1. Introduction
Deciphering the genetic code has led to the elucidation of the fundamental mechanisms underlying the translation of the information encoded within genes to proteins. More recently, as studies were undertaken by different researchers to understand the complex molecular underpinnings of development in an organism, striking variations in gene expression were observed in different cell types of the same individual. These mitotically inheritable modifications are essentially modulated by epigenetic regulators, which include alterations to histone proteins along with the DNA sequence in the genome. Apart from DNA methylation, other epigenetic regulators such as methylation, acetylation, ubiquitination and the phosphorylation of histones have a profound effect on the modulation of gene expression (1). Moreover, studies using yeast and mice have demonstrated that the chromatin remodeling complexes, which utilize energy derived from ATP hydrolysis for the movement of nucleosomes, thus affecting histone-DNA interaction, are responsible for changes in chromatin compaction and subsequently, have a profound effect on the gene transcription machinery (2). Different proteins, which include histones, nuclear receptors, transcription factors and enzymes, undergo acetylation at the lysine residues, a common post-translational modification, which helps control their functions within cells (3,4). Previous research has indicated that histone acetylation is linked to increased transcriptional activity, whereas histone deacetylation can lead to the downregulation of gene expression or gene silencing (5).
Histone deacetylases (HDACs) are typically categorized into class I (includes HDAC1, 2, 3, 8 and 11) and class II (includes HDAC4, 5, 6, 7, 9 and 10) and are instrumental in the dissociation of acetyl groups from the lysine residues located on N-terminal ends of histone proteins in nucleosomes (5,6). Histone acetyltransferases (HATs) are enzymes involved in histone acetylation by the transfer of acetyl groups from acetyl-CoA to lysine residues located on the histones (7). They are primarily categorized into the Type-A and Type-B superfamilies. While Type-A HATs are involved in the acetylation of histone proteins in nuclear chromatin, Type-B HATs play a role in the acetylation of free histones in the cytoplasm (8). HATs and HDACs, conserved in yeast, flies, worms and humans, are tightly co-regulated, and this has been found to be an indicator of cell fate during proliferation and differentiation (9-12).
An equilibrium can be observed between histone acetylation and deacetylation in cells, and a perturbations in the balance can lead to development of a wide array of inflammatory disorders. In a previous study by Tsaprouni et al (13), the authors reported an upregulation of histone acetylation in murine models of inflammatory bowel disease (IBD), wherein the localization of histone proteins with acetylated lysine residues 8 and 12 was detected in the inflamed mucosal tissue of trinitrobenzene sulphonic acid (TNBS)-induced mice (that mimic the immunopathological properties of Crohn's disease); they also detected the localization of acetylated histone H4 in inflamed tissue and Peyer's patches in dextran sodium sulfate (DSS)-treated murine models (that mimic the immunopathological features of ulcerative colitis) (13). Smith et al (14) outlined the plausible signaling pathways involved in neuronal degeneration mediated by pro-inflammatory Th1 cytokines, such as interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α and interferon (IFN)-γ upon inflammation, injury, infection, etc. caused to microglial cells in the central nervous system (CNS). This thus indicated new possibilities of targeting these cytokines for the amelioration of neurodegenerative diseases (14). Previously published studies have demonstrated that upon the alternative activation of primary monocytes by Th2 cytokines, such as IL-4 and IL-13, some specific genes, namely monoamine oxidase A (MAO-A), 15-lipoxygenase (15-LOX-1), etc. are induced, which are associated with several cardiovascular and inflammation-related disorders (15-17). Moreover, a co-induction of the MAO-A and 15-LOX-1 genes has been found in human monocytes, as well as in A549 lung cancer cells following treatment with IL-4/IL-13(17). These observations have opened avenues for the investigation of the basic mechanisms involved in the epigenetic control of inflammation. In the present review article, the epigenetic regulation of the differential expression of genes which are crucial for inflammation is discussed, with a focus on histone acetylation/deacetylation through HATs and HDACs. In addition, the present review also discusses into the latest developments in therapeutic strategies that harness the histone acetylation machinery to combat a wide range of inflammatory disorders.
2. Role of HAT and HDAC in regulating pro-inflammatory gene expression
HATs and HDACs are prominent players involved in regulating the expression of a vast array of genes, which are widely expressed during inflammation in the human genome, leading to altered immune responses. Through the enhanced recruitment of transcription factors to the gene promoters or through nucleosome remodeling, these enzymes perform their regulatory functions. A detailed study of these gene expression control strategies may help to identify potent intermediates in the signaling cascade, leading to the expression and activity of these genes, which may help to design possible methods of manipulation of these pathways or manipulate potent signaling intermediates for therapeutic application against inflammatory disorders. Among the numerous genes that play a role in promoting inflammation, TNF-α, IL-6 and nuclear factor (NF)-κB are important candidates, which have been implicated in the pathogenesis of myriad diseases. In this section, the authors aim to discuss some of the molecular underpinnings of HAT and HDAC-mediated expression of these genes during inflammation.
TNF-α
TNF-α is produced predominantly from myeloid cells and T-cells (18) and acts as a key regulator of an array of biological processes, which typically include inflammation, proliferation and apoptosis of macrophages, osteoclast formation, etc. (19,20). The lipopolysaccharide (LPS)-induced release of TNF-α in primary human monocytes, as well as in the human monocytic cell line, THP-1, has been reported by Schildberger et al (21), and is considered to be a well-accepted model system for studying the regulation of TNF-α gene expression.
Previous studies have demonstrated that the development of cells of myeloid lineage from embryonic stem cells to monocytes is accompanied by alterations in the epigenome at the TNF-α locus through changes in histone 3 lysine 4 methylation and histone 4 acetylation (22). A plausible explanation for the increase in histone acetylation leading to the enhanced expression of TNF-α in cells may be due to the fact that transcription factors are recruited preferentially to the TNF-α promoter or through the action of chromatin-remodeling complexes, which can identify acetylated histone tails and subsequently modulate their interaction with the transcription machinery to control gene expression. Studies have identified nuclear factor of activated T cells (NFAT), Ets-1, Elk-1, Sp1, Egr-1 and ATF-2-Jun binding sites within the TNF-α promoter for the assembly of transcription factors (23-25). Different HATs act as co-activators of TNF-α expression, which include cAMP response element-binding protein (CREB) and P300 (23-25).
The HAT-mediated transcriptional activation of TNF-α in LPS-treated macrophages in vivo requires the binding of ATF-2-c-jun, Sp1, Egr-1, Elk-1 and Ets-1 factors to their respective binding sites within the TNF-α promoter. CBP and p300 enzymes, containing transactivation domains, have been reported to play a pivotal role in mediating LPS-induced TNF-α expression in macrophages via the co-activation of the complex formed by the transcription factors mentioned above (24), which is illustrated in Fig. 1. Further investigation may help speculate the mechanistic details of transactivation-dependent TNF-α gene expression by HATs. The study by Lee et al (26) suggested that the treatment of monocytic cell lines with HDAC inhibitors (HDACis) resulted in a profound increase in TNF-α production. Their study further observed that the increase in TNF-α production in less mature cell lines required treatment of the cells with multiple HDACis, whereas the most mature cells could be induced to produce TNF-α upon induction by only a single HDACi. This observation coupled with ChIP assays suggested that the developmental stage of the cells may play a role in determining the acetylation profile of the histone proteins, which in turn may influence TNF-α gene expression in those cells (26). The findings by Mahlknecht et al (27) shed light onto the control of TNF-α production both at mRNA and protein level, wherein HDAC3 was implicated in downregulating TNF-α expression in LPS-treated monocytic cells by repressing the phosphorylation of MAPK11 and interfering with the binding of ATF-2 and NF-κB transcription factors to respective binding sites in the TNF-α promoter. The nucleo-cytoplasmic transport of TNF-α mRNA is controlled by a MEK/ERK dependent pathway in LPS-treated mouse macrophages and it has been found that this is crucial for the post-transcriptional expression of TNF-α (28). Notably, tumor progression locus 2 (Tpl2), a pro-inflammatory serine/threonine kinase kinase kinase 8 (MAP3K8) (29), has been observed to mediate TNF-α expression through the ERK pathway upon induction by LPS (28).
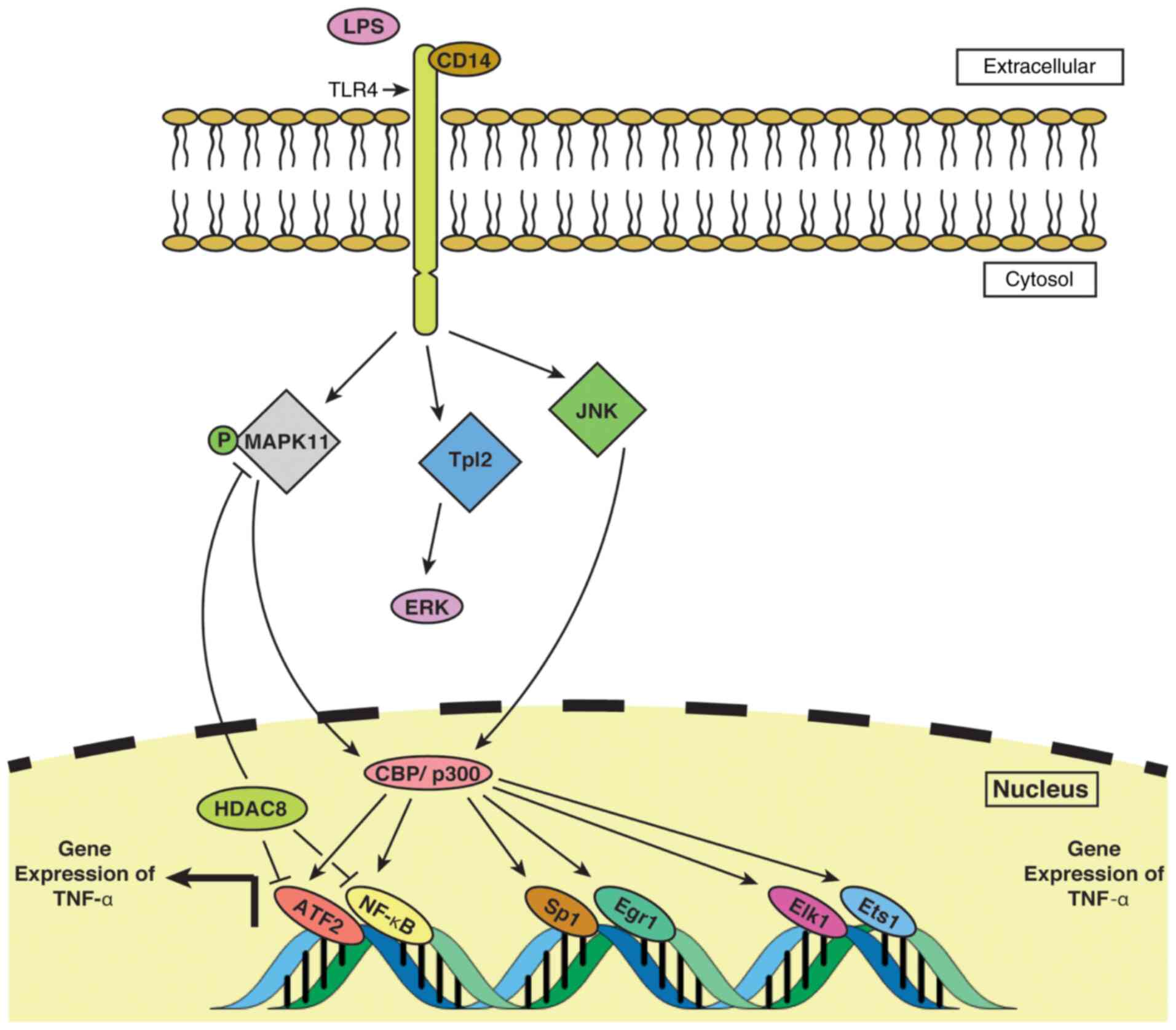 |
Figure 1
TNF-α expression is regulated by HATs/HDACs in monocytes/macrophages. LPS-mediated signal is carried to the cell by TLR4 via the CD14 protein. Signal transduces into the nucleus through i) MAPK11; ii) JNK; and iii) ERK-based pathways. The ERK pathway, mediated by Tpl2, a pro-inflammatory MAP3K8, along with the other two pathways, transmit the LPS signal to the nucleus. CBP/p300-based HAT activity induces binding of ATF2, NF-κB, Sp1, Egr1, Elk1 and Ets1 to their respective binding sites within the TNF-α promoter, which results in an increased expression of TNF-α. HDAC8, a member of the family of HDACs, blocks the binding of ATF2 and NF-κB to their respective positions within TNF-α and also inhibits phosphorylation of MAPK11, together resulting in a downregulation of TNF-α expression. HAT, histone acetyltransferase; HDAC, histone deacetylase; TLR4, Toll-like receptor 4; Tpl2, tumor progression locus 2; LPS, lipopolysaccharide.
|
IL-6
IL-6, a cytokine known primarily for promoting inflammation, helps in the modulation of immune response to injury and inflammation by promoting B cell differentiation, the activation of macrophages, thymocytes and natural killer (NK) cells, as well as by stimulating T-cell differentiation (30). Studies have observed that IL-6 signaling through classical pathway occurs by involving transmembrane IL-6 receptor (IL-6R) that regulate the anti-inflammatory response and are typically characterized by the regeneration of epithelial cells in the intestine and repression of epithelial apoptosis (31,32).
The expression of IL-6, which has been implicated in the pathogenesis of paraquat (1,1'-dimethyl-4, 4'-bipyridinium)-induced pulmonary fibrosis, has been observed to be stimulated by the HAT activity of p300 protein, along with the detection of acetylated lysine residues on histones (H3K9ac) at the IL-6 promoter in the presence of HDACi. Moreover, a reduction in IL-6 transcription by the CRISPR-ON transcription activation system in the presence of a HAT inhibitor and increased transcription in presence of an HDACi has been observed (33). These findings, taken together, provide evidence for a HAT/HDAC-mediated epigenetic regulation of IL-6 expression. Kruppel-like factor 4 (KLF4), a zinc-finger transcription factor, activated by p300/CREB-mediated acetylation (34) is involved in regulating the expression of IL-6 by binding with the IL-6 promoter at specific sites (35). Their study also reported that KLF4-deficient cells exhibited a decrease in acetylation, which led to the conclusion that this transcription factor regulates IL-6 expression by chromatin remodeling at the IL-6 promoter (35).
The overexpression of IL-6 has been detected in osteoarthritis-affected human synovial fibroblasts stimulated by CCN4 (also known as WNT-inducible signaling pathway protein-1; WSP-1) (36). Another study by Yang et al (37) reported the increased expression of IL-6 in synovial fibroblasts from osteoarthritis patients. An investigation into the epigenetic mechanisms involved in the regulation of IL-6 in osteoarthritis has uncovered some interesting observations. The same study reported the presence of hyperacetylated histones (H3K9/K14 and H4K12) in the IL-6 promoter region in osteoarthritis synovial fibroblasts compared to normal fibroblasts (37). This finding, coupled with the observation that the treatment of the synovial fibroblasts with anacardic acid, a common HAT inhibitor, led to an inhibition of histone acetylation and a decrease in binding of HAT1 and CREB binding protein (CBP) to IL-6 promoter binding sites (37), provide evidence for the involvement of HATs in modulating IL-6 expression in human synovial fibroblasts of an osteoarthritic model.
In contrast to other findings that demonstrate the upregulation of IL-6 expression by HDAC inhibition, Makki and Haqqi (38) reported the downregulation of IL-6 expression in IL-1β-treated chondrocytes from osteoarthritis-affected individuals by suberoylanilide hydroxamic acid (SAHA, a pan-HDAC inhibitor). The study proposed that HDACi suppressed IL-6 expression by inducing the expression of monocyte chemotactic protein-induced protein-1 (MCPIP1), which is a negative regulator of IL-6 transcription (38,39). This was achieved through the enhanced interaction of CCAAT/enhancer-binding protein α (CEBPα) transcription factor with the MCPIP1 promoter and through the attenuation of the inhibitory effect of miR-9 on MCPIP1 expression (38). The controversial results for the regulation of IL-6 expression is context-dependent and in the majority of the cases, HDAC inhibition results in the induction of positive regulators of IL-6 transcription, whereas in this particular case, IL-6 transcription is downregulated due to the induction of a negative regulator as discussed above (38,39). Moreover, HDACis have been shown to inhibit IL-6R expression and IL-6 production in LPS-treated macrophages in murine models of experimental colitis (40). This has been shown to be accompanied by a lowered count of Th17 cells, owing to the altered polarization of CD4+ cells downstream of IL-6R. The reduced expression of IL-6R resulted in decreased signal transducer and activator of transcription (STAT)3 phosphorylation and the downregulation of RAR-related orphan receptor γ (RORγT), a transcription factor essential for polarization into Th17 cells (40). A diagrammatic representation of the putative HAT/HDAC-mediated molecular underpinnings of IL-6 expression is presented in Fig. 2.
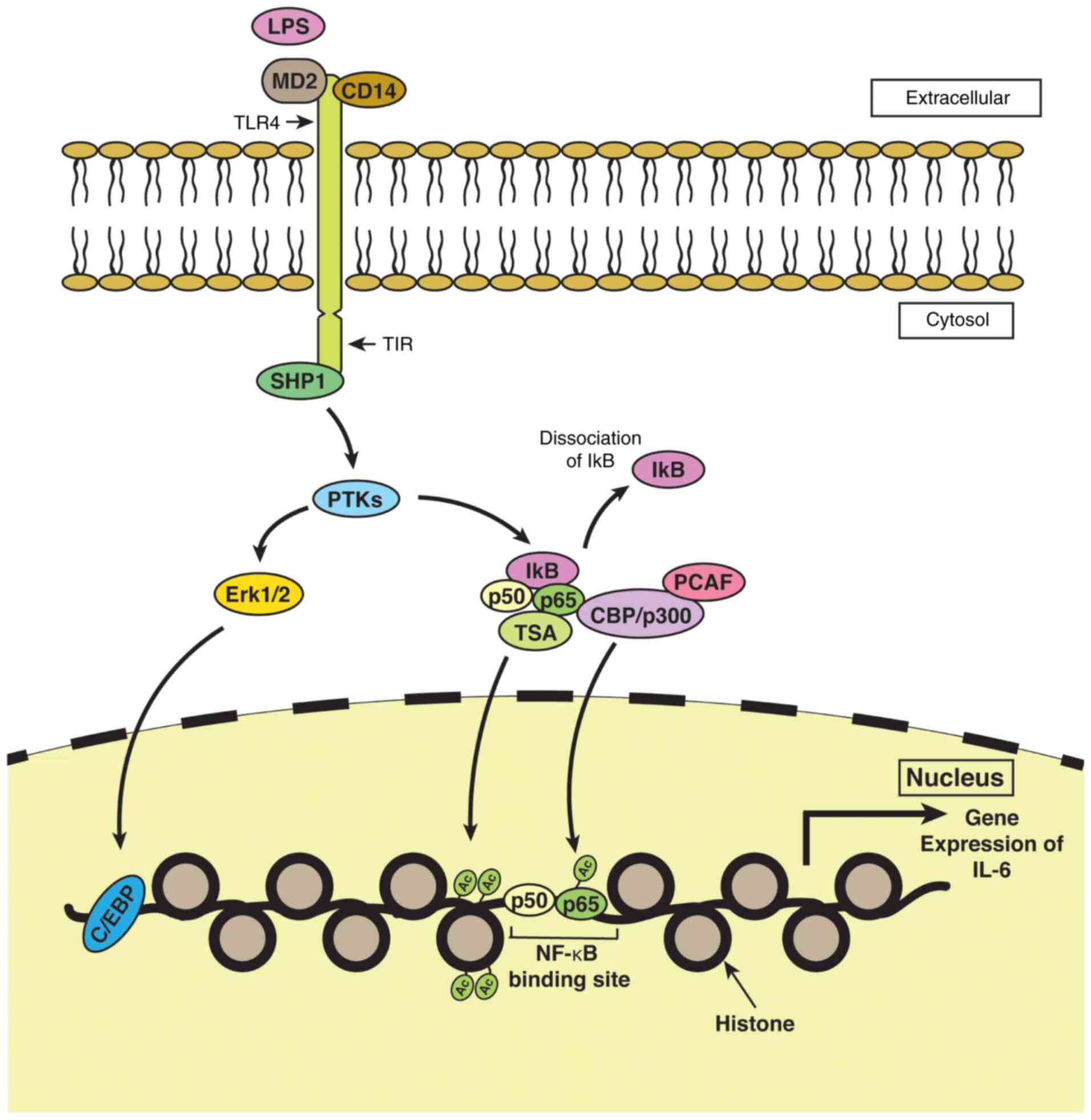 |
Figure 2
Epigenetic regulation of IL-6 expression in monocytes/macrophages. MD2 and CD14 proteins help to relay LPS-induced signals to TLR4 located within the plasma membrane of monocytes/macrophages. i) SHP1 binds to TIR, the cytoplasmic domain of TLR4, and exhibits PTK activity. The phosphorylation of Erk1/2 by PTKs helps in transcriptional co-activation of the IL-6 gene by C/EBP. ii) PTKs induce the phosphorylation and subsequent dissociation of IκB from p50 and p65 subunits of NF-κB, facilitated by CBP/p300 bound to PCAF and/or by TSA. The binding of acetylated p50 and p65 to NF-κB binding sites within the DNA, besides acetylation of histones within the nucleus by the action of TSA, promotes the expression of the IL-6 gene. MD2, myeloid differentiation factor 2; CD14, cluster of differentiation 14; LPS, lipopolysaccharide; TLR4, Toll-like receptor 4; PTK, phosphotyrosine kinase.
|
NF-κB
NF-κB is a eukaryotic transcription factor that plays a central role in regulating inflammatory responses to injury or microbial infection through the recruitment of macrophages and phagocytic leukocytes. Studies have documented the connection between the onset of NF-κB activation and inflammation-associated metabolic disorders, such as atherosclerosis and rheumatoid arthritis (41). An understanding of the key regulatory networks that control NF-κB expression and activity is essential to comprehending the mechanisms through which this transcription factor functions in the pathogenesis of different disease states.
The acetylation of the RelA (also known as p65) subunit of NF-κB, involved in NF-κB heterodimer formation and its activation, has been shown to occur at lysine residues 218, 221 and 310 by the HAT activity of p300/CREB-binding protein (CBP) in vivo (42). Chen et al (42) further reported that the acetylation of lysine 221 was crucial for the DNA binding of homodimeric RelA and mutation in the lysine 221 residue resulted in the significant attenuation of NF-κB binding with the κB enhancer. It was also observed that the acetylation of lysine 221 coupled with lysine 218 upregulated NF-κB expression by weakening the NF-κB-IκBα assembly (42). In addition, acetylated lysine 310 is known to be an important player in NF-κB transcription, wherein Brd4, a member of the bromodomain and extra-terminal domain (BET) family of proteins acts as a co-activator by binding to RelA following TNF-α stimulation (43). In addition, P300/CBP-associated factor (PCAF) along with p300, acetylates lysine 122 and 123 on the p50 subunit of NF-κB (44). Furia et al (45) demonstrated the acetylation of the NF-κB-p50 subunit alone or in association with p65 by p300/CBP, under the influence of the HIV-1 Tat protein. The acetylation of lysine 410 and 413 residues within DNA binding sites of the STAT1 transcription factor has been found to be crucial for the STAT1-mediated transcription of NF-κB p65(46).
Apart from HATs, the expression of NF-κB is profoundly influenced by HDAC activity, a putative mechanistic model of which is depicted in Fig. 3. Members of the HDAC family, namely HDAC1(47), HDAC2(47) and HDAC3(48) play important roles in modulating NF-κB levels. Ashburner et al (47) suggested that HDAC1 attenuated TNF-α-induced NF-κB expression via its interaction with the p65 subunit of NF-κB, while HDAC2 exerted the same effect, although indirectly through binding with HDAC1. Another study by Leus et al (48) indicated that in RAW 264.7 murine macrophages stimulated with LPS/IFN-γ, treatment with the HDAC3-specific inhibitor, RGFP966, led to the decreased transcription of p65, without an alteration in the acetylation of p65. It was previously reported that resveratrol (a SIRT1 activator), suppressed the transcriptional activity of the cIAP-2 gene, a crucial regulator of NF-κB expression upon induction by TNF-α (49), by binding to its promoter (49). The study further mentioned that the interaction of SIRT1 deacetylase with the cIAP-2 promoter was accompanied by a loss of acetylated histone (H3K14) marks in cells treated with TNF-α and resveratrol (49,50). Moreover, a reduction in the level of p300 protein, which can acetylate lysine 310 residue on RelA protein, coupled with a loss of acetylation at RelA lysine 310 upon treatment with resveratrol, along with the other results, indicated that SIRT1 protein played a role in controlling the expression of NF-κB (50). SIRT6, another NAD+-dependent deacetylase, which is crucial for maintaining the hypoacetylated chromatin profile at telomeres through the removal of histone 3 lysine 9 (H3K9) mark (51), has been implicated in the repression of genes regulated by NF-κB activity by binding to their respective promoters, following TNF-α treatment (52). It has also been demonstrated that SIRT6 occupies RelA-binding sites in the promoters of these genes and that it plays a role in the deacetylation of nucleosomes, leading to the decreased affinity of RelA for the nucleosomes, thus implying that SIRT6 is a potent regulator of NF-κB transcription by modulation of the chromatin state (52).
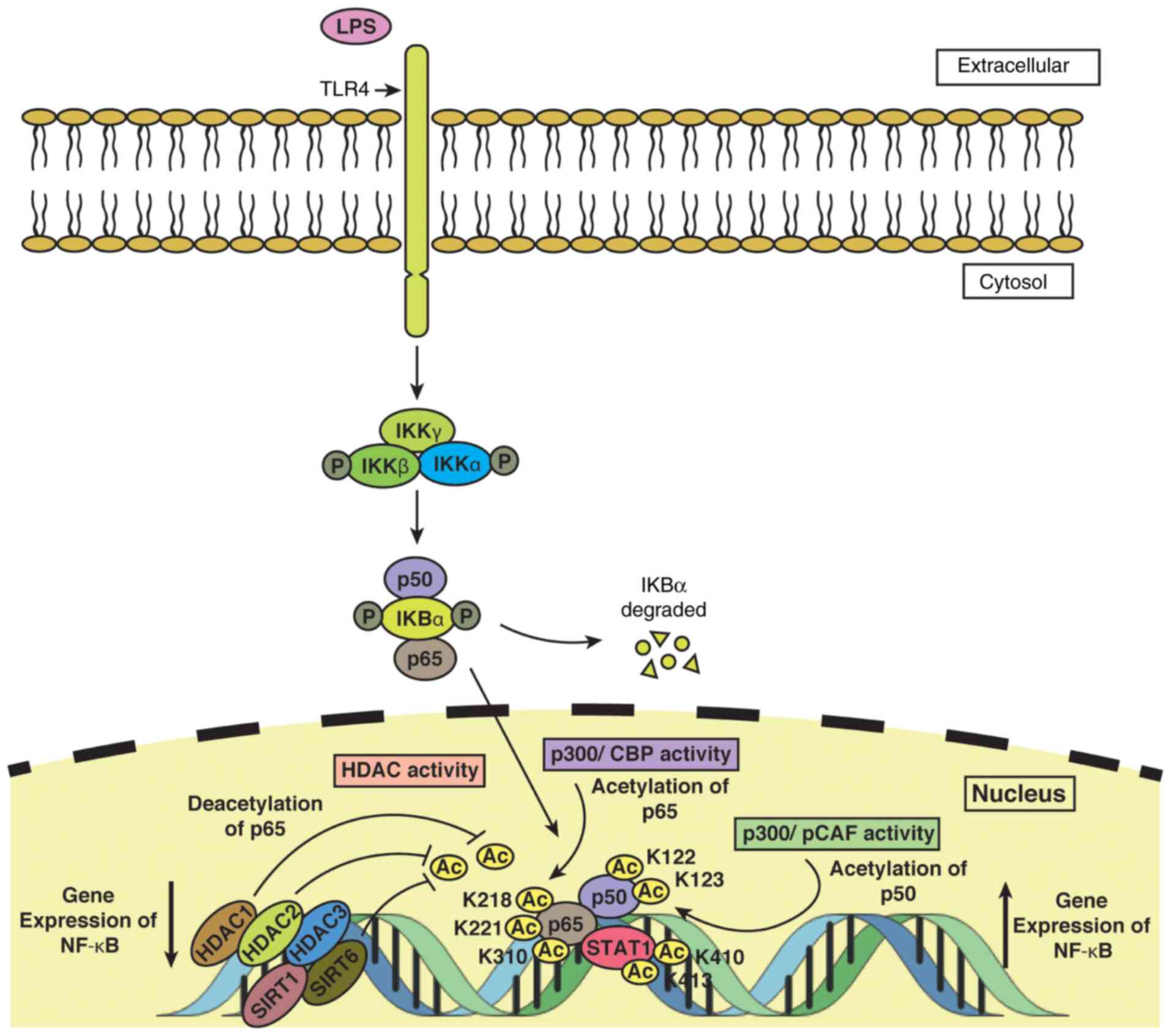 |
Figure 3
Epigenetic regulation of NF-κB in monocytes/macrophages. TLR4 located within the monocyte/macrophage plasma membrane transmits the LPS-mediated signal to the cell. The association of IKBα with the p50 and p65 subunits of NF-κB and the phosphorylation of IKBα leads to degradation of IKBα. i) Acetylation of p65 is induced by p300/CBP activity at lysine residues 218, 221, and 310 and of p50 by p300/PCAF activity at lysine residues 122 and 123. STAT1 acetylation at lysine residues 410 and 413, coupled with acetylation of p50 and p65, triggers expression of NF-κB. ii) Deacetylation of p65 is induced by HDAC1 and by HDAC2 by indirectly binding to HDAC1, thus leading to downregulation in NF-κB expression. HDAC3, besides SIRT1 and SIRT6, are among other HDACs that help to reduce the transcriptional level of NF-κB. TLR4, Toll-like receptor 4; LPS, lipopolysaccharide; HDAC, histone deacetylase; PCAF, P300/CBP-associated factor.
|
A detailed investigation into the regulatory mechanisms of some of these genes involved in inflammation, namely TNF-α, IL-6, IFN-γ, TGF-β, etc. may help to identify potent signaling pathways that maybe manipulated to treat inflammatory disorders and may thus aid in the design of novel therapeutic strategies and/or drugs.
3. Role of HAT and HDAC in regulating anti-inflammatory gene expression
Macrophages act as the first line of defense in organisms against pathogens and have a distinctive capability to change their phenotype based on cytokine microenvironment. During alternative activation, macrophages shift to an M2 phenotype following activation with TH2 cytokines (IL-4/IL-13) (53). Alternatively activated macrophages play a critical role in the protection of the host during any damage by decreasing/suppressing inflammatory response and promoting tissue repair and wound healing. A number of genes involved in inflammatory resolution are overexpressed by these cytokines in monocytes, such as 15-LOX-1, fibronectin (FN), MAO-A, coagulation factor XIII (FXIII), heat shock protein (HSP8), etc. (54). Among these, 15-LOX-1, MAO-A, scavenger receptor CD36, FN and FXIII genes with potential anti-inflammatory properties are highly upregulated during alternative activation (54). Since HATs and HDACs plays a significant role in the epigenetic regulation of several genes and are also involved in progression of a number of diseases, such as cancer and neurodegenerative disorders, the present review article mainly focuses on 15-LOX-1 and MAO-A gene expression, and their epigenetic regulation by HATs and HDACs.
15-LOX-1, the lipid peroxidative enzyme
The human ALOX15 gene has two isoenzymes; Reticulocyte-type (15-LOX-1) and epidermis-type 15-lipoxygenase-2 (15-LOX-2) (55). The reticulocyte-type 15-lipoxygenase (15-LOX-1) is an enzyme that peroxidises lipids and acts on various free/esterified polyunsaturated fatty acids specifically linoleic acid and arachidonic acid and converts them to their hydroperoxy derivatives (56). This enzyme is known to be involved in various physiological processes, such as membrane remodeling, cell differentiation, inflammation and apoptosis (57). The deregulated expression of this gene may be involved in various types of cancers and inflammation-related diseases. The elevation in the expression of the 15-LOX-1 gene may lead to oxidative stress and membrane degradation. Thus, this gene is expressed only in specific human cells and its expression is highly controlled (57).
The ALOX15 gene is present on chromosome 17, p13.3 locus and consists of 14 exons. This enzyme is present in the cytosol and maybe associated with some organelles, such as mitochondrial membranes and endoplasmic reticulum (58). Previous research has revealed the downregulation of the 15-LOX-1 gene in a number of types of cancer, such as colon, esophageal, breast, pancreatic and lung cancer (59).
i) Regulation of 15-LOX-1 expression by Th2 cytokines. It has already been well-established, that IL-4/IL-13 induces 15-LOX-1 expression through STAT6 in eosinophils, mast cells, dendritic cells, reticulocytes, activated monocytes and bronchial epithelial cells (60). In human monocytes, 15-LOX-1 induction by IL-13 requires the activation of JAK2 and TYK2 tyrosine kinases along with STAT6 dimerization and nuclear import (61). Additionally, the phosphorylation of STAT1 and STAT3 by p38 MAPK activation has been demonstrated to be significant for IL-13-induced 15-LOX-1 expression in primary human monocytes (16).
ii) Regulation of 15-LOX-1 expression by HAT/HDAC activity. In a number of types of cancer, 15-LOX-1 is transcriptionally repressed and treatment with HDACis reactivates its induction and restores apoptosis by inhibiting tumor cell growth through its metabolites, 13-S-HODE and 15-HETE. Non-steroidal anti-inflammatory drugs (NSAIDs) and other non-specific HDACis [e.g., sodium butyrate and suberoylanilide hydroxamic acid (SAHA)] increased 15-LOX-1 expression in colorectal cancer cells (62). In addition, SAHA has been shown to induce 15-LOX-1 gene in various colorectal cancer cells, such as Caco 2, SW 480 and HCT 116(63). Sodium butyrate, an HDACi, has also been reported to induce 15-LOX-1 expression in A549 cells (64). Similar results were observed in the study by Kamitani et al (65), where they demonstrated an upregulation in 15-LOX-1 expression in colorectal cancer cells treated with sodium butyrate and that this upregulation was linked to the state of histone acetylation (65). This observation suggests that deacetylation processes are more prevalent than acetylation in resting cancer cells, leading to the suppression of 15-LOX-1 gene transcription.
A recent study by Ho et al (66) elucidated that HDAC and HAT inhibitors act as epigenetic regulators of 15-LOX-1 expression in SH-SY5Y human neuroblastoma cells. Treatment with HDAC inhibitors, such as trichostatin A (TSA) or sodium butyrate significantly enhanced 15-LOX-1 expression at the mRNA level in SH-SY5Y cells. The results were consistent when they used specific class I HDAC inhibitors, such as MS-275 and depsipeptide, indicating a role of class I HDACs such as HDACs 1, 2, 3 and 8 in regulating 15-LOX-1 expression in these cells (66). However, treatment with other HDACis, such as TMP 195 (class IIa-specific HDACi) and tubacin (class IIb HDAC6 inhibitor) did not exert any effect on 15-LOX-1 mRNA expression in these cells, suggesting that they were not involved in 15-LOX-1 gene expression (66). They also demonstrated the involvement of p300 HAT and perhaps Tip60 HAT, but not GCN5 HAT in increasing 15-LOX-1 expression at the mRNA level in SH-SY5Y cells (66). In eukaryotes, HATs and HDACs play a significant role in several aspects of cellular homeostasis and also regulate the expression of different genes.
Transcription factors, such as STAT6, NF-1, GATA, activator protein 2 (AP-2) and p-1 have binding sites at the 5'-flanking region of the 15-LOX-1 gene promoter (66). p53, GATA1 and E2F1 are among few transcription factors which are acetylated outside their DNA binding site, resulting in enhanced DNA binding. It has also been demonstrated that STAT6 interacts with CREB-binding protein and an associated protein, named p300 exhibiting HAT activity, thus acetylating STAT6(64). 15-LOX-1 gene is an alluring molecular target, and its re-expression/reactivation by modulating the acetylation and deacetylation of certain histones, DNA, or proteins in cancer cells can inhibit tumorigenesis. These results suggest the involvement of both HAT and HDAC in 15-LOX-1 gene expression; however, the mechanisms involved are not yet well understood. Based on previous studies, the mechanisms involved in 15-LOX-1 expression involving HATs and HDACs are discussed below.
iii) Role of transcription factor STAT6. The study by Shankaranarayanan et al (64) demonstrated that STAT6 acetylation (in addition to phosphorylation) was required for 15-LOX-1 gene transcription in IL-4-treated A549 cells. IL-4 treatment increased the activity of cellular acetyltransferases, particularly of CBP/p300, which is responsible for the acetylation of nuclear histones H3, as well as the transcription factor STAT6, leading to 15-LOX-1 gene transcription (64). In another study, it was demonstrated that SAHA, an HDACi, induced 15-LOX-1 transcription in colorectal cancer cells (63), whereas in another study, it was demonstrated that it decreased the expression of STAT6 and phospho-STAT6 in cutaneous T-cell lymphoma cell lines (67).
This observation raises a question about the contribution of STAT6 in 15-LOX-1 gene transcription, as SAHA decreases STAT6 expression, whereas it induces 15-LOX-1 expression. However, as demonstrated in the study by Zuo et al (68), STAT-6 was necessary for IL-4-induced but not for depsipeptide (HDACi)-induced 15-LOX-1 transcriptional activation in colon cancer cell lines, suggesting that IL-4 and HDACis function via different pathways. It was also demonstrated that the activation of 15-LOX-1 transcription by depsipeptide occurred much earlier than that by IL-4 in A549 lung cancer cells (68).
iv) Chromatin remodeling (histone acetylation and demethylation by HDACi). HDAC inhibitors, such as SAHA or MS-275 have been reported to increase acetylation of specific sites on the core of H3 and H4 histones (69). Depsipeptide, (a specific HDAC1 and HDAC2 inhibitor), activates 15-LOX-1 transcription by inducing 15-LOX-1 promoter chromatin remodeling, that is, histone H3 and H4 acetylation by KAT3B (p300). As previously demonstrated, the knockdown of p300 (histone acetyltransferase) by siRNA attenuated H3 and H4 acetylation in the 15-LOX-1 promoter and thus suppressed the activation of 15-LOX-1 expression by depsipeptide (68). Furthermore, the binding of KDM3A (a demethylase of H3K9me2) with 15-LOX-1 promoter was enhanced significantly by depsipeptide, leading to the activation of 15-LOX-1 transcription in Caco-2 cells (68). Thus, both the acetylation, as well as methylation of histones, which are independent of STAT6, act as critical epigenetic regulators for 15-LOX-1 transcription.
v) Role of transcription factor GATA-6. GATA is a family of six (1-6) transcriptional regulation proteins with significant contributions in controlling cellular differentiation during vertebrate embryogenesis; GATA expression is enhanced at the time of proliferation and is reduced during the differentiation of intestinal cells (69). The expression of GATA-6 is high in a number of cancer cell lines, such as gastric, colon and lung cancer cells (70-73). The presence of a GATA binding site at -240 of the 5'-flanking region in the 15-LOX-1 promoter of Caco-2 cells was confirmed by Kamitani et al (74). A high expression of GATA-6 and its binding to the GATA site was detected in Caco-2 cells, while it was not present in cells treated with NaBT (a HDACi), indicating that the decreased expression of GATA-6 was very likely linked with the regulation of 15-LOX-1 expression and GATA-6 probably acted as the negative regulator (repressor) for the 15-LOX-1 gene (74). The downregulation of GATA-6 during the transcriptional activation of 15-LOX-1 by sodium butyrate and NSAIDs emphasizes the fact that GATA-6 functions as a transcriptional repressor by binding at the 15-LOX-1 promoter. Furthermore, it was found that the downregulation of specific GATA-6 through a siRNA approach inhibited its binding to the 15-LOX-1 promoter, but could not induce 15-LOX-1 transcription (72). These observations indicate that the downregulation of GATA-6 alone is not sufficient for the induction of the 15-LOX-1 gene; however, it does augment the potential of HDACi and NSAIDs to express 15-LOX-1(72).
Based on these studies, it can be concluded that the transcriptional repression of 15-LOX-1 in cancer cells is multi-factorial and does not solely depend on GATA-6. Most probably, GATA-6 contributes to the transcriptional silencing of 15-LOX-1 by inhibiting the binding of activator proteins, indicating that there may be a possibility of some uncharacterized proteins of the GATA family or related proteins, that bind to the GATA site and induce the 15-LOX-1 transcription.
vi) Recruitment of the nucleosome remodeling and deacetylase (NURD) complex. The involvement of specific HDACS (HDAC1 and HDAC2) in 15-LOX-1 gene upregulation has been shown by using inhibitors and siRNA knockdown experiments. However, HDAC3 (a member of class1 HDAC) does not have a significant effect on 15-LOX-1 gene regulation. These outcomes indicate a mechanistic link between HDAC1 and HDAC2 up-regulation and 15-LOX-1 downregulation (75).
Since HDACs do not bind with DNA directly and can be recruited to promoters as a part of transcription repression complex, the same group of researchers further proposed the involvement of NuRD, a repression complex recruited to the 15-LOX-1 promoter region between -215 and -283 bp, leading to 15-LOX-1 transcriptional repression in colon cancer cells; HDACis can activate gene transcription via NuRD dissociation from a promoter (75). HDAC1, HDAC2, Mi2, metastasis-associated proteins (MTA1 and MTA2) are the key components of this complex, and all MTA family members consists of a GATA zinc finger DNA binding domain (76). In various human cancers, such as colon cancer, MTA1 expression is increased at the mRNA level (77), and promotes lymphoma and breast tumorigenesis in the mouse model (76). An upregulation in 15-LOX-1 expression was observed when the essential components of the NuRD complex, such as HDAC1, HDAC2 and MTA1 were downregulated via the siRNA approach (75). These studies led to the conclusion that HDAC1 and HDAC2, together with MTA1, act as part of the NuRD complex and suppress 15-LOX-1 transcription, contributing to colonic tumorigenesis.
vii) Role of DNA methylation on 15-LOX-1 gene expression. It is well known that there is anomalous promoter methylation during tumorigenesis, leading to the silencing of several tumor suppressor genes. The 15-LOX-1 gene promoter has CpG islands which are methylated in various types of cancer, such as lung, epidermoid, cervical, lymphoma and prostate cancer (60,78). Thus, to establish an association between DNA methylation and 15-LOX-1 transcription, Liu et al (60) demonstrated that the disruption of DNA methylation by 5-aza-2-deoxycytidine (5-aza-dc), a DNA methyltransferase (DNMT) inhibitor, facilitated histone acetylation at the 15-LOX-1 promoter by TSA or IL-4, leading to a significant increase in 15-LOX-1 expression in L428 lymphoma cells. However, the hypomethylating agent, 5-aza-dc, has been shown to induce 15-LOX-1 expression in colon cancer cells (79); these findings are contradictory to those of the study by Kamitani et al (65), who demonstrated that 5-aza-dc could not activate 15-LOX-1 transcription in the same cell lines.
In another study, it was shown that 15-LOX-1 expression was not associated with 15-LOX-1 promoter DNA demethylation and thus, promoter demethylation by DNMT1 and DNMT3b double knockout did not restore 15-LOX-1 expression in colorectal cancer cell lines (80). However, the dissociation of DNMT1 by a HDACi (SAHA) from the 15-LOX-1 promoter was essential for 15-LOX-1 induction (80). It can thus be concluded that 15-LOX-1 promoter methylation does not play a predominant role in the silencing of the gene. DNMT1 however, seems to play a significant role as a co-repressor of 15-LOX-1 transcription, as it may bind with the same region in the 15-LOX-1 promoter, where HDAC1 and HDAC2 are recruited as components of the NURD complex (80,81).
MAO-A, a pro-oxidative enzyme is involved in several pathological disorders
MAO is a flavoenzyme, present on the outer membrane of the mitochondria, responsible for catalyzing the oxidative deamination of biogenic and dietary amines in the brain and peripheral tissues, changing them to their corresponding aldehydes and generating hydrogen peroxide (H2O2) as the by-product (82,83). MAO exists in two isoforms, MAO-A and MAO-B, both of them present on the X-chromosome, and have specificities for substrate and inhibitors (82).
MAO-A is overexpressed in alternatively activated monocytes/macrophages and the enzyme has been employed in the breakdown of certain neurotransmitters, such as epinephrine, norepinephrine, serotonin and dopamine (84). It has also been reported that MAO-A is involved in reactive oxygen species (ROS) generation in alternatively activated monocytes/macrophages (84). The high production of ROS has been implicated in the pathogenesis of several human diseases involving cancer, as well neurodegenerative, cardiovascular, inflammation- and ageing-related disorders (85-87). The downregulation of MAO-A has been observed in various types of cancer cells, such as cholangiocarcinoma, esophageal squamous cell carcinoma and hepatocellular carcinoma (88,89). In addition, a previous study demonstrated that MAO-A expression was suppressed in hepatocellular carcinoma via epigenetic modulations, such as methylation and histone acetylation (89). Of note, the overexpression of MAO-A has been observed in certain types of cancer, such as renal cell carcinoma and prostate cancer, suggesting that the regulatory pathway and role of MAO-A differs in various cancer types (90-92).
i) Role of HATs and HDACs in MAO-A expression. In a previous study, valproic acid (VPA), an HDAC inhibitor, was shown to play a prominent role in the regulation of chromatin remodeling and gene expression, and activated MAO-A gene expression via the activation of AKT/FOXO1 signaling pathway in the human neuroblastoma BE (2) C cell line (93). VPA demonstrated the ability to activate selective signaling pathways, such as PI3K/AKT and some other extracellular kinase pathways. Cells pre-treated with LY294002, a potent PI3K inhibitor, exhibited a deceased induction of MAO-A by VPA indicating a regulatory role of AKT. The induction of MAO-A by VPA was also attenuated by the knockdown of endogenous FOXO1 by the corresponding siRNA (93).
In addition to PI3K and AKT, VPA treatment was previously shown to increase the association of Sp1 (a pivotal transcriptional activator of MAO-A) with the histone acetyltransferase p300(94). It was also found that the catalytic and promoter activities of MAO-A were increased by 2-fold by both sodium butyrate and TSA (HDAC inhibitors) in human neuroblastoma BE (2) C cell lines, which supports the hypothesis that the induction of MAO-A by VPA was possibly due to modulation of acetylation status of MAO-A promoter (93).
As an upregulation of MAO-A gene expression by HDAC inhibition was observed in the human neuroblastoma BE (2)C cell line, it can be hypothesized that the treatment of A549 lung carcinoma cells with the HDAC inhibitor SAHA or transfection of A549 cells with HDAC esiRNA will enhance the expression of the MAO-A gene (Fig. 4). However, further experiments are required to confirm the involvement of HDAC in MAO-A gene expression.
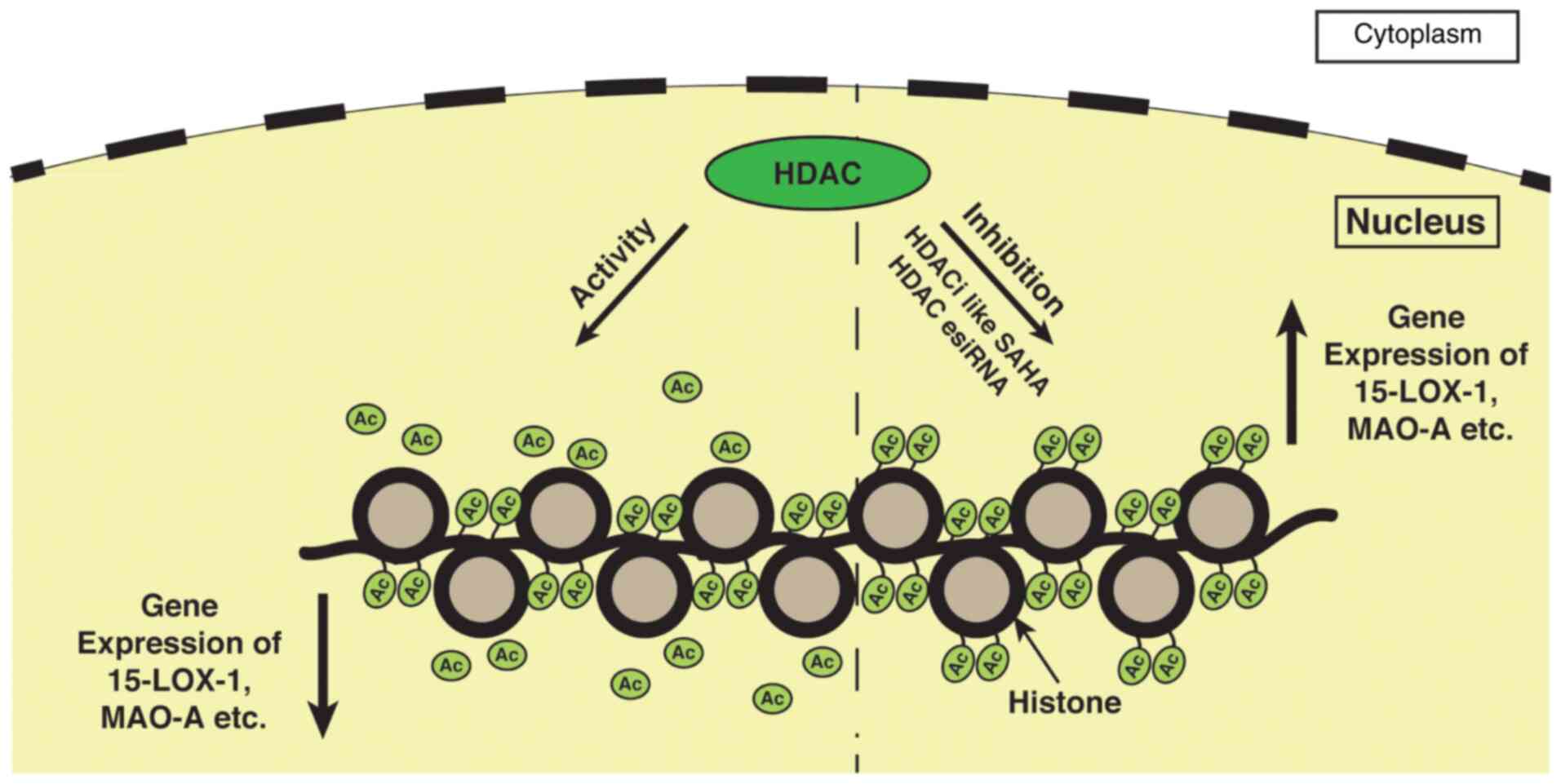 |
Figure 4
Inhibition of HDAC activity facilitates various gene transcription such as 15-lipoxygenase, MAO-A, etc. in cancer cells. During the resting stage, HDACs have an increased expression in many types of cancers, including ovarian, breast, bladder, and other cancers. Thus, in this hypothetical model, it can be deduced that the increased HDAC activity removes acetyl groups from histones which are hyperacetylated initially and suppress gene transcription in cancer cells. However, whenever the HDAC activity is inhibited by using HDACis, such as SAHA the histone again becomes hyperacetylated leading to the expression of these genes. Similar to SAHA, HDAC esiRNA has a similar effect on the expression of these genes, showing its involvement in 15-LOX-1 and MAO-A gene transcription. HDAC, histone deacetylase; HDACis, histone deacetylase inhibitors; 15-LOX-1, 15-lipoxygenase-1; MAO-A, monoamine oxidase-A; SAHA, suberoylanilide hydroxamic acid.
|
ii) Role of STAT3 acetylation. MAO-A is expressed through the JAK-STAT pathway involving STAT3 as one of the major components in monocytes and A549 lung cancer cells (17). It has been reported that the acetylation status of STAT3 has a marked impact on the epigenetic regulation of cancer cells (95). Hence, it can be hypothesized at this stage that there may be a link between STAT3 acetylation and 15-LOX-1 and MAO-A gene expression in monocytes and A549 lung cancer cells. However, since there is a co-induction of 15-LOX-1 and MAO-A gene expression following IL-13 stimulation in A549 cell lines, a decreased 15-LOX-1 and MAO-A gene expression may occur, as well following the downregulation of HAT activity by HDACi treatment. A plausible diagrammatic representation of the JAK/STAT signaling pathway, where the acetylation of STATs leads to the enhanced expression of genes, such as MAO-A, 15-LOX-1, etc. is presented in Fig. 5.
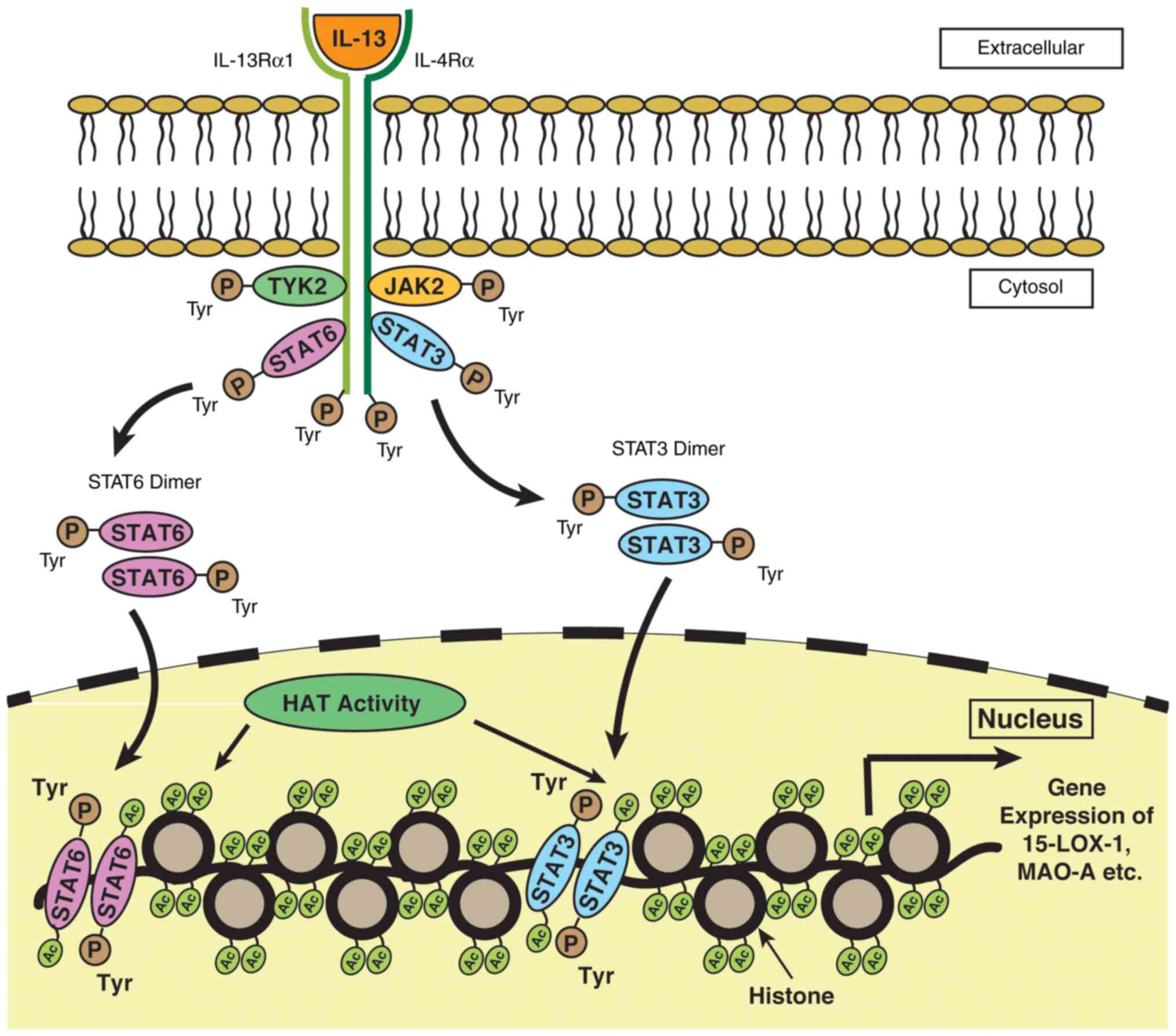 |
Figure 5
A plausible signaling pathway depicting the role of STAT3 acetylation on MAO-A and 15-LOX-1 gene expression in monocytes and A549 lung cancer cells. The binding of IL-13 with the receptor leads to the activation of a number of signaling proteins, including JAK kinases such as JAK2 and TYK2 and transcription factors such as STATs. The activation of JAK phosphorylates the intracellular domains of the receptor, resulting in the phosphorylation of STATs. Following phosphorylation, STAT3 and STAT6 become dimerized and translocate to the nucleus, where they are acetylated by HAT. Acetylated STATs bind to the specific binding site present on the DNA facilitating 15-LOX-1 and MAO-A gene transcription. STAT, signal transducer and activator of transcription; MAO-A, monoamine oxidase-A; 15-LOX-1, 15-lipoxygenase-1.
|
4. Conclusion and future perspectives
The human immune system can play significant pro- and anti-tumorigenic roles at all stages of tumorigenesis, and it has become evident that an inflammatory microenvironment is an essential component of almost all tumors. Histone acetylation and deacetylation are epigenetic modulators of inflammatory gene expression, which are instrumental in controlling immune responses that the body exhibits against cancer and autoimmune disorders (96). The present review discussed the roles of HATs and HDACs in regulating the expression of key pro-inflammatory genes, namely TNF-α, IL-6 and NF-κB, along with anti-inflammatory genes, such as 15-LOX-1 and MAO-A.
The tumor-inducing potential of TNF-α has been well-elucidated in the study by Wu and Zhou (97), wherein it was suggested that this cytokine played crucial role to play in tumor initiation, cell proliferation, and survival as well as tumor angiogenesis. NF-κB activation was reported to be pivotal for TNF-α mediated cell proliferation (97). The experimental findings by Dobreva et al (98) provide evidence of the decreased TNF-α production in SAHA (HDAC inhibitor)-treated human PBMCs following administration with LPS. Taken together, these reports suggest that the inhibition of HDAC activity may hold great therapeutic potential in combating cancer. Moreover, the role of IL-6 in cancer metastasis via the induction of STAT3 phosphorylation has been previously studied (99). In alternatively activated macrophages and A549 cells, the decrease in acetylation at the IL-6 promoter region has been reported to have led to a decline in IL-6 transcription (100). These results, coupled with the findings of the study by Zhao et al (99), indicate that the modulation of IL-6 activity may be targeted to treat certain types of cancer like lung adenocarcinoma.
Anti-inflammatory genes are found to be suppressed in a number of cancer types. Thus, the re-expression of these genes by manipulating HATs or HDACs activity/expression may be a novel approach for combating cancer. The present review article aimed to discuss the mechanisms through which HDACs and HATs regulate 15-LOX-1 and MAO-A gene expression. HDACis, such as SAHA, TSA, or sodium butyrate induce 15-LOX-1 gene transcription in various types of cancer, such as colorectal, lung carcinoma and neuroblastoma (63-66). Depsipeptide, a specific HDAC1 and HDAC2 inhibitor, also alters the chromatin remodeling by histone (H3 and H4) acetylation and significantly increases the recruitment of KDM3A, a demethylase of H3K9me2, to the 15-LOX-1 gene promoter leading to increased expression of 15-LOX-1 in Caco-2 cells (68). Thus, it can be concluded that both histone acetylation, as well as 15-LOX-1 promoter demethylation, are necessary requirements for its expression. Furthermore, various previous studies performed on colon cancer cells, revealed that 15-LOX-1 suppression in cancer cells was complex and seemed to involve numerous layers of repression mechanisms involving GATA-6 (72,74), DNMT-1 (80,81), the NURD complex (75) and histone methylation, specifically the H3K9me2 repression modification (68). However further studies are warranted to explore the interaction between the various repression mechanisms to get a clear picture of the pathway.
In addition to histone acetylation, the present review also discusses the acetylation of transcription factors, STAT3 and STAT6. Previously it was demonstrated that the acetylation of STAT6 by CBP/p300 in IL-4 stimulated A549 lung cancer cells played a role in 15-LOX-1 gene expression (64).
Similarly, STAT3 plays a major role in the epigenetic repression and de-repression of numerous genes. Thus, the present review aimed to elucidate its role in anti-inflammatory genes, such as 15-LOX-1 or MAO-A and hypothesized that IL-13 may induce HAT activity, leading to an increase in STAT3 acetylation which may play a role in 15-LOX-1 and MAO-A gene transcription. However further investigations are required to confirm this hypothesis.
In the human neuroblastoma BE(2)C cell line, the MAO-A gene was induced by valproic acid, a HDACi via the stimulation of the AKT/FOXO1 signaling pathway (93). Similar results were found with sodium butyrate and TSA in these cell lines (93). It was shown in a study that the induction of MAO-A by VPA was due to the increased association of Sp1, a key transcriptional activator of MAO-A with the histone acetyltransferase p300 in human neuroblastoma cells (94).
These observations and results suggest that epigenetic modifications, such as the acetylation/deacetylation of histones, DNA methylation, phosphorylation, etc. has a profound effect on the regulation of genes involved in inflammation, and alterations in the expression of these genes by epigenetic defects may lead to cancer development and progression. However, these epigenetic abnormalities can be reversed by the use of epigenetic drugs, such as HDACis or DNMT inhibitors, etc. A combination of surgery, epigenetic drugs, radiotherapy, immunotherapy, chemotherapy and targeted therapies have emerged as more effective methods for the treatment of cancer (101-103). Even combination epigenetic therapy involving two or more different types of the epigenetic drug has proven to be a promising approach in the treatment of cancer with less side-effects (104,105).
Acknowledgements
Not applicable.
Funding
The present study was funded by the research grant of Ramalingaswami fellowship awarded to Ashish Bhattacharjee by the Department of Biotechnology, Govt. of India (BT/RLF/Re‑entry/56/2011) and by the research grant to A.B. from the Ministry of Science and Engineering Research Board SERB, Govt. of India (SERB/SR/SO/HS‑093/2013).
Availability of data and materials
Not applicable.
Authors' contributions
SS and ABa contributed to the writing, editing and revising of the manuscript. ABa prepared the models for the review. ABh provided critical feedback and helped edit the manuscript to its final version and also guided the design of the models. ABh and SS confirm the authenticity of all the raw data. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Zhao Z and Shilatifard A: Epigenetic modifications of histones in cancer. Genome Biol. 20(245)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
De la Serna IL, Ohkawa Y and Imbalzano AN: Chromatin remodeling in mammalian differentiation: Lessons from ATP-dependent remodellers. Nat Rev Genet. 7:461–473. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV and Mann M: Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 325:834–840. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wapenaar H and Dekker FJ: Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin Epigenetics. 8(59)2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Barnes PJ, Adcock IM and Ito K: Histone acetylation and deacetylation: Importance in inflammatory lung diseases. Eur Respir J. 25:552–563. 2005.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gallinari P, Di Marco S, Jones P, Pallaoro M and Steinkühler C: HDACS, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 17:195–211. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sun XJ, Man N, Tan Y, Nimer SD and Wang L: The role of histone acetyltransferases in normal and malignant hematopoiesis. Front Oncol. 5(108)2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bannister AJ and Kouzarides T: Regulation of chromatin by histone modifications. Cell Res. 21:381–395. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Marmorstein R and Roth SY: Histone acetyltransferases: Function, structure, and catalysis. Curr Opin Genet Dev. 11:155–161. 2001.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM and Marsh JL: Inhibition of specific HDACS and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum Mol Genet. 17:3767–3775. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Legube G and Trouche D: Regulating histone acetyltransferases and deacetylases. EMBO Rep. 4:944–947. 2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Peserico A and Simone C: Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol. 2011(371832)2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tsaprouni LG, Ito K, Powell JJ, Adcock IM and Punchard N: Differential patterns of histone acetylation in inflammatory bowel diseases. J Inflamm (Lond). 8(1)2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Smith JA, Das A, Ray SK and Banik NL: Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 87:10–20. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Conrad DJ, Kuhn H, Mulkins M, Highland E and Sigal E: Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc Natl Acad Sci USA. 89:217–221. 1992.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Xu B, Bhattacharjee A, Roy B, Xu HM, Anthony D, Frank DA, Feldman GM and Cathcart MK: Interleukin-13 induction of 15-lipoxygenase gene expression requires p38 mitogen-activated protein kinase-mediated serine 727 phosphorylation of Stat1 and Stat3. Mol Cell Biol. 23:3918–3928. 2003.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Dhabal S, Das P, Biswas P, Kumari P, Yakubenko VP, Kundu S, Cathcart MK, Kundu M, Biswas K and Bhattacharjee A: Regulation of monoamine oxidase A (MAO-A) expression, activity, and function in IL-13-stimulated monocytes and A549 lung carcinoma cells. J Biol Chem. 293:14040–14064. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Beutler B and Brown T: A CAT reporter construct allows ultrasensitive estimation of TNF synthesis, and suggests that the TNF gene has been silenced in non-macrophage cell lines. J Clin Invest. 87:1336–1344. 1991.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Feng X: Regulatory roles and molecular signaling of TNF family members in osteoclasts. Gene. 350:1–13. 2005.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Parameswaran N and Patial S: Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. 20:87–103. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Schildberger A, Rossmanith E, Eichhorn T, Strassl K and Weber V: Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflamm. 2013(697972)2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Sullivan KE, Reddy ABM, Dietzmann K, Suriano AR, Kocieda VP, Stewart M and Bhatia M: Epigenetic regulation of tumor necrosis factor alpha. Mol Cell Biol. 27:5147–5160. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Falvo JV, Tsytsykova AV and Goldfeld AE: Transcriptional control of the TNF gene. Curr Dir Autoimmun. 11:27–60. 2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Tsai EY, Falvo JV, Tsytsykova AV, Barczak AK, Reimold AM, Glimcher LH, Fenton MJ, Gordon DC, Dunn IF and Goldfeld AE: A lipopolysaccharide-specific enhancer complex involving Ets, Elk-1, Sp1, and CREB binding protein and p300 is recruited to the tumor necrosis factor alpha promoter in vivo. Mol Cell Biol. 20:6084–6094. 2000.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Falvo JV, Jasenosky LD, Kruidenier L and Goldfeld AE: Epigenetic control of cytokine gene expression: Regulation of the TNF/LT locus and T helper cell differentiation. Adv Immunol. 118:37–128. 2013.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lee JY, Kim NA, Sanford A and Sullivan KE: Histone acetylation and chromatin conformation are regulated separately at the TNF-alpha promoter in monocytes and macrophages. J Leukoc Biol. 73:862–871. 2003.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Mahlknecht U, Will J, Varin A, Hoelzer D and Herbein G: Histone deacetylase 3, a class I histone deacetylase, suppresses MAPK11-mediated activating transcription factor-2 activation and represses TNF gene expression. J Immunol. 173:3979–3990. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Dumitru CD, Ceci JD, Tsatsanis C, Kontoyiannis D, Stamatakis K, Lin JH, Patriotis C, Jenkins NA, Copeland NG, Kollias G and Tsichlis PN: TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell. 103:1071–1083. 2000.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Senger K, Pham VC, Varfolomeev E, Hackney JA, Corzo CA, Collier J, Lau VWC, Huang Z, Hamidzhadeh K, Caplazi P, et al: The kinase TPL2 activates ERK and p38 signaling to promote neutrophilic inflammation. Sci Signal. 10(eaah4273)2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Huang M, Yang D, Xiang M and Wang J: Role of interleukin-6 in regulation of immune responses to remodeling after myocardial infarction. Heart Fail Rev. 20:25–38. 2015.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Scheller J, Chalaris A, Schmidt-Arras D and Rose-John S: The pro-and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 1813:878–888. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Schett G: Physiological effects of modulating the interleukin-6 axis. Rheumatology (Oxford). 57 (suppl_2):ii43–ii50. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hu L, Yu Y, Huang H, Fan H, Hu L, Yin C, Li K, Fulton DJ and Chen F: Epigenetic regulation of interleukin 6 by histone acetylation in macrophages and its role in paraquat-induced pulmonary fibrosis. Front Immunol. 7(696)2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Evans PM, Zhang W, Chen X, Yang J, Bhakat KK and Liu C: Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J Biol Chem. 282:33994–34002. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Rosenzweig JM, Glenn JD, Calabresi PA and Whartenby KA: KLF4 modulates expression of IL-6 in dendritic cells via both promoter activation and epigenetic modification. J Biol Chem. 288:23868–23874. 2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Hou CH, Tang CH, Hsu CJ, Hou SM and Liu JF: CCN4 induces IL-6 production through αvβ5 receptor, PI3K, Akt, and NF-κB singling pathway in human synovial fibroblasts. Arthritis Res Ther. 15(R19)2013.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yang F, Zhou S, Wang C, Huang Y, Li H, Wang Y, Zhu Z, Tang J and Yan M: Epigenetic modifications of interleukin-6 in synovial fibroblasts from osteoarthritis patients. Sci Rep. 7(43592)2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Makki MS and Haqqi TM: Histone deacetylase inhibitor vorinostat (SAHA, MK0683) perturb miR-9-MCPIP1 axis to block IL-1β-induced IL-6 expression in human OA chondrocytes. Connect Tissue Res. 58:64–75. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Jura J, Skalniak L and Koj A: Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochim Biophys Acta. 1823:1905–1913. 2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Glauben R, Sonnenberg E, Wetzel M, Mascagni P and Siegmund B: Histone deacetylase inhibitors modulate interleukin 6-dependent CD4+ T cell polarization in vitro and in vivo. J Biol Chem. 289:6142–6151. 2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Baker RG, Hayden MS and Ghosh S: NF-κB, inflammation, and metabolic disease. Cell Metab. 13:11–22. 2011.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Chen LF, Mu Y and Greene WC: Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 21:6539–6548. 2002.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Huang B, Yang XD, Zhou MM, Ozato K and Chen LF: Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol. 29:1375–1387. 2009.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Kiernan R, Brès V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, Jin DY, Emiliani S and Benkirane M: Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. 278:2758–2766. 2003.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Furia B, Deng L, Wu K, Baylor S, Kehn K, Li H, Donnelly R, Coleman T and Kashanchi F: Enhancement of nuclear factor-kappa B acetylation by coactivator p300 and HIV-1 Tat proteins. J Biol Chem. 277:4973–4980. 2002.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Krämer OH, Baus D, Knauer SK, Stein S, Jäger E, Stauber RH, Grez M, Pfitzner E and Heinzel T: Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev. 20:473–485. 2006.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ashburner BP, Westerheide SD and Baldwin AS Jr: The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 21:7065–7077. 2001.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Leus NG, van der Wouden PE, van den Bosch T, Hooghiemstra WTR, Ourailidou ME, Kistemaker LE, Bischoff R, Gosens R, Haisma HJ and Dekker FJ: HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuatiy. Biochem Pharmacol. 108:58–74. 2016.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J and Korneluk RG: Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA. 105:11778–11783. 2008.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA and Mayo MW: Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23:2369–2380. 2004.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, et al: SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 452:492–496. 2008.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, mcCord RA, Ongaigui KC, Boxer LD, Chang HY and Chua KF: SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 136:62–74. 2009.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Varin A and Gordon S: Alternative activation of macrophages: Immune function and cellular biology. Immunobiology. 214:630–641. 2009.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Chaitidis P, O'Donnell V, Kuban RJ, Bermudez-Fajardo A, Ungethuem U and Kühn H: Gene expression alterations of human peripheral blood monocytes induced by medium-term treatment with the TH2-cytokines interleukin-4 and-13. Cytokine. 30:366–377. 2005.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Schewe T: 15-lipoxygenase-1: A prooxidant enzyme. Biol Chem. 383:365–374. 2002.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Lee YW, Kuhn H, Kaiser S, Hennig B, Daugherty A and Toborek M: Interleukin 4 induces transcription of the 15-lipoxygenase I gene in human endothelial cells. J Lipid Res. 42:783–791. 2001.PubMed/NCBI
|
|
57
|
Liu C, Xu D, Han H, Fan Y, Schain F, Xu Z, Claesson HE, Björkholm M and Sjöberg J: Transcriptional regulation of 15-lipoxygenase expression by histone h3 lysine 4 methylation/demethylation. PLoS One. 7(e52703)2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Çolakoğlu M, Tunçer S and Banerjee S: Emerging cellular functions of the lipid metabolizing enzyme 15-Lipoxygenase-1. Cell Prolif. 51(e12472)2018.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Moussalli MJ, Wu Y, Zuo X, Yang XL, Wistuba II, Raso MG, Morris JS, Bowser JL, Minna JD, Lotan R and Shureiqi I: Mechanistic contribution of ubiquitous 15-lipoxygenase-1 expression loss in cancer cells to terminal cell differentiation evasion. Cancer Prev Res (Phila). 4:1961–1972. 2011.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Liu C, Xu D, Sjöberg J, Forsell P, Björkholm M and Claesson HE: Transcriptional regulation of 15-lipoxygenase expression by promoter methylation. Exp Cell Res. 297:61–67. 2004.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Roy B and Cathcart MK: Induction of 15-lipoxygenase expression by IL-13 requires tyrosine phosphorylation of Jak2 and Tyk2 in human monocytes. J Biol Chem. 273:32023–32029. 1998.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Shureiqi I, Chen D, Lee JJ, Yang P, Newman RA, Brenner DE, Lotan R, Fischer SM and Lippman SM: 15-LOX-1: A novel molecular target of nonsteroidal anti-inflammatory drug-induced apoptosis in colorectal cancer cells. J Natl Cancer Inst. 92:1136–1142. 2000.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Hsi LC, Xi X, Lotan R, Shureiqi I and Lippman SM: The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis via induction of 15-lipoxygenase-1 in colorectal cancer cells. Cancer Res. 64:8778–8781. 2004.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Shankaranarayanan P, Chaitidis P, Kühn H and Nigam S: Acetylation by histone acetyltransferase CREB-binding protein/p300 of STAT6 is required for transcriptional activation of the 15-lipoxygenase-1 gene. J Biol Chem. 276:42753–42760. 2001.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Kamitani H, Taniura S, Ikawa H, Watanabe T, Kelavkar UP and Eling TE: Expression of 15-lipoxygenase-1 is regulated by histone acetylation in human colorectal carcinoma. Carcinogenesis. 22:187–191. 2001.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Ho CF, Bon CP, Ng YK, Herr DR, Wu JS, Lin TN and Ong WY: Expression of DHA-metabolizing enzyme Alox15 is regulated by selective histone acetylation in neuroblastoma cells. Neurochem Res. 43:540–555. 2018.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Zhang C, Richon V, Ni X, Talpur R and Duvic M: Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: Relevance to mechanism of therapeutic action. J Invest Dermatol. 125:1045–1052. 2005.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Zuo X, Morris JS and Shureiqi I: Chromatin modification requirements for 15-lipoxygenase-1 transcriptional reactivation in colon cancer cells. J Biol Chem. 283:31341–31347. 2008.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Shureiqi I, Jiang W, Fischer SM, Xu X, Chen D, Lee JJ, Lotan R and Lippman SM: GATA-6 transcriptional regulation of 15-lipoxygenase-1 during NSAID-induced apoptosis in colorectal cancer cells. Cancer Res. 62:1178–1183. 2002.PubMed/NCBI
|
|
70
|
Al-azzeh ED, Fegert P, Blin N and Gött P: Transcription factor GATA-6 activates expression of gastroprotective trefoil genes TFF1 and TFF2. Biochim Biophys Acta. 1490:324–332. 2000.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Akiyama Y, Watkins N, Suzuki H, Jair KW, van Engeland M, Esteller M, Sakai H, Ren CY, Yuasa Y, Herman JG and Baylin SB: GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric cancer. Mol Cell Biol. 23:8429–8439. 2003.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Shureiqi I, Zuo X, Broaddus R, Wu Y, Guan B, Morris JS and Lippman SM: The transcription factor GATA-6 is overexpressed in vivo and contributes to silencing 15-LOX-1 in vitro in human colon cancer. FASEB J. 21:743–753. 2007.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Guo M, Akiyama Y, House GM, Hooker CM, Heath E, Gabrielson E, Yang SC, Han Y, Baylin SB, Herman JG and Brock MV: Hypermethylation of the GATA genes in lung cancer. Clin Cancer Res. 10:7917–7924. 2004.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Kamitani H, Kameda H, Kelavkar UP and Eling TE: A GATA binding site is involved in the regulation of 15-lipoxygenase-1 expression in human colorectal carcinoma cell line, caco-2. FEBS Lett. 467:341–347. 2000.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Zuo X, Morris JS, Broaddus R and Shureiqi I: 15-LOX-1 transcription suppression via the NuRD complex in colon cancer cells. Oncogene. 28:1496–1505. 2009.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Manavathi B and Kumar R: Metastasis tumor antigens, an emerging family of multifaceted master coregulators. J Biol Chem. 282:1529–1533. 2007.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Giannini R and Cavallini A: Expression analysis of a subset of coregulators and three nuclear receptors in human colorectal carcinoma. Anticancer Res. 25:4287–4292. 2005.PubMed/NCBI
|
|
78
|
Kelavkar UP, Harya NS, Hutzley J, Bacich DJ, Monzon FA, Chandran U, Dhir R and O'Keefe DS: DNA methylation paradigm shift: 15-lipoxygenase-1 upregulation in prostatic intraepithelial neoplasia and prostate cancer by atypical promoter hypermethylation. Prostaglandins Other Lipid Mediat. 82:185–197. 2007.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Hsi LC, Xi X, Wu Y and Lippman SM: The methyltransferase inhibitor 5-aza-2-deoxycytidine induces apoptosis via induction of 15-lipoxygenase-1 in colorectal cancer cells. Mol Cancer Ther. 4:1740–1746. 2005.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Zuo X, Shen L, Issa JP, Moy O, Morris JS, Lippman SM and Shureiqi I: 15-Lipoxygenase-1 transcriptional silencing by DNA methyltransferase-1 independently of DNA methylation. FASEB J. 22:1981–1992. 2008.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Fuks F, Burgers WA, Brehm A, Hughes-Davies L and Kouzarides T: DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 24:88–91. 2000.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Grimsby J, Chen K, Wang LJ, Lan NC and Shih JC: Human monoamine oxidase A and B genes exhibit identical exon-intron organization. Proc Natl Acad Sci USA. 88:3637–3641. 1991.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Zhang J and Piantadosi C: Prevention of H2O2, generation by monoamine oxidase protects against CNS 02 toxicity. J Appl Physiol (1985). 71:1057–1061. 1991.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Cathcart MK and Bhattacharjee A: Monoamine oxidase A (MAO-A): A signature marker of alternatively activated monocytes/macrophages. Inflamm Cell Signal. 1(e161)2014.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Sosa V, Moliné T, Somoza R, Paciucci R, Kondoh H and LLeonart ME: Oxidative stress and cancer: An overview. Ageing Res Rev. 12:376–390. 2013.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Mittal M, Siddiqui MR, Tran K, Reddy SP and Malik AB: Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 20:1126–1167. 2014.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Datta C and Bhattacharjee A: Role of monoamine oxidase A (MAO-A) in cardiac aging. J Cardiolcardiovasc Sci. 4:31–40. 2020.
|
|
88
|
Rybaczyk LA, Bashaw MJ, Pathak DR and Huang K: An indicator of cancer: Downregulation of monoamine oxidase-A in multiple organs and species. BMC Genomics. 9(134)2008.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Li J, Yang XM, Wang YH, Feng MX, Liu XJ, Zhang YL, Huang S, Wu Z, Xue F, Qin WX, et al: Monoamine oxidase A suppresses hepatocellular carcinoma metastasis by inhibiting the adrenergic system and its transactivation of EGFR signaling. J Hepatol. 60:1225–1234. 2014.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Hodorová I, Rybárová S, Vecanová J, Solár P, Domorákova I, Adamkov M and Mihalik J: Comparison of expression pattern of monoamine oxidase A with histopathologic subtypes and tumour grade of renal cell carcinoma. Med Sci Monit. 18:BR482–BR486. 2012.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Wu JB, Shao C, Li X, Li Q, Hu P, Shi C, Li Y, Chen YT, Yin F, Liao CP, et al: Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J Clin Invest. 124:2891–2908. 2014.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Bhattacharjee A, Biswas P, Dhabal S, Das P, Das P, Swaroop S, Prasad T, Dhanalakshmi J and Indhumathi S: Role of monoamine oxidase a (MAO-A) in cancer progression and metastasis. Cancer Cell Microenviron. 5(e1623)2018.
|
|
93
|
Wu JB and Shih JC: Valproic acid induces monoamine oxidase A via Akt/forkhead box O1 activation. Mol Pharmacol. 80:714–723. 2011.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S, Leeds P and Chuang DM: Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: A potential role of Sp1 acetylation. J Neurochem. 111:976–987. 2009.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Dutta P, Sabri N, Li J and Li WX: Role of STAT3 in lung cancer. JAKSTAT. 3(e999503)2015.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Khan ANH and Tomasi TB: Histone deacetylase regulation of immune gene expression in tumor cells. Immunol Res. 40:164–178. 2008.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Wu Y and Zhou BP: TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer. 102:639–644. 2010.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Dobreva ZG, Grigorov BG and Stanilova SA: Effect of a histone deacetylases inhibitor of IL-18 and TNF-alpha secretion in vitro. Open Access Maced J Med Sci. 6:269–273. 2018.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Zhao Z, Cheng X, Wang Y, Han R, Li L, Xiang T, He L, Long H, Zhu B and He Y: Metformin inhibits the IL-6-induced epithelial-mesenchymal transition and lung adenocarcinoma growth and metastasis. PLoS One. 9(e95884)2014.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Wang YC, Wu YS, Hung CY, Wang SA, Young MJ, Hsu TI and Hung JJ: USP24 induces IL-6 in tumor-associated microenvironment by stabilizing p300 and β-TrCP and promotes cancer malignancy. Nat Commun. 9(3996)2018.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Sánchez-Gundín J, Fernández-Carballido AM, Martínez Valdivieso L, Barreda-Hernández D and Torres Suárez AI: New trends in the therapeutic approach to metastatic colorectal cancer. Int J Med Sci. 15:659–665. 2018.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Licht JD and Bennett RL: Leveraging epigenetics to enhance the efficacy of immunotherapy. Clin Epigenetics. 13(115)2021.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Wang L, Yang W, Read P, Larner J and Sheng K: Tumor cell apoptosis induced by nanoparticle conjugate in combination with radiation therapy. Nanotechnology. 21(475103)2010.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, et al: Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 1:598–607. 2011.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Young CS, Clarke KM, Kettyle LM, Thompson A and Millis KI: Decitabine-Vorinostat combination treatment in acute myeloid leukemia activates pathways with potential for novel triple therapy. Oncotarget. 8:51429–51446. 2017.PubMed/NCBI View Article : Google Scholar
|














