Introduction
Genomic variability is a key aspect of human
genetics contributing to various types of diseases and cancer. One
gene implicated in diseases and cancer predisposition is partner
and localizer of BRCA2 (PALB2). PALB2 gene, also
known as FANCN, is located on chromosome 16 and has three
structural domains: N-terminal, central and C-terminal. It serves a
key role in tumour suppression through homologous recombination
(HR) repair of DNA double-strand breaks. This is achieved by
mediating the recruitment of polymerase (Pol η), BRCA2 and RAD51
recombinase to the site of DNA damage forming a D-loop
intermediate, in addition to interaction with several other tumour
suppressors, including BRCA1and KEAP1(1). These tumour suppressors exert a
synergistic effect on DNA repair through PALB2. For example,
reactive oxygen species (ROS), natural byproducts of cellular
metabolism, are highly reactive substances containing oxygen
radicals; it accumilation leads to a detrimental effects, including
DNA damage. KEAP1 causes downregulation of NRF2, a master
transcriptional factor for antioxidant genes; PALB2 competes with
NRF2 to bind KEAP1, thereby disturbing KEAP1-NRF2 complexes.
PALB2, therefore, coordinates DNA repair via ROS
detoxification (1).
PALB2 interacts with BRCA1 and BRCA2 to activate and
maintain DNA damage G2/M checkpoints and protect active genes from
genotoxic stress (2,3). To date, 604 different PALB2
variants have been identified; only 140 variants are considered
pathogenic, while ~400 have unclear significance (4). The presence of a heterozygous germline
mutation in PALB2 has been linked to the emergence of
breast, pancreatic and ovarian cancer (5). A study in Saudi Arabia linked
association of PALB2 pathological variants (PVs) with
familial cancer in patients with breast and colon cancer and
relatives with a family history of cancer (5). Similarly, an international study
(6) analysed >500 families from
21 countries with PALB2 PVs and reported the link between
PALB2 PVs with breast, ovarian and pancreatic cancer;
however, prostate and colorectal cancer were independent of
PALB2. Moreover, PALB2 is included in the breast
cancer gene panels based on guidelines of the UK Cancer Genetics
Group (7).
Fanconi anemia (FA) is a rare autosomal and X-linked
genetic condition characterised by bone marrow failure, physical
abnormality, early onset cancer and premature death (3,8,9).
Patients with FA have bi-allelic inheritance of pathogenic
variants, either homozygous or compound heterozygous. A total of 22
genes are confirmed as FA genes, including PALB2/Fanconi
Anemia, Complementation Group N (FANCN) (10). Among these genes, FANCA,
FANCC and FANCG are the most frequently mutated genes
in FA (11). FA genes encode
proteins that participate in the FA pathway of interstrand
crosslink (ICL) DNA repair and genome maintenance when cell DNA is
damaged (12). An essential step in
the FA pathway is monoubiquitination of upstream FA protein, FANCI
and FANCD2, forming ID2 complex; this step depends on other FA
proteins. Ubiquitinated ID2 complex orchestrates the actions of
downstream repair proteins, such as BRCA2, BRCA1 Interacting
Protein C-terminal Helicase 1(BRIP1), PALB2 (FANCN), RAD51C and
FANCP, which are necessary for later stages of homologous
recombination (12,13). FA proteins also stimulate stem cell
function, inhibit inaccurate repair and prevent cancer (12). PALB2/FANCN-deficient cells
exhibit reduced levels of BRAC2 due to the role of PALB2 in
stabilising BRAC2 (14,15). This indicates the key role of PALB2
for HR and tumour suppressive functions of BARC2. In a case series
from Saudi Arabia, ten children were newly diagnosed with FA; two
infants had homozygous PALB2 mutation at c.3425del
p.Leu1142Tyrfs*21, and one presented with early onset of cancer
(16). In Turkey, 20 FA cases with
PVs in PALB2/FANCN were found in 2.5% of the subjects with a
paternal novel heterozygous gross deletion of exon 5-6(17).
In Saudi Arabia, the prevalence of PALB2
mutations in breast and ovarian cancer is relatively low (0.65%),
as found in a cohort of 918 patients (18). Prevalence of PALB2 mutations
is 5.2% among 879 Taiwanese patients with breast cancer (19) and 4.4% along with other PVs in
ataxia-Telangiectasia Mutated, BRCA1, BRCA2, Checkpoint
Kinase 2 (CHEK2), or PALB2 in USA (20). These figures suggest that
PALB2 mutations are generally rare, especially in Saudi
Arabia. FA may be underdiagnosed in Saudi Arabia due to unique
mutation patterns observed in Saudi patients compared with Europe
and North America (16). FA due to
PALB2 mutation is the least reported genotype (16). Furthermore, consanguinity plays a
key role in the inheritance of diseases. Saudi Arabia,
characterised by a high rate of consanguineous marriages across
tribes, exhibits an increased prevalence of genetic disease
(21). This highlights the need for
further regional genetic studies to understand the full scope of
PALB2-associated conditions, including its role in FA.
Materials and methods
Ethics approval and sample
collection
The present study was approved by the local ethical
committee (approval no. 013-CEGMR-02-ETH of the Center of
Excellence in Genomic Medicine Research, King Abdulaziz University
Jeddah (Jeddah, Saudi Arabia). Written informed consent for
laboratory and genetic testing was obtained from the patient's
legal guardians. A total of 2 ml of blood samples were collected
from four family members, including the father 42 yeas old, mother
37 year, the 11 years old brother and the patient. The study was
performed in accordance with the Declaration of Helsinki 2013.
Blood samples were collected and DNA was extracted from blood
stored in the EDTA tubes (Roche Life Science), as previously
described (22). Nanodrop
2000/2000c spectrophotometer was used for DNA concentration and
quality check.
Clinical report of the patient
The proband (IV-2) was a 2-month-old female who was
admitted to the King Abdulaziz University hospital Jeddah Saudi
Arabia in December 2019 with multiple physical abnormalities after
birth. FA was diagnosed 2 months later based on mild pulmonary
valve stenosis, dysmorphic features, lymphangiectasia, non-immune
hydrops fetalis and right-sided pleural effusion. The prenatal
course was complicated preterm; serious developmental concerns were
noted as she was born at 32 weeks gestational age with non-immune
hydrops fetalis, right-sided pleural effusion, mild pulmonary valve
stenosis and lymphangiectasis and was underweight along with
dysmorphic features.
The family had a history of three generations of
consanguine marriages. The grandparents and parents were first
cousins. Whole exome sequencing (WES) was performed for the
affected proband at the age of 6 months. No previous history of the
disease was reported in the other family members.
WES
Extracted DNA from the proband was enriched for the
coding region and splice site junctions of genes. hg19, GRCH37/UCSC
(https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&chromInfoPage=)
for reference sequences based on human genome build were used.
Capillary sequencing was used to assess variants with clinical or
uncertain significance. All sequence alterations were defined using
the Human Genome Variation Society nomenclature guidelines
(23). Data were analysed using
gene-specific filtering. The products were sequenced on an Illumina
NextSeq instrument with 2x76 paired-end reads, as previously
described (24,25).
Following WES, the generated files (FASTQ) were
further converted to BAM and variant call format (vcf) files
containing all identified variants. Moreover, obtained variants
were used for identification of variants leading to the disease
phenotype established through rare ulta rare common/novel (minor
allele frequency) (MAF+0.01%) frequency, homozygous or heterozygous
conditions along with structure and function [predicted damage by
Polyphen-2 (v2.2.2) polyphen/sorting intolerant from tolerant;
faculty.washington.edu/wjs18/GS561/cSNPs_lab.html],
pathogenicity, genomic position, protein damaging effect and
linkage with disease phenotype. Reference sequences were aligned
using the GRCh37 database (ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.13/.
The list of obtained variants was filtered to determine the disease
linked with the identified variants in public databases, such as
1000 genomes (internationalgenome.org/) and Genome Aggregation
Database (gnomAD) for allele frequencies <5.0% (gnomad.broadinstitute.org/); frameshift,
nonsense, and splice-site variants in disease-associated genes with
a minor allele frequency ≤1.0% were observed in gnomAD. College of
American Pathologists (26) were
used for variant classification. Deleterious effects and
abnormalities were identified using bionormatics tools such as
Mutation Assessor version 2.0 (mutationassessor.org/r3/, Exome Aggregation Consortium
Version 0.3.1 https://ngdc.cncb.ac.cn/databasecommons/database/id/3774,
SIFT version 2.1 sans.org/tools/sift-workstation/, PolyP
(phyloP46way_placental) epd.expasy.org/mga/hg19/phylop/phylop.html for
identified variant leading to disease. Mutation Tester (mutationtaster.org/) also predicted the variant as a
disease-causing variant.
Sanger sequencing
Sanger sequencing was performed for a novel
homozygous missense variant, NM_024675.3: c.3296C>G
(p.Thr1099Arg) in PALB2 gene (OMIM: 610355) based on WES
results. Primers were designed using the online free software
Primer 3 (https://bioinfo.ut.ee/primer3-0.4.0/) for PCR. Sanger
sequencing was performed for all affected and normal family
members. Thermocycling conditions were as follows: Initial
denaturation at 95˚C for 15 min, followed by 30 cycles 58˚C for 30
sec and 1 min extention at 72˚C. Primer sequences were as follows:
Forward, 5'AGCCTATCGGTCATTGCTTT3' and reverse,
5'AGGGAATCTGGGGTTTGACT3'. Sequencing files were obtained from the
AB1 sequencing unit. The obtained files were aligned with the
wild-type reference sequence using BioEdit version 7.2 (Informer
Technologies, Inc.). Further validation was performed by using 100
normal control samples from Saudi Arabia to confirm that the
identified variant is not present in the in the population.
Results
WES identified a novel homozygous missense variant,
NM_024675.3: c.3296C>G in PALB2 gene. The patient was
homozygous for the PALB2 variant c.3296C>G, where
p.Thr1099Arg was a change in the protein. Both parents were
identified to have heterozygous missense variant, NM_024675.3:
c.3296C>G (p.Thr1099Arg) in PALB2 gene, while a male
sibling was healthy without PALB2 gene mutation (Table I). The parents were heterozygous
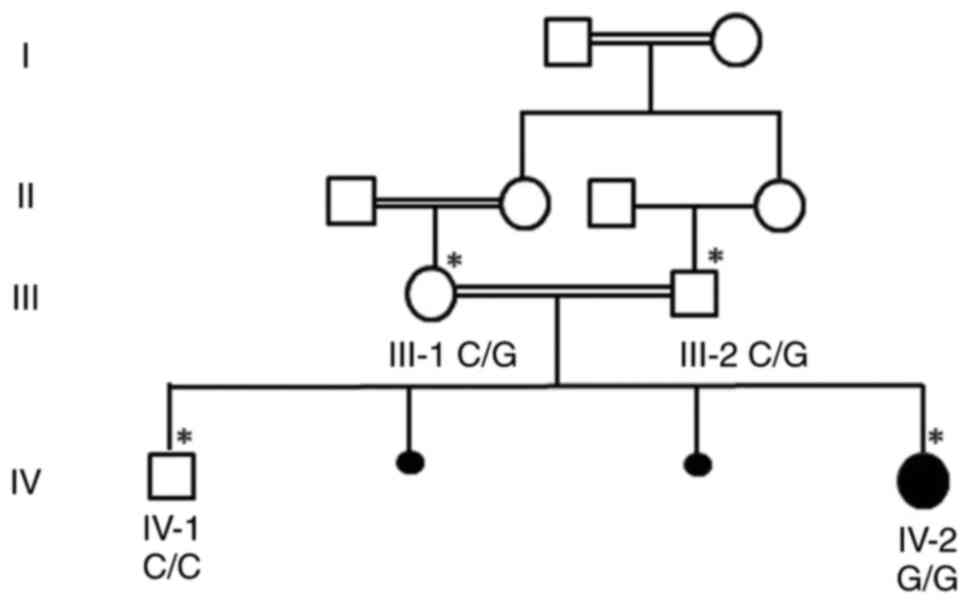
carriers who were first cousins, (Fig.
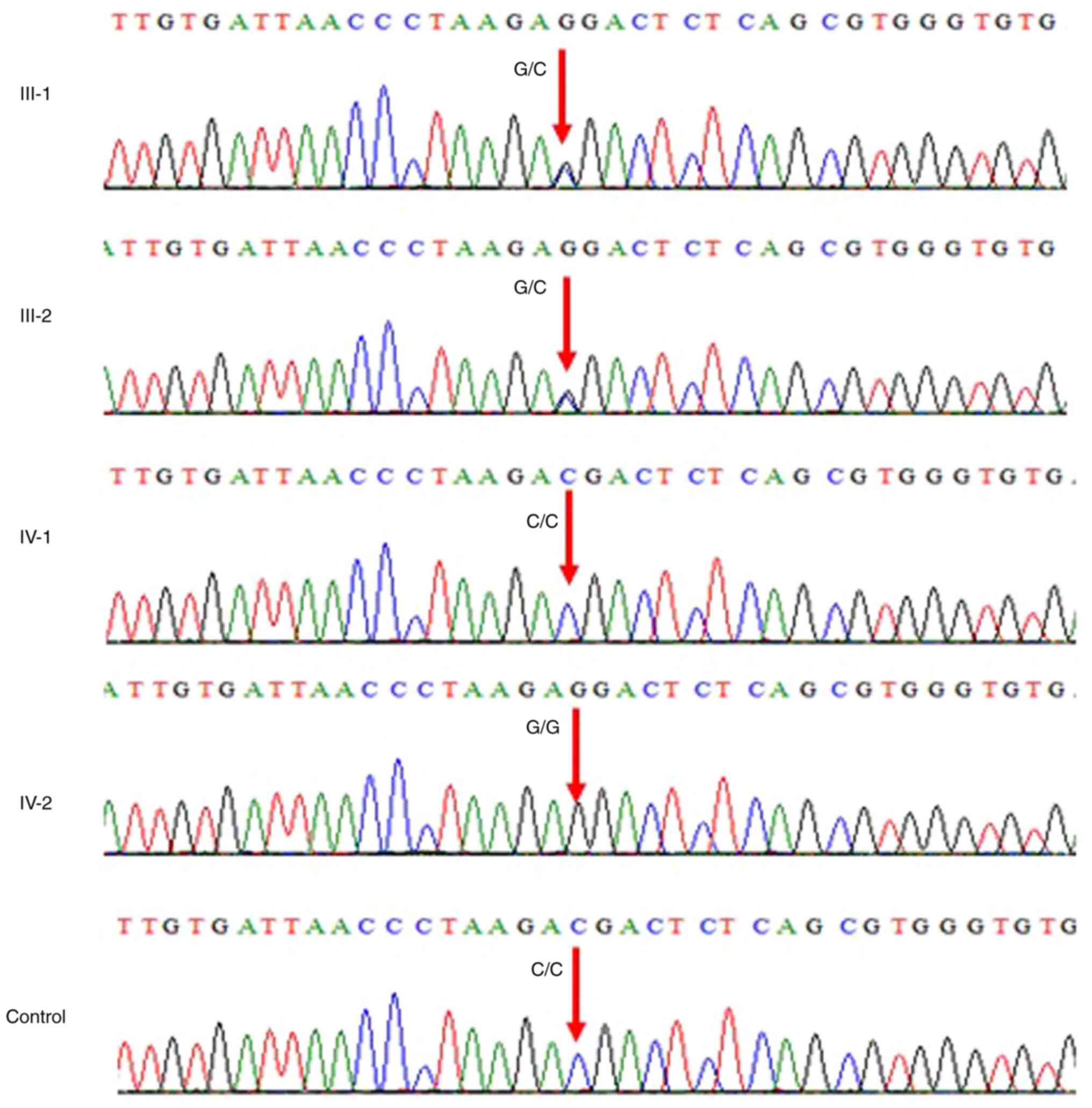
1). Sanger sequencing chromatogram showed that both the parents
III-1 and III-2 were heterozygous carriers having G/C on both
alleles; sibling IV-1 was wild-type C/C and IV-2 was homozygous G/G
(Fig. 2). Table I lists known variants in
homozygous/heterozygous state of PALB2 gene.
 | Table IPathogenic mutations in partner and
localiser of BRCA2 gene with Fanconi anaemia. |
Table I
Pathogenic mutations in partner and
localiser of BRCA2 gene with Fanconi anaemia.
| No. | First author,
year | Mutation | Result | State | Origin | (Refs.) |
|---|
| 1 | Abdulkareem et
al, 2024 | c.3296C>G | p.Thr1099Arg | Homozygous | Saudi Arabian | Present study |
| 2 | Viakhireva et
al, 2020 |
c.172_175delTTGT | p.Gln60Argfs | Homozygous | Russian | (40) |
| 3 | Byrd et al,
2016 | c.1676_1677
delAAinsG | p.Gln559
ArgfsTer2 | Homozygous | Serbian | (32) |
| 4 | | c.2586
+1G>A |
p.Thr839_Lys862del | Homozygous | | |
| 5 | Ghazwani et
al, 2016 | c.3425del | p.Leu1142
Tyrfs*21 | Homozygous | Saudi Arabian | (16) |
| 6 | Serra et al,
2012 | c.1676_c1677
delAAinsG | p.Gln526
ArgfsX1 | Homozygous | German | (41) |
| 7 | Reid et al,
2007 | 395delT | V132fs | Biallelic (not
specified) | Albanian | (9) |
| 8 | | c.3113+5G>C |
r.2835_3113del279/A946fs | | Moroccan | |
| 9 | | 757_758delCT | L253fs | | German | |
| 10 | | 3294_3298
delGACGA | K1098fs | | German | |
| 11 | | 2257C-T | R753X | | Hispanic | |
| 12 | | c.3549C>A | p.Tyr1183X | | North American | |
| 13 | | 2393_2394insCT | T799fs | | German | |
| 14 | | 3350+4A-G |
r.3350insGCAG/F1118fs | | German | |
| 15 | | 2521delA | T841fs | | North American | |
| 16 | | 3323delA | Y1108fs | | African | |
| 17 | | 2962C-T | Q988X | | British | |
| 18 | | c.3549C>G | p.Tyr1183X | | British North
American | |
| 19 | | 3116delA | N1039fs | | North American | |
Moreover, the variant was not reported in gnomAD
exomes and 1000 genomes. There was a moderate physiochemical
difference between thiamin and arginine acid. Furthermore, in
silico tools used to aid in interpreting sequence variants
identified the variant as disease-causing. Moreover, protein
alignment showed highly conserved amino acids between different
species (Fig. 3).
Discussion
FA is a rare genetic disease characterised by bone
marrow failure and several physical anomalies. The estimated
worldwide incidence is 1 in 360,000 live births, while the
prevalence of carriers is estimated to be 1 in 181(8). FA was first described in 1927 by the
Swiss paediatrician Guido Fanconi when 3/5 brothers died from
severe conditions resembling pernicious anaemia associated with
physical birth defects (27). A
total of 22 genes responsible for FA have been identified and
labelled FANCA-W (10). The
most commonly affected gene is FANCA, followed by
FANC-C, -G,-D2 and -P (11,26).
The major function of the FA genes pathway is to orchestrate DNA
repair ICL. During the final phase of ICL, seven downstream FA
proteins, including PALB2, combine to repair broken DNA via HR
(8). Therefore, mutations in the FA
pathway lead to defective HR, increasing dependence on error-prone
non-homologous DNA end-joining repair pathway (28).
Bi-allelic mutations in PALB2 account for FA,
which is a rare autosomal and X-linked disorder characterised by
severe bone marrow failure, developmental and physical abnormality
and susceptibility to early-onset cancers. A total of >20
distinct genes have been reported to cause FA. ≈The present study
reports a case of a preterm female baby with FA associated with
mild pulmonary valve stenosis, dysmorphic features,
lymphangiectasia, non-immune hydrops fetalis and right-sided
pleural effusion. To the best of our knowledge, the present study
is the first to reported FA with lymphangiectasia, a dilation of
lymphatic blood vessels leading to oedema (29). The typical reported clinical
features of FA are distinctive for each FA gene; they generally
include growth retardation, facial abnormality, radial ray defects,
hyper-hypo pigmentation, renal malformation and microcephaly
(9,30). For example, in a recent Chinese
study of 148 patients with FA, finger deformities and café au lait
spots were associated with FANCA, while FANC,-D2, G,
I and -P presented mostly with cardiovascular deformity
(31). Fiesco-Roa et al
(30) reported that PALB2
abnormality is associated with anal congenetal abnormalities due to
a defect in the downstream DNA repair pathway. Ghazwani et
al (16) reported 10 cases of
FA, two with PALB2 genotype, one of which exhibited a novel
mutation c.3425del (p.Leu1142Tyrfs*21) with multiple congenital
anomalies, metastatic Wilms tumour and stage I neuroblastoma in the
first year of life and one had café au lait spots and mosaic
duplication of the long arm of chromosome 17 (band q21.2). The
present patient exhibited two typical features of FA
(cardiovascular abnormality and facial deformity); to the best of
our knowedge, however, lymphangiectasia, non-immune hydrops fetalis
and right-sided pleural effusion have not been reported before.
Similarly, a previous study reported atypical features of FA with
cancer (32) in two sisters (aged
12.0 and 3.5 years at time of FA diagnosis) with PALB2
mutations, one truncating, c.1676_1677delAAinsG;
(p.Gln559ArgfsTer2), and c.2586 +1G>A; p.Thr839_Lys862del
causing frame skip of exon 6 (24 amino acids), resulting in B cell
non-Hodgkin lymphoma and atypical features of FA including learning
difficulty and dyslexia without developmental defect. Similarly, an
in silico study linked PALB2 genotype with tumours of
embryonal origin, including medulloblastoma, Wilms tumour and
neuroblastoma (11). Notably,
PALB2 mutations differ from other subtypes of FA owing to
their association with high-risk paediatric malignancy similar to
those induced by BRCA1 and BRCA2 (10,33).
This highlights the key interaction of PALB2 in the localisation
and stability of BRCA2, which facilitates BRCA2 functions in DNA
repair, and interaction with several other tumour suppressors,
facilitating the role of PALB2 as a typical cancer
suppressor gene (3).
In the present study, WES results validated by
Sanger sequencing identified a homozygous missense variant,
NM_024675.3: c.3296C>G (p.Thr1099Arg) in PALB2 gene in a
preterm female baby. The parents were first cousins and
heterozygous carriers and the brother was healthy. In
silico, a total of 84 PALB2 variants with uncertain
significance, including the variant identified in the present
study, were assessed; four variants (p.L24S,c.71T>C;
p.L35P,c.104T>C; pI944N,c.2831T>A and p.L1070P,c.3209T>C)
were associated with disrupted PALB2-mediated homology-directed DNA
repair (34). Although the
aforementioned study did not list the present variant as a PV, the
present study confirms the pathological features of the
variant.
FA is an autosomal recessive genetic disease with
chromosomal instability. The present patient was a product of a
consanguineous marriage (three generations). Consanguinity is a
practice in several tribes in Saudi Arabia and other countries,
resulting in disease in the offspring of carrier families (35,36).
WES is a diagnostic tool to identify molecular defects in patients
with suspected genetic disorders (37-39).
Identifying deleterious PVs facilitates genetic counselling,
prenatal testing and development of therapeutic strategies.
The present study reported a Saudi family with
PALB2 with a novel homozygous missense variant, c.3296C>G
(p.Thr1099Arg). The present results contribute to the mutation
spectrum of PALB2-associated diseases, which will help to
manage this disease in the future. To the best of our knowledge,
the present study is the first report of this rare genetic variant
related to the PALB2 gene in Saudi Arabia with other
symptoms that have not been reported previously. PALB2
variant carries important clinical implications, including an
increased risk for certain types of cancer including breast and
pancreatic cancers (6). This
necessitates monitoring and potential adjustments in patient
management strategies. Moreover, it underscores the need for
thorough genetic counselling for affected individuals and their
family members, given the potential hereditary nature of the risk
associated with this variant. WES testing of patients of
consanguineous marriages with a strong family history of genetic
disease is advisable.
Acknowledgements
Not applicable.
Funding
Funding: The authors extend their appreciation to the King
Salman Center for Disability Research for funding this work through
Research Group no. KSRG-2023-024.
Availability of data and materials
The data generated in the present study are not
publicly available due to refusal of consent by the parents of the
patient but may be requested from the corresponding author.
Authors' contributions
MIN and AAA designed and performed the experiments.
BHS and HAB designed experiments and collected clinical details and
samples. MIN, AH and AAA analysed the data. MIN, BHS and HAB wrote
the manuscript. AAA, HAB, AH, BHS and MIN revised the manuscript.
MIN, HAB and BHS confirm the authenticity of all the raw data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Ethics approval was obtained from the Ethical
Committee (approval no. 013-CEGMR-02-ETH) of the Center of
Excellence in Genomic Medicine Research, King Abdulaziz University
Jeddah (Jeddah, Saudi Arabia). Written informed consent was
obtained from the parents.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Park JY, Zhang F and Andreassen PR: PALB2:
The hub of a network of tumor suppressors involved in DNA damage
responses. Biochim Biophys Acta. 1846:263–275. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Simhadri S, Vincelli G, Huo Y, Misenko S,
Foo TK, Ahlskog J, Sørensen CS, Oakley GG, Ganesan S, Bunting SF
and Xia B: PALB2 connects BRCA1 and BRCA2 in the G2/M checkpoint
response. Oncogene. 38:1585–1596. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hanenberg H and Andreassen PR: PALB2
(partner and localizer of BRCA2). Atlas Genet Cytogenet Oncol
Haematol. 22:484–290. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hamdan O and Nowak KM: Gene of the month:
PALB2. J Clin Pathol. 76:73–75. 2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
AlHarbi M, Mobark NA, AlJabarat WAR,
ElBardis H, AlSolme E, Hamdan AB, AlFakeeh AH, AlMushawah F,
AlHarthi F, AlSharm AA, et al: Investigating the prevalence of
pathogenic variants in Saudi Arabian patients with familial cancer
using a multigene next generation sequencing panel. Oncotarget.
14:580–594. 2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yang X, Leslie G, Doroszuk A, Schneider S,
Allen J, Decker B, Dunning AM, Redman J, Scarth J, Plaskocinska I,
et al: Cancer risks associated with germline PALB2 pathogenic
variants: An international study of 524 families. J Clin Oncol.
38:674–685. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Taylor A, Brady AF, Frayling IM, Hanson H,
Tischkowitz M, Turnbull C and Side L: UK Cancer Genetics Group
(UK-CGG). Consensus for genes to be included on cancer panel tests
offered by UK genetics services: Guidelines of the UK cancer
genetics group. J Med Genet. 55:372–377. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Mamrak NE, Shimamura A and Howlett NG:
Recent discoveries in the molecular pathogenesis of the inherited
bone marrow failure syndrome Fanconi anemia. Blood Rev. 31:93–99.
2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Reid S, Schindler D, Hanenberg H, Barker
K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, et al:
Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and
predispose to childhood cancer. Nat Genet. 39:162–164.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Fang CB, Wu HT, Zhang ML, Liu J and Zhang
GJ: Fanconi anemia pathway: Mechanisms of breast cancer
predisposition development and potential therapeutic targets. Front
Cell Dev Biol. 8(160)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
McReynolds LJ, Biswas K, Giri N, Sharan SK
and Alter BP: Genotype-cancer association in patients with Fanconi
anemia due to pathogenic variants in FANCD1 (BRCA2) or FANCN
(PALB2). Cancer Genet. 258-259:101–109. 2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kottemann MC and Smogorzewska A: Fanconi
anaemia and the repair of Watson and Crick DNA crosslinks. Nature.
493:356–363. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Bick G, Zhang F, Meetei AR and Andreassen
PR: Coordination of the recruitment of the FANCD2 and PALB2 Fanconi
anemia proteins by an ubiquitin signaling network. Chromosoma.
126:417–430. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu
J, Christ N, Liu X, Jasin M, Couch FJ and Livingston DM: Control of
BRCA2 cellular and clinical functions by a nuclear partner, PALB2.
Mol Cell. 22:719–729. 2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Xia B, Dorsman JC, Ameziane N, de Vries Y,
Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, et al:
Fanconi anemia is associated with a defect in the BRCA2 partner
PALB2. Nat Genet. 39:159–161. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Ghazwani Y, AlBalwi M, Al-Abdulkareem I,
Al-Dress M, Alharbi T, Alsudairy R, Alomari A, Aljamaan K, Essa M,
Al-Zahrani M and Alsultan A: Clinical characteristics and genetic
subtypes of Fanconi anemia in Saudi patients. Cancer Genet.
209:171–176. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Toksoy G, Uludağ Alkaya D, Bagirova G,
Avcı Ş, Aghayev A, Günes N, Altunoğlu U, Alanay Y, Başaran S,
Berkay EG, et al: Clinical and molecular characterization of
Fanconi anemia patients in Turkey. Mol Syndromol. 11:183–196.
2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Siraj AK, Bu R, Parvathareddy SK, Iqbal K,
Azam S, Qadri Z, Al-Rasheed M, Haqawi W, Diaz M, Victoria IG, et
al: PALB2 germline mutations in a large cohort of Middle Eastern
breast-ovarian cancer patients. Sci Rep. 13(7666)2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cheng HF, Tsai YF, Liu CY, Hsu CY, Lien
PJ, Lin YS, Chao TC, Lai JI, Feng CJ, Chen YJ, et al: Prevalence of
BRCA1, BRCA2, and PALB2 genomic alterations among 924 Taiwanese
breast cancer assays with tumor-only targeted sequencing: Extended
data analysis from the VGH-TAYLOR study. Breast Cancer Res.
25(152)2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Yadav S, Boddicker NJ, Na J, Polley EC, Hu
C, Hart SN, Gnanaolivu RD, Larson N, Holtegaard S, Huang H, et al:
Contralateral breast cancer risk among carriers of germline
pathogenic variants in ATM, BRCA1, BRCA2, CHEK2, and PALB2. J Clin
Oncol. 41:1703–1713. 2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Alfares A, Alfadhel M, Wani T, Alsahli S,
Alluhaydan I, Al Mutairi F, Alothaim A, Albalwi M, Al Subaie L,
Alturki S, et al: A multicenter clinical exome study in unselected
cohorts from a consanguineous population of Saudi Arabia
demonstrated a high diagnostic yield. Mol Genet Metab. 121:91–95.
2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Abdulkareem AA, Abulnaja KO, Jan MM, Karim
S, Rasool M, Ansari SA, Chaudhary AG, Al-Qahtani MH and Naseer MI:
A novel homozygous nonsense mutation in CCDC88A gene cause
PEHO-like syndrome in consanguineous Saudi family. Neurol Sci.
40:299–303. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bruford EA, Braschi B, Denny P, Jones TEM,
Seal RL and Tweedie S: Guidelines for human gene nomenclature. Nat
Genet. 52:754–758. 2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Naseer MI, Abdulkareem AA, Pushparaj PN,
Bibi F and Chaudhary AG: Exome analysis identified novel homozygous
splice site donor alteration in NT5C2 gene in a Saudi family
associated with spastic diplegia cerebral palsy, developmental
delay, and intellectual disability. Front Genet.
11(14)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Naseer MI, Abdulkareem AA, Muthaffar OY,
Sogaty S, Alkhatabi H, Almaghrabi S and Chaudhary AG: Whole exome
sequencing identifies three novel mutations in the ASPM gene from
Saudi families leading to primary microcephaly. Front Pediatr.
8(627122)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lobitz S and Velleuer E: Guido Fanconi
(1892-1979): A jack of all trades. Nat Rev Cancer. 6:893–898.
2006.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Adamo A, Collis SJ, Adelman CA, Silva N,
Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ and La Volpe A:
Preventing nonhomologous end joining suppresses DNA repair defects
of Fanconi anemia. Mol Cell. 39:25–35. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lymphangiectasia. In: Rheumatology and
Immunology Therapy. Abbott JD, Ball G, Boumpas D, Bridges SL,
Chatham W, Curtis J, Daniel C, Hughes LB, Kao AH, Langford C, et
al (eds). Springer Berlin Heidelberg, Berlin, Heidelberg,
pp555-556, 2004.
|
|
30
|
Fiesco-Roa MO, Giri N, McReynolds LJ, Best
AF and Alter BP: Genotype-phenotype associations in Fanconi anemia:
A literature review. Blood Rev. 37(100589)2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chang L, Zhang L, An W, Wan Y, Cai Y, Lan
Y, Zhang A, Liu L, Ruan M, Liu X, et al: Phenotypic and genotypic
correlation evaluation of 148 pediatric patients with Fanconi
anemia in a Chinese rare disease cohort. Clin Chim Acta. 539:41–49.
2023.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Byrd PJ, Stewart GS, Smith A, Eaton C,
Taylor AJ, Guy C, Eringyte I, Fooks P, Last JI, Horsley R, et al: A
hypomorphic PALB2 allele gives rise to an unusual form of FA-N
associated with lymphoid tumour development. PLoS Genet.
12(e1005945)2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ng PS, Pan JW, Ahmad Zabidi MM, Rajadurai
P, Yip CH, Reuda OM, Dunning AM, Antoniou AC, Easton DF, Caldas C,
et al: Characterisation of PALB2 tumours through whole-exome and
whole-transcriptomic analyses. NPJ Breast Cancer.
7(46)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wiltshire T, Ducy M, Foo TK, Hu C, Lee KY,
Belur Nagaraj A, Rodrigue A, Gomes TT, Simard J, Monteiro ANA, et
al: Functional characterization of 84 PALB2 variants of uncertain
significance. Genet Med. 22:622–632. 2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Siddiqui F, Ansari S, Agha A, Nusrat N,
Munzir S, Shan S, Hanifa A, Farzana T Sr, Taj M, Borhany M, et al:
Chromosomal breakage in Fanconi anemia and consanguineous
marriages: A social dilemma for developing countries. Cureus.
12(e10440)2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Ben Haj Ali A, Messaoud O, Elouej S,
Talmoudi F, Ayed W, Mellouli F, Ouederni M, Hadiji S, De
Sandre-Giovannoli A, Delague V, et al: FANCA gene mutations in
North African Fanconi anemia patients. Front Genet.
12(610050)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yang Y, Muzny DM, Reid JG, Bainbridge MN,
Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al:
Clinical whole-exome sequencing for the diagnosis of mendelian
disorders. N Engl J Med. 369:1502–1511. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lee H, Deignan JL, Dorrani N, Strom SP,
Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M,
et al: Clinical exome sequencing for genetic identification of rare
Mendelian disorders. JAMA. 312:1880–1887. 2014.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Yang Y, Muzny DM, Xia F, Niu Z, Person R,
Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al: Molecular
findings among patients referred for clinical whole-exome
sequencing. JAMA. 312:1870–1879. 2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Viakhireva I, Musatova E, Bessonova L,
Shcherbatyuk Y, Korobkov S, Zhikriveckaya S, Sofronova Y, Mironova
I, Khmelkova D, Konovalov F, et al: Novel intronic variant in PALB2
gene and effective prevention of Fanconi anemia in family. Fam
Cancer. 19:241–246. 2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Serra A, Eirich K, Winkler AK, Mrasek K,
Göhring G, Barbi G, Cario H, Schlegelberger B, Pokora B, Liehr T,
et al: Shared copy number variation in simultaneous nephroblastoma
and neuroblastoma due to Fanconi anemia. Mol Syndromol. 3:120–130.
2012.PubMed/NCBI View Article : Google Scholar
|