229–W235.
2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lin PH, Lee SH, Su CP and Wei YH:
Oxidative damage to mitochondrial DNA in atrial muscle of patients
with atrial fibrillation. Free Radic Biol Med. 35:1310–1318.
2003.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Movassaghi S, Jafari S, Falahati K, Ataei
M, Sanati MH and Jadali Z: Quantification of mitochondrial DNA
damage and copy number in circulating blood of patients with
systemic sclerosis by a qPCR-based assay. An Bras Dermatol.
95:314–319. 2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Suissa S, Wang Z, Poole J, Wittkopp S,
Feder J, Shutt TE, Wallace DC, Shadel GS and Mishmar D: Ancient
mtDNA genetic variants modulate mtDNA transcription and
replication. PLoS Genet. 5(e1000474)2009.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Fan W, Waymire KG, Narula N, Li P, Rocher
C, Coskun PE, Vannan MA, Narula J, Macgregor GR and Wallace DCA:
mouse model of mitochondrial disease reveals germline selection
against severe mtDNA mutations. Science. 319:958–962.
2008.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Komar AA: Genetics. SNPs, silent but not
invisible. Science. 315:466–467. 2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Shabalina SA, Spiridonov NA and Kashina A:
Sounds of silence: Synonymous nucleotides as a key to biological
regulation and complexity. Nucleic Acids Res. 41:2073–2094.
2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE,
Calcagno AM, Ambudkar SV and Gottesman MM: A ‘silent’ polymorphism
in the MDR1 gene changes substrate specificity. Science.
315:525–528. 2007.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Du J, Yu S, Wang D, Chen S, Chen S, Zheng
Y, Wang N, Chen S, Li J and Shen B: Germline and somatic mtDNA
mutation spectrum of rheumatoid arthritis patients in the Taizhou
area, China. Rheumatology (Oxford). 59:2982–2991. 2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Schaefer C and Rost B: Predict impact of
single amino acid change upon protein structure. BMC Genomics. 13
(Suppl 4)(S4)2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Kauzmann W: Some factors in the
interpretation of protein denaturation. Adv Protein Chem. 14:1–63.
1959.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Aznauryan M, Nettels D, Holla A, Hofmann H
and Schuler B: Single-molecule spectroscopy of cold denaturation
and the temperature-induced collapse of unfolded proteins. J Am
Chem Soc. 135:14040–14043. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Karamitros CS, Murray K, Winemiller B,
Lamb C, Stone EM, D'Arcy S, Johnson KA and Georgiou G: Leveraging
intrinsic flexibility to engineer enhanced enzyme catalytic
activity. Proc Natl Acad Sci USA. 119(e2118979119)2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Counago R, Wilson CJ, Pena MI,
Wittung-Stafshede P and Shamoo Y: An adaptive mutation in adenylate
kinase that increases organismal fitness is linked to
stability-activity trade-offs. Protein Eng Des Sel. 21:19–27.
2008.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Jaenicke R: Protein stability and
molecular adaptation to extreme conditions. Eur J Biochem.
202:715–728. 1991.PubMed/NCBI View Article : Google Scholar
|
|
46
|
DePristo MA, Weinreich DM and Hartl DL:
Missense meanderings in sequence space: A biophysical view of
protein evolution. Nat Rev Genet. 6:678–687. 2005.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Bjorklund P, Lindberg D, Akerstrom G and
Westin G: Stabilizing mutation of CTNNB1/beta-catenin and protein
accumulation analyzed in a large series of parathyroid tumors of
Swedish patients. Mol Cancer. 7(53)2008.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Song W, Patel A, Qureshi HY, Han D,
Schipper HM and Paudel HK: The Parkinson disease-associated A30P
mutation stabilizes alpha-synuclein against proteasomal degradation
triggered by heme oxygenase-1 over-expression in human
neuroblastoma cells. J Neurochem. 110:719–733. 2009.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Hirst J, King MS and Pryde KR: The
production of reactive oxygen species by complex I. Biochem Soc
Trans. 36:976–980. 2008.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Malfatti E, Bugiani M, Invernizzi F, de
Souza CF, Farina L, Carrara F, Lamantea E, Antozzi C, Confalonieri
P, Sanseverino MT, et al: Novel mutations of ND genes in complex I
deficiency associated with mitochondrial encephalopathy. Brain.
130:1894–1904. 2007.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Rodenburg RJ: Mitochondrial complex
I-linked disease. Biochim Biophys Acta. 1857:938–945.
2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Hofhaus G and Attardi G: Lack of assembly
of mitochondrial DNA-encoded subunits of respiratory NADH
dehydrogenase and loss of enzyme activity in a human cell mutant
lacking the mitochondrial ND4 gene product. EMBO J. 12:3043–3048.
1993.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Bai Y and Attardi G: The mtDNA-encoded ND6
subunit of mitochondrial NADH dehydrogenase is essential for the
assembly of the membrane arm and the respiratory function of the
enzyme. EMBO J. 17:4848–4858. 1998.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Bai Y, Hu P, Park JS, Deng JH, Song X,
Chomyn A, Yagi T and Attardi G: Genetic and functional analysis of
mitochondrial DNA-encoded complex I genes. Ann N Y Acad Sci.
1011:272–283. 2004.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Muller FL, Liu Y and Van Remmen H: Complex
III releases superoxide to both sides of the inner mitochondrial
membrane. J Biol Chem. 279:49064–49073. 2004.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Bleier L and Drose S: Superoxide
generation by complex III: From mechanistic rationales to
functional consequences. Biochim Biophys Acta. 1827:1320–1331.
2013.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Keightley JA, Anitori R, Burton MD, Quan
F, Buist NR and Kennaway NG: Mitochondrial encephalomyopathy and
complex III deficiency associated with a stop-codon mutation in the
cytochrome b gene. Am J Hum Genet. 67:1400–1410. 2000.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Carossa V, Ghelli A, Tropeano CV,
Valentino ML, Iommarini L, Maresca A, Caporali L, La Morgia C,
Liguori R, Barboni P, et al: A novel in-frame 18-bp microdeletion
in MT-CYB causes a multisystem disorder with prominent exercise
intolerance. Hum Mutat. 35:954–958. 2014.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Emmanuele V, Sotiriou E, Rios PG, Ganesh
J, Ichord R, Foley AR, Akman HO and Dimauro S: A novel mutation in
the mitochondrial DNA cytochrome b gene (MTCYB) in a patient with
mitochondrial encephalomyopathy, lactic acidosis, and strokelike
episodes syndrome. J Child Neurol. 28:236–242. 2013.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Amaral-Fernandes MS, Marcondes AM, do Amor
Divino Miranda PM, Maciel-Guerra AT and Sartorato EL: Mutations for
Leber hereditary optic neuropathy in patients with alcohol and
tobacco optic neuropathy. Mol Vis. 17:3175–3179. 2011.PubMed/NCBI
|
|
62
|
Li Y, Park JS, Deng JH and Bai Y:
Cytochrome c oxidase subunit IV is essential for assembly and
respiratory function of the enzyme complex. J Bioenerg Biomembr.
38:283–291. 2006.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Jonckheere AI, Smeitink JA and Rodenburg
RJ: Mitochondrial ATP synthase: Architecture, function and
pathology. J Inherit Metab Dis. 35:211–225. 2012.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Dautant A, Meier T, Hahn A,
Tribouillard-Tanvier D, di Rago JP and Kucharczyk R: ATP synthase
diseases of mitochondrial genetic origin. Front Physiol.
9(329)2018.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Coto-Segura P, Santos-Juanes J, Gomez J,
Alvarez V, Díaz M, Alonso B, Corao AI and Coto E: Common European
mitochondrial haplogroups in the risk for psoriasis and psoriatic
arthritis. Genet Test Mol Biomarkers. 16:621–623. 2012.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Gomes TM, Ng YS, Pickett SJ, Turnbull DM
and Vincent AE: Mitochondrial DNA disorders: From pathogenic
variants to preventing transmission. Hum Mol Genet. 30:R245–R253.
2021.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Negishi Y, Hattori A, Takeshita E, Sakai
C, Ando N, Ito T, Goto Y and Saitoh S: Homoplasmy of a
mitochondrial 3697G>A mutation causes Leigh syndrome. J Hum
Genet. 59:405–407. 2014.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Al-Kafaji G, Alharbi MA, Alkandari H,
Salem AH and Bakhiet M: Analysis of the entire mitochondrial genome
reveals Leber's hereditary optic neuropathy mitochondrial DNA
mutations in an Arab cohort with multiple sclerosis. Sci Rep.
12(11099)2022.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Crispim D, Estivalet AAF, Roisenberg I,
Gross JL and Canani LH: Prevalence of 15 mitochondrial DNA
mutations among type 2 diabetic patients with or without clinical
characteristics of maternally inherited diabetes and deafness. Arq
Bras Endocrinol Metabol. 52:1228–1235. 2008.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Keogh MJ and Chinnery PF: Mitochondrial
DNA mutations in neurodegeneration. Biochim Biophys Acta.
1847:1401–1411. 2015.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Zhan D, Tanavalee A, Tantavisut S,
Ngarmukos S, Edwards SW and Honsawek S: Relationships between blood
leukocyte mitochondrial DNA copy number and inflammatory cytokines
in knee osteoarthritis. J Zhejiang Univ Sci B. 21:42–52.
2020.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Al-Kafaji G, Bakheit HF, Alharbi MA,
Farahat AA, Jailani M, Ebrahin BH and Bakhiet M: Mitochondrial DNA
copy number in peripheral blood as a potential non-invasive
biomarker for multiple sclerosis. Neuromolecular Med. 22:304–313.
2020.PubMed/NCBI View Article : Google Scholar
|