1. Introduction
Depression is a mood disorder characterized by an
enduring feeling of sadness and interest loss. According to the
Diagnostic and Statistical Manual of Mental Disorders- 5th Edition
(DSM-5) (1), depressive disorders
can be classified into different groups, including major depressive
disorder (MDD) and dysthymia (2).
As of 2019, depressive disorders are the leading cause of non-fatal
disease worldwide (3). The burden
of mental disorders, such as depression is prevalent throughout the
entire lifespan in both sexes and across multiple locations
(4). Therefore, it is not
surprising that depression is of increasing concern, since it
negatively affects the quality of life of affected individuals on a
global scale (5).
Depression is a multifactorial disease with a
relatively complex etiology and great variability in presentation.
For this reason, treatment, which includes pharmacotherapy,
psychotherapy, or a combination of both, is a complex issue
(3,5). Antidepressant drugs, the most widely
used and effective form of treatment, still do not lead to complete
remission in a considerable percentage of patients, and showcase a
delayed clinical onset, which may vary from 2 to 4 weeks (6,7). The
variable effects of drugs may be due to several reasons, including
but not limited to, drug interactions, disease-related mechanisms,
complex pathophysiology and genetics (8). Emerging evidence, though, highlights
that epigenetics also play a pivotal role in psychiatric disorders,
such as depression and appear to affect the responses of patients
to the drugs (9). The present
review aims to accumulate information from the currently available
literature in order to highlight the mechanisms through which
epigenetics may affect depression and the response of patients to
antidepressants.
2. Depression
The pathogenesis of mood disorders is not yet fully
understood; however, several theories have arisen regarding
depression, such as the monoamine and cytokine hypotheses, plus
hypotheses based on the dysfunction of the
hypothalamus-pituitary-adrenal gland (HPA) axis (10,11).
The most commonly accepted theory regarding the
pathogenesis of depression is the monoamine hypothesis, which
states that the decrease in monoamines, such as serotonin (5HT),
noradrenaline (NA) and dopamine (DA) in synaptic gaps can lead to
depression (10). This hypothesis
emerged when the anti-hypertensive drug, reserpine, caused the
depletion of monoamines and, subsequently, depression in patients
who did not suffer from the mentioned disorder prior to drug
administration (12). The monoamine
hypothesis is supported by the fact that currently used
antidepressants, such as tricyclic antidepressants (TCAs),
monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake
inhibitors (SSRIs), and serotonin and noradrenaline reuptake
inhibitors (SNRIs) are considered to function by increasing
monoamine levels (13). Moreover,
an extensive number of studies have focused on the role of
serotonin in depression, with many reporting low 5-HT levels and an
altered 5-HT receptor expression in depressed individuals (14). Nevertheless, the response to such
antidepressants is extremely varied and several studies have
questioned the importance of monoamine dysregulation in depression
(15).
Stress is also known to play a critical role in the
emergence of major depressive disorder (16) and the possible role the HPA axis
hyperactivity may have on depression pathophysiology has gained an
increasing amount of scientific interests (10). All organisms are programmed to
maintain an inner equilibrium for optimal organism function termed
homeostasis and stress refers to the state of threatened or
perceived as such homeostasis. The stress response system is a
sophisticated regulatory system, whose role is to maintain or
re-establish homeostasis (17). The
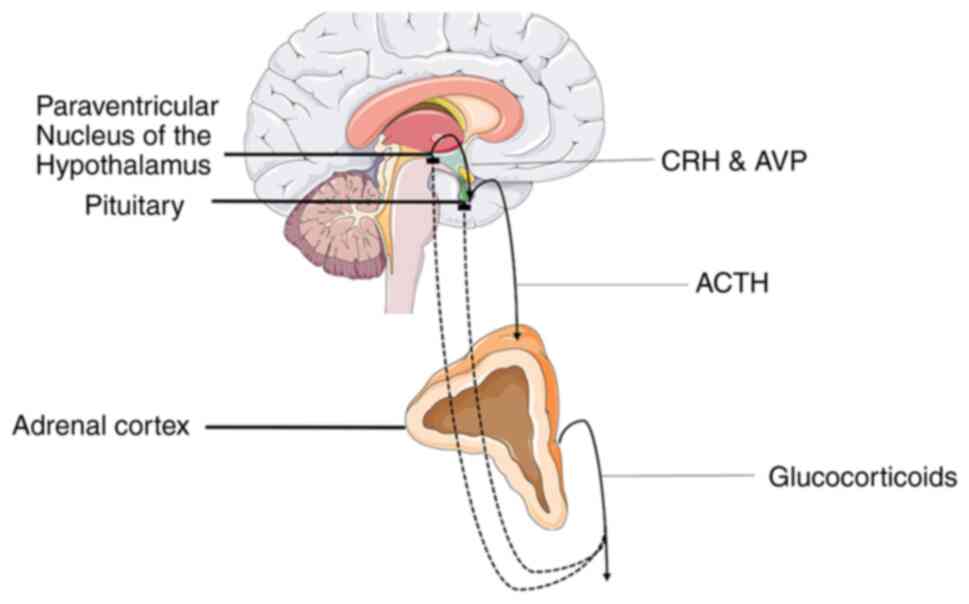
HPA axis is a vital component of the fight-or-flight response that
regulates the production of glucocorticoids (GCs), which are main
mediators of the stress response system. Specifically, a stressful
stimulus triggers the synthesis and secretion of arginine
vasopressin (AVP) and corticotropin-releasing hormone (CRH) by the
paraventricular nucleus of the hypothalamus (PVN), with the
mentioned hormones eliciting the secretion of adrenocorticotropic
hormone (ACTH) in the anterior pituitary, which in turn acts on the
adrenal glands to promote the release of GCs (18). Elevated glucocorticoid levels then
suppress CRH and ACTH secretion through a negative feedback loop by
acting on glucocorticoid receptors (GRs) in the hippocampus and
thus reversing their levels to normal (Fig. 1) (19). It is considered that chronic stress
leads to the dysfunction of the HPA axis, causing an abnormal
increase in GC levels that in turn induce a decrease in the volume
of the hippocampus, which is a main characteristic of MDD. These
elevated GC levels may promote atrophy in hippocampal mature
neurons and/or decrease hippocampal neurogenesis (10). A potential mediator of the effect of
stress on hippocampus is the brain-derived neurotrophic factor
(BDNF). GR appears to downregulate BDNF expression, an event that
may lead to negative morphological changes in hippocampal neurons
(10,20). This theory is supported by the fact
that depressed patients display lower BDNF serum levels and
antidepressants can recover stress-related morphological changes in
the hippocampus by increasing BDNF expression (10). The hypothesis of the dysfunction of
the HPA axis is reinforced by findings on SSRIs and TCAs, which
appear to affect GC signaling (21,22).
The cytokine theory hypothesis claims that
depression is dependent on the activation of the inflammatory
response system and altered levels of immunomodulatory molecules
cause the various symptoms observed in this disorder (11). Specifically, cytokines, which are a
category of small proteins that regulate the immune response, have
been implicated in the pathogenesis of MDD (23). Cytokines can be grouped into
pro-inflammatory and anti-inflammatory cytokines, with
pro-inflammatory cytokines, such as interleukin (IL)-1, IL-6 and
tumor necrosis factor α (TNFα), being directly or indirectly
involved in the inflammatory process and anti-inflammatory
cytokines, such as IL-4 and IL-10 suppressing the immune response
(11). Research has demonstrated
that non-depressed individuals may display symptoms similar to
depression following exposure to pro-inflammatory cytokines, an
effect that can be ameliorated by antidepressant treatment
(24). Moreover, patients with MDD
exhibit higher levels of TNFα, which can be significantly decreased
following the administration of an SNRI antidepressant, such as
venlafaxine. A possible mechanism through which inflammation causes
depression involves microglia activation (25). Chronic stress induces microglia
activation (26), which in turn
causes the production of IL-1 and TNFα. These pro-inflammatory
cytokines can hinder long-term potentiation (LTP) induction, which
can lead to symptoms characteristic of MDD (25). There exist several other mechanisms
in which cytokines may cause depression. One such mechanism
includes the activation of indoleamine-2,3-dioxygenase by IL-6 and
TNFα, which results in serotonin reduction and changes in monoamine
oxidase production (27). Moreover,
cytokines such as IL-6 and TNFα may prevent the entry of the GR
complex into the neuronal nucleus and inhibit its binding to DNA,
thus promoting the hyperactivity of the HPA axis and the loss of
its negative feedback loop (28).
3. Antidepressants
As aforementioned, the currently used
antidepressants include TCAs, MAOIs, selective SSRIs and SNRIs.
These drugs are considered to mainly function through mechanisms
supportive of the monoamine hypothesis.
TCAs and MAOIs were the first antidepressant classes
discovered and were the sole medication for depression for ~30
years (29). TCAs achieve their
effect by acting on distinct neurotransmitter pathways.
Specifically, they block the reuptake of serotonin and
noradrenaline in the synaptic cleft, increasing the concentrations
of mentioned monoamines and exerting an antidepressant effect
(30). TCAs function mainly by
targeting the serotonin transporter (SERT) and the norepinephrine
transporter (NET), but also influence other neurotransmitter
systems such as cholinergic, adrenergic, muscarinic and
histaminergic receptors (30-32).
MAOIs achieve their antidepressant effects by blocking monoamine
oxidase function. This enzyme is responsible for breaking down
neurotransmitters, such as serotonin, noradrenaline and dopamine in
the brain. The use of MAOIs suppresses the breakdown of the
aforementioned neurotransmitters, thus increasing their levels and
exerting an antidepressant effect (33). Both TCAs and MAOIs are associated
with severe side-effects, a fact that has led to their decreased
prescription in favor of more modern types of antidepressants, such
as SSRIs and SNRIs which are associated with less severe
side-effects. In particular, MAOIs may cause a possibly fatal
hypertensive crisis after excessive tyramine consumption, while
TCAs may cause cardiac sodium channel blockage and arrhythmia
(34). Nonetheless, these drugs
continue to play a key role in battling depression, particularly in
treatment-resistant patients (29).
SSRIs along with SNRIs are among the most commonly
prescribed drugs in the USA (35).
The function of SSRIs, as their name suggests, is based on the
inhibition of serotonin reuptake, and more precisely by inhibiting
SERT at the presynaptic axon terminal, therefore increasing the
amount of 5-HT in the synaptic cleft. The side-effects of SSRIs are
fewer than those of TCAs and MAOIs, since they have a minimal
effect on other monoamines, such as dopamine and noradrenaline, and
do not affect the functions of histaminergic, adrenergic and
cholinergic receptors (36). SNRIs,
as their name also suggests, function by inhibiting the presynaptic
neuronal uptake of 5-HT and noradrenaline, and act on SERT and NERT
in a specific manner (37). These
drugs are considered to have a dual effect, though the precise
degree of serotonin or adrenaline inhibition is both agent- and
dose-dependent (38). Selective
serotonin and noradrenaline reuptake inhibitors have been shown to
exert their antidepressant effect more rapidly than SSRIs in a
clinical setting (39). SNRIs have
similar side-effects with SSRIs; however, due to their enhancement
of noradrenergic activity, they may also increase blood pressure
and heart rate (40).
Lastly, studies published over the last decade have
highlighted the potential use of ketamine, an
N-methyl-D-aspartate (NMDA) glutamate receptor antagonist,
as an antidepressant (41). Chronic
stress has been shown to increase glutamate release, and
subsequently impair LTP and promote the atrophy of apical dendrites
in the hippocampus (42). As an
NMDA receptor antagonist, ketamine functions by inhibiting the
action of glutamate. In contrast to generally used antidepressants,
which require 2 to 4 weeks to ameliorate depressive symptoms, a
single intravenous administration of ketamine ameliorates
depressive symptoms in 1 to 3 days, and displays a long-lasting
effect (10,42). However, the use of ketamine is
associated with severe adverse effects, including psychotomimetic
effects and dissociative properties, and may lead to drug abuse.
Therefore, other research has focused on ketamine enantiomers and
metabolites as potential antidepressants (43).
4. Epigenetics
Epigenetics, i.e., heritable and stable structural
and biochemical alterations of the chromosome that are not
associated with DNA sequence alterations, have been associated with
numerous physiological and pathological processes, such as
metabolic disorders, autoimmune diseases, cancer and
neuropsychiatric disorders (44-50).
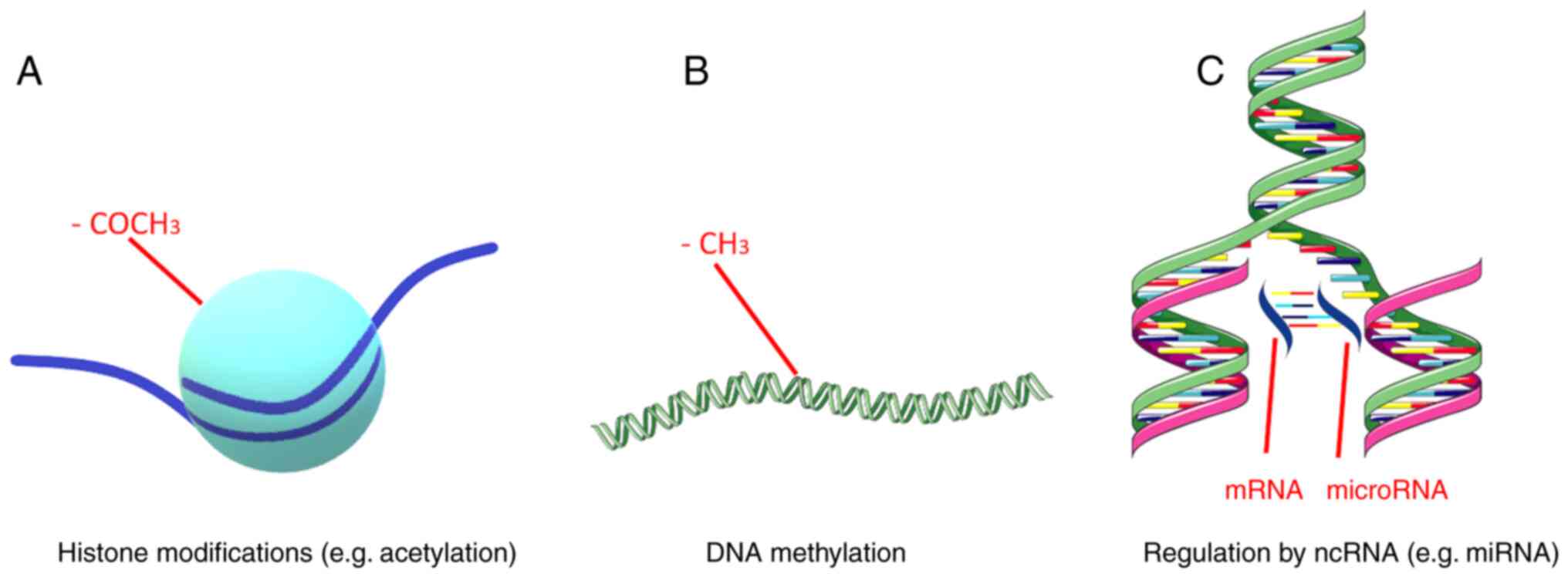
Epigenetic mechanisms include DNA methylation, regulation by
non-coding RNAs (ncRNAs) and histone modifications (Fig. 2). These mechanisms are responsible
for fine-tuning gene expression (51). Gene expression, which refers to the
production of a functional gene product using the information
provided by the DNA sequence, is a quintessential process in all
living organisms, since it allows them to adjust the amount and
type of gene product in response to different environmental factors
(52). A main part of gene
expression is achieved at the transcriptional level, although
several post-transcriptional events also play a key role, such as
the aforementioned histone modifications and regulation by ncRNAs
(53,54).
DNA methylation includes the extensively studied
attachment of a methyl group to the carbon-5 position of cytosine
(m5C) and the lesser-studied linkage of a methyl group to the
adenosine base at the nitrogen-6 position of deoxyadenosine (m6dA)
(55). The methylation of m5C is
considered the predominant form of DNA methylation and occurs on
DNA regions known as CpG islands. These genomic regions are at
least 500 bp in length and display a high content of cytosine and
guanine nucleotides (>55%). The methylation of CpG islands is
performed by DNA methyltransferases (DNMTs), while the
demethylation of 5-methylcytosine makes use of TET methylcytosine
dioxygenases 1, 2, and 3, and leads to the production of m5C
oxidative derivatives. The methylation and demethylation of these
sites alteres the expression of nearby genes (56). DNA methylation may function either
as a repressive or activating mark for gene transcription.
Specifically, methylation can make transcription machinery binding
more difficult or create a landscape prime for transcription
(57).
ncRNAs are RNA molecules that are incapable of
protein coding (58). Non-coding
RNAs can be classified into two major categories, those with a
length of <200 nucleotides, which are termed small ncRNAs, and
those with a length >200 nucleotides, which create a distinct
category known s long ncRNAs (lncRNAs). Small ncRNAs include RNA
types, such as microRNAs (miRNAs/miRs), small interfering RNAs
(siRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs
(snoRNAs) and PIWI-interacting RNAs (piRNAs) (59). The majority of ncRNAs have been
found to be associated with the regulation of gene expression
(60). miRNAs are short
single-stranded RNA molecules with a length of ~22 nucleotides
(59). Generally, miRNAs silence
gene expression by RNA-induced silencing at a post-transcriptional
level. lncRNAs can regulate pre-mRNA splicing, inhibit mRNA
translation or promote chromatin remodeling. siRNAs are
double-stranded RNAs that function by dividing into their single
strands and binding to a distinct target mRNA to suppress gene
expression (61). snRNAs, which are
~150 nucleotides in length, are primarily found in the splicing
regions of eukaryotic cell nuclei, and thus can affect pre-miRNA
splicing. snoRNAs are generally located in the nucleoli of
eukaryotic cells and play a role in rRNA processing.
PIWI-interacting RNAs bind to PIWI proteins, a subfamily of
ARGONAUTE proteins, to influence chromatin function (60). Lastly, lncRNAs have the ability to
interact with mRNA, DNA, several protein complexes and miRNAs and
affect gene expression at multiple regulatory levels (62).
In eukaryotic cells, genomic DNA is organized into
chromatin, a polymer whose main structural unit is the nucleosome.
The nucleosome core consists of a histone octamer composed of two
copies of core histones, H2A, H2B, H3, H4 and 146 DNA base pairs
wrapped around it (63). Chromatin
condensation influences gene expression, since a highly condensed
structure hinders DNA accessibility and thus interferes with gene
transcription. Histone modifications can affect chromatin
condensation and organize the genome into transcriptionally active
or inactive regions (64). These
modifications include methylation, acetylation, phosphorylation and
the ubiquitination of histone proteins, plus chromatin remodeling
and regulation by ncRNAs, piRNAs and lncRNAs (65,66).
Histone modifications occur predominantly, although not
exclusively, at the N-terminal tails of histone proteins and can
alter gene expression (66).
Histone methylation usually includes the addition of methyl groups
at lysine (K) residues of histones H3 and H4. Histone lysine
residues can be mono-, di- and tri-methylated in order to act as
repressive or active marks of gene expression, a process mediated
by the histone methyltransferase (HMT) group of enzymes (65). Histone acetylation occurs on lysine
residues via the addition of an acetyl group from an
acetyl-coenzyme A donor to an ε-amino group of a lysine side chain
(67). This modification is
considered an active gene expression mark and is regulated by the
equilibrium between histone acetyltransferases and histone
deacetylases (HDACs) (65).
Specifically, the addition of an acetyl group weakens the positive
charge of a lysine residue, thus reducing the tail's affinity for
chromatin and leaving the underlying DNA exposed (67). Histone ubiquitination involves the
covalent attachment of ubiquitin, a small 76 amino-acid protein, to
a ε-amino group of a lysine residue. This process is catalyzed by
E1 ubiquitin activating enzymes, E2 ubiquitin conjugating enzymes,
and E3 ubiquitin ligases (68).
Histone ubiquitination is a reversible process with
deubiquitinating enzymes, a family of proteases and
metalloproteases, being able to remove ubiquitin moieties from
histones (69). The effect histone
ubiquitination has on gene expression depends on the number of
ubiquitin moieties being added to the lysine residue and which
specific core histone is being altered. Histone phosphorylation is
regulated by two enzymes of opposing function, specifically kinases
which add phosphate groups and phosphatases which remove phosphate
groups. Histone phosphorylation mostly funcionts in conjunction
with other histone modifications creating a complex regulating
network with varied effects on gene expression (66). Chromatin remodeling is the process
of dynamic changes on chromatin structure that alters how condensed
or uncondensed a chromatin region is, thereby influencing the
exposure of the underlying DNA and subsequently gene expression.
This process in undertaken by chromatin-remodeling protein
complexes (65). Lastly, as
aforementioned, ncRNAs, i.e., RNA molecules that do not code a
protein product, can be mediators of histone modification. Specific
cases include piRNAs that can bind to PIWI proteins and recruit
histone methyltransferases to influence chromatin function and
lncRNAs that can change chromatin status by recruiting protein
complexes that influence histone methylation and acetylation
(65).
5. Epigenetics and depression
Epigenetic mechanisms appear to play a vital role in
depression and may help provide a biological framework in which
genetic and environmental factors interact and influence disease
pathogenesis and pathophysiology (70).
Epigenetics can regulate the expression of
antidepressant molecular targets, which are also main participants
of monoamine signaling (71). SERT
is coded by the SLC6A gene and sustained alterations on its gene
expression profile have been implicated in depression. These
alterations may emerge due to epigenetic modifications in response
to stressful events (72). Indeed,
the differential methylation of the SLC6A4 gene has been shown to
be associated with a risk of mental illness, including depression
(73). A previous study on the
SLC6A2 gene, which codes for NET, also demonstrated epigenetic
modifications on the mentioned gene, and more specifically DNA
acetylation, which may be responsible for a mechanism underlying
depression in conjunction with hypertension (74). Lastly, epigenetic alterations on the
monoamine oxidase A (MAOA) gene may be a factor associated with the
pathogenesis of depression. Specifically, the hypomethylation of
the MAOA gene may increase monoamine oxidase expression and
subsequently, its activity, thereby reducing monoamine utilization
by the brain. This increased monoamine oxidase activity has been
detected in patients with depressive symptoms and is in accordance
with the monoamine hypothesis and the use of MAOIs as
antidepressants (75).
Epigenetic alterations on genes regulating HPA
function may play a crucial role in disease pathogenesis and onset
age (76). A previous study on mice
demonstrated that early-life stress (ELS) affected AVP expression
in the PVN through methyl CpG binding protein 2 phosphorylation and
AVP enhancer hypomethylation, which in turn promoted neuroendocrine
and behavioral features that are present in depression (77). Moreover, animal models which
associate ELS with depression have shown that CRH, ACTH are also
hypomethylated while the gene encoding GR (NR3C1) is
hypermethylated (78). Thus,
epigenetics may provide a framework in which HPA axis dysfunction
may promote depression.
Genome-wide methylation analysis has identified
differentially methylated regions that are associated both with the
pathogenesis of depression and immune dysfunction (79). Another study has also proposed that
IL-6 methylation may be used as a biomarker for depression
(80). Particularly, depressed
patients display IL-6 hypomethylation in peripheral tissue. This
epigenetic modification possibly leads to a higher IL-6 expression
(80) which in turn may play a role
in the pathology of depression or even its pathogenesis, through
some of the mechanisms mentioned in the previous chapters. These
findings give credence to the cytokine hypothesis. Thereby, the
role of DNA methylation in depression has both been extensively
observed and can be described through the view of all major
hypotheses of disease pathogenesis.
Histone methylation has been found to be associated
with depression in genome-wide association studies. Studies on mice
have demonstrated than chronic social defeat stress can
downregulate HMTs, specifically G9α and G9α-like protein, which
catalyze the dimethylation of the lysine 9 residue of H3 (H3K9me2).
H3k9me2 is known to be a major repressive mark in the NAc of the
hypothalamus (81). This brain
region is essential in the regulation of reward behavior (82). On the other hand, G9α overexpression
in the NAc exerts antidepressant-like effects, and increases in
H3K9me2 at distinct gene promoters may be some of the mechanisms of
action of fluoxetine, an SSRI. Thus, it is possible that stress
leads to maladaptive alterations in the specific brain action via
the repression of histone methylation, a process that can be
ameliorated by the use of antidepressants (81). Chronic social defeat stress also
transiently suppresses histone acetylation in the mouse NAc, with
HDAC inhibition resulting in antidepressant effects (82). Studies on the direct association
between histone phosphorylation and depression are limited. These
studies have focused on the stress response and have detected that
H3 phosphorylation is increased in the hippocampus and prefrontal
cortex of mice and rats, respectively that have been subjected to
stress (83). Research on histone
ubiquitination and its role in depression is also limited. A
previous study demonstrated that the UBE2A gene, which is an E2
ubiquitin conjugating enzyme is upregulated in the post-mortem
dorsolateral prefrontal cortex of MDD patients. Histone
ubiquitination by the UBE2A protein is considered a transcriptional
activation tag. However, it should be mentioned that this protein
also participates in the ubiquitin-proteasome system, which is the
main mechanism of protein catabolism, and this process has already
been associated with MDD (84). As
regards chromatin remodeling, several types of stress have been
demonstrated to induce repressive chromatin complexes in the mouse
NAc. The same complexes are induced in the NAc of depressed humans
(81). The effects of lncRNAs on
chromatin have also been associated with MDD. A prime example is
the BDNF antisense RNA (BDNF-AS) that functions as a scaffold to
recruit chromatin modifiers to act on the BDNF gene promoter and
repress its expression, a process implicated in MDD (85).
Multiple studies have highlighted the fact that the
dysregulation of ncRNAs is present in depressed patients and in
animal models of depression (86,87).
miRNAs and lncRNAs are the most extensively studied type of ncRNAs
in MDD (88). miRNAs are considered
to affect the pathogenesis of depression via the regulation of
monoamine and glutamate signaling (86). Numerous miRNAs have displayed
altered expression levels in depressed patients. It has been
demonstrated the downregulation of several miRNAs in the prefrontal
cortex of patients with MDD post-mortem (86). Some of these miRNAs are known to
target depression-associated mRNAs. In particular, miR-20α,
miR-20b, miR-34α, and miR-34b target VEGF, whose protein levels are
elevated in the peripheral blood of patients with MDD, miR-34α
targets BCL2, whose protein levels are downregulated in depressed
patients, and miR-148b targets DNMT3B, whose protein levels are
downregulated in depressed individuals. Additionally, microarray
studies on patients with MDD have demonstrated the upregulation of
miRNAs, such as let-7d-5p and let-7f-5p, whose expression can be
influenced by antidepressant treatment (86). The most well-known
depression-associated lncRNA is the aforementioned BDNF-AS. Apart
from BDNF-AS, other lncRNAs have also been implicated in the
pathogenesis of depression, with a prime example being the growth
arrest specific 5 (GAS5). GAS5 is upregulated in the hippocampal
tissues of mice with depressive-like behaviors, and its silencing
appears to eliminate such behaviors. It appears that GAS5
influences early growth response gene 1 via miR-26α binding, and
promotes the release of inflammatory factors and the apoptosis of
hippocampal neurons in mice with depressive-like behaviors
(89). Additionally, the
overexpression of GAS5 may lead to the increased expression of the
type 1 receptor of CRH and a subsequent long-term activation of the
HPA axis, a common feature of depression (90).
6. Epigenetics and antidepressants
Current antidepressants may exert some of their
effects via epigenetic mechanisms, while differences in gene
expression in patients due to epigenetic alterations may be one of
the underlying causes of variations in drug responses (50,91).
Classical antidepressants, such as TCAs and SSRIs have an indirect
effect on DNA methylation and chromatin structure (82). Imipramine, a TCA, reverses changes
induced by social defeat stress in H3 methylation in the mouse NAc.
Furthermore, chronic imipramine treatment decreases histone
methylation, increases histone acetylation at BDNF promoters and
downregulates HDAC5 in the mouse hippocampus (92). Citalopram, a commonly used SSRI,
affects DNA methylation on a large scale and influences the gene
expression of a large set of genes that are involved in depression
pathogenesis and pathology (93).
Moreover, some data suggest that paroxetine, another SSRI, has the
ability to affect DNMT activity and thus influence DNA methylation.
On the other hand, DNA methylation itself can influence drug
response. The promoter methylation of depression-associated genes,
such as BDNF, HTR1A, HTR1B, SLC6A4 and IL11 appear to be predictive
of an antidepressant response (91). Some examples include the
hypermethylation of the SLC6A4 promoter, which is a predictor for
an improved SSRI drug response, and the hypomethylation of the BDNF
promoter, which is a predictor of non-responsiveness to TCAs,
MAOIS, SSRIs and SNRIs (92).
7. Conclusions and future perspectives
Epigenetics appear to play a significant role in the
pathology and pathogenesis of depression, while also influencing
the response of patients to antidepressants and vice versa. Further
research on the epigenetics of depression may help to elucidate the
molecular peculiarities of depression, while it may also help to
predict the response of patients to antidepressants. The latter is
of immense interest, since it can help clinicians tailor therapy to
each individual patient.
Acknowledgements
Parts of the figures were drawn using images from
Servier Medical Art. Servier Medical Art by Servier is licensed
under a Creative Commons Attribution 3.0 Unported License
(https://creativecommons.org/licenses/by/3.0/).
Funding
Funding: The authors would like to acknowledge funding from
‘MilkSafe: A novel pipeline to enrich formula milk using omics
technologies’, a research co-financed by the European Regional
Development Fund of the European Union and Greek national funds
through the Operational Program Competitiveness, Entrepreneurship
and Innovation, under the call RESEARCH-CREATE-INNOVATE (project
code: T2EDK-02222).
Availability of data and materials
Not applicable.
Authors' contributions
All authors (TM, EP and DV) contributed to the
conceptualization, design, writing, drafting, revising, editing and
reviewing of the manuscript. All authors have read and approved the
final manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DV is an Editor of the journal. However, he had no
personal involvement in the reviewing process, or any influence in
terms of adjudicating on the final decision, for this article. The
other authors declare that they have no competing interests.
References
|
1
|
American Psychiatric A, American
Psychiatric Association DSMTF (eds): Diagnostic and statistical
manual of mental disorders: DSM-5. American Psychiatric
Association, Arlington, VA, 2013.
|
|
2
|
Chand SP and Arif H: Depression.
StatPearls (Internet): StatPearls Publishing, Treasure Island, FL,
2022.
|
|
3
|
Mekonen T, Chan GCK, Connor JP, Hides L
and Leung J: Estimating the global treatment rates for depression:
A systematic review and meta-analysis. J Affect Disord.
295:1234–1242. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Global prevalence and burden of depressive
and anxiety disorders in 204 countries and territories in 2020 due
to the COVID-19 pandemic. Lancet. 398:1700–1712. 2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Proudman D, Greenberg P and Nellesen D:
The growing burden of major depressive disorders (MDD):
Implications for researchers and policy makers. Pharmacoeconomics.
39:619–625. 2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Marasine NR, Sankhi S, Lamichhane R,
Marasini NR and Dangi NB: Use of antidepressants among patients
diagnosed with depression: A scoping review. Biomed Res Int.
2021(6699028)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Karrouri R, Hammani Z, Benjelloun R and
Otheman Y: Major depressive disorder: Validated treatments and
future challenges. World J Clin Cases. 9:9350–9367. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Roden DM, McLeod HL, Relling MV, Williams
MS, Mensah GA, Peterson JF and Van Driest SL: Pharmacogenomics.
Lancet. 394:521–532. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhou J, Li M, Wang X, He Y, Xia Y, Sweeney
JA, Kopp RF, Liu C and Chen C: Drug response-related DNA
methylation changes in schizophrenia, bipolar disorder, and major
depressive disorder. Front Neurosci. 15(674273)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Boku S, Nakagawa S, Toda H and Hishimoto
A: Neural basis of major depressive disorder: Beyond monoamine
hypothesis. Psychiatry Clin Neurosci. 72:3–12. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Shadrina M, Bondarenko EA and Slominsky
PA: Genetics factors in major depression disease. Front Psychiatry.
9(334)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Boas GR, de Lacerda RB, Paes MM, Gubert P,
da Cruz AWL, Rescia VC, de Carvalho PMG, de Carvalho AAV and
Oesterreich SA: Molecular aspects of depression: A review from
neurobiology to treatment. Eur J Pharmacol. 851:99–121.
2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Marathe SV, D'Almeida PL, Virmani G,
Bathini P and Alberi L: Effects of monoamines and antidepressants
on astrocyte physiology: Implications for monoamine hypothesis of
depression. J Exp Neurosci. 12(1179069518789149)2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tian H, Hu Z, Xu J and Wang C: The
molecular pathophysiology of depression and the new therapeutics.
MedComm (2020). 3(e156)2022.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Chávez-Castillo M, Núñez V, Nava M, Ortega
Á, Rojas M, Bermúdez V and Rojas-Quintero J: Depression as a
neuroendocrine disorder: Emerging neuropsychopharmacological
approaches beyond monoamines. Adv Pharmacol Sci.
2019(7943481)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Richter-Levin G and Xu L: How could stress
lead to major depressive disorder? IBRO Rep. 4:38–43.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tsigos C, Kyrou I, Kassi E and Chrousos
GP: Stress: Endocrine physiology and pathophysiology. In: Feingold
KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et
al., (eds). Endotext. South Dartmouth (MA): MDText.com, Inc. Copyright© 2000-2023, MDText.com, Inc.; 2020.
|
|
18
|
Menke A: Is the HPA axis as target for
depression outdated, or is there a new hope? Front Psychiatry.
10(101)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Nicolaides NC, Pavlaki AN, Maria Alexandra
MA, Chrousos GP, Feingold KR, Anawalt B, Blackman MR, Boyce A,
Chrousos G, Corpas E, et al: Glucocorticoid therapy and adrenal
suppression. Copyright © 2000-2023, MDText.com, Inc.;
2018.
|
|
20
|
Chen H, Amazit L, Lombès M and Le Menuet
D: Crosstalk between glucocorticoid receptor and early-growth
response protein 1 accounts for repression of brain-derived
neurotrophic factor transcript 4 expression. Neuroscience.
399:12–27. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Budziñski ML, Sokn C, Gobbini R, Ugo B,
Antunica-Noguerol M, Senin S, Bajaj T, Gassen NC, Rein T, Schmidt
MV, et al: Tricyclic antidepressants target FKBP51 SUMOylation to
restore glucocorticoid receptor activity. Mol Psychiatry.
27:2533–2545. 2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ronaldson A, Carvalho LA, Kostich K,
Lazzarino AI, Urbanova L and Steptoe A: The effects of six-day SSRI
administration on diurnal cortisol secretion in healthy volunteers.
Psychopharmacology (Berl). 235:3415–3422. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Roohi E, Jaafari N and Hashemian F: On
inflammatory hypothesis of depression: What is the role of IL-6 in
the middle of the chaos? J Neuroinflammation. 18(45)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Miller AH and Raison CL: The role of
inflammation in depression: From evolutionary imperative to modern
treatment target. Nat Rev Immunol. 16:22–34. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Innes S, Pariante CM and Borsini A:
Microglial-driven changes in synaptic plasticity: A possible role
in major depressive disorder. Psychoneuroendocrinology.
102:236–247. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Schramm E and Waisman A: Microglia as
central protagonists in the chronic stress response. Neurol
Neuroimmunol Neuroinflamm. 9(e200023)2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Arčan IS, Kouter K and Paska AV:
Depressive disorder and antidepressants from an epigenetic point of
view. World J Psychiatry. 12:1150–1168. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Grygiel-Górniak B, Limphaibool N and
Puszczewicz M: Cytokine secretion and the risk of depression
development in patients with connective tissue diseases. Psychiatry
Clin Neurosci. 73:302–316. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chockalingam R, Gott BM and Conway CR:
Tricyclic antidepressants and monoamine oxidase inhibitors: Are
they too old for a new look? Handb Exp Pharmacol. 250:37–48.
2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Moraczewski J and Aedma KK: Tricyclic
Antidepressants. StatPearls. Treasure Island (FL): StatPearls
Publishing Copyright©. 2022, StatPearls Publishing LLC.; 2022.
|
|
31
|
Andersen J, Stuhr-Hansen N, Zachariassen
L, Toubro S, Hansen SM, Eildal JN, Bond AD, Bøgesø KP,
Bang-Andersen B, Kristensen AS and Strømgaard K: Molecular
determinants for selective recognition of antidepressants in the
human serotonin and norepinephrine transporters. Proc Natl Acad Sci
USA. 108:12137–12142. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Cottingham C, Percival S, Birky T and Wang
Q: Tricyclic antidepressants exhibit variable pharmacological
profiles at the α(2A) adrenergic receptor. Biochem Biophys Res
Commun. 451:461–466. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Laban TS and Saadabadi A: Monoamine
oxidase inhibitors (MAOI). StatPearls. Treasure Island (FL):
StatPearls Publishing Copyright ©. 2022, StatPearls Publishing
LLC.; 2022.
|
|
34
|
Edinoff AN, Akuly HA, Hanna TA, Ochoa CO,
Patti SJ, Ghaffar YA, Kaye AD, Viswanath O, Urits I, Boyer AG, et
al: Selective serotonin reuptake inhibitors and adverse effects: A
narrative review. Neurol Int. 13:387–401. 2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Fuentes AV, Pineda MD and Venkata KCN:
Comprehension of top 200 prescribed drugs in the US as a resource
for pharmacy teaching, training and practice. Pharmacy (Basel).
6(43)2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chu A and Wadhwa R: Selective serotonin
reuptake inhibitors. Statpearls. treasure island (FL): StatPearls
publishing copyright©. 2022, StatPearls Publishing LLC.; 2022.
|
|
37
|
Takano A, Halldin C and Farde L: SERT and
NET occupancy by venlafaxine and milnacipran in nonhuman primates:
A PET study. Psychopharmacology (Berl). 226:147–153.
2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Fanelli D, Weller G and Liu H: New
serotonin-norepinephrine reuptake inhibitors and their anesthetic
and analgesic considerations. Neurol Int. 13:497–509.
2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Li J, Lu C, Gao Z, Feng Y, Luo H, Lu T,
Sun X, Hu J and Luo Y: SNRIs achieve faster antidepressant effects
than SSRIs by elevating the concentrations of dopamine in the
forebrain. Neuropharmacology. 177(108237)2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Haller E, Geier M and Finley P:
Antidepressants, pharmacology of. In: Aminoff MJ, Daroff RB,
editors. Encyclopedia of the Neurological Sciences (Second
Edition). Oxford: Academic Press; 2014. p. 219-23.
|
|
41
|
Onaolapo AY and Onaolapo OJ: Glutamate and
depression: Reflecting a deepening knowledge of the gut and brain
effects of a ubiquitous molecule. World J Psychiatry. 11:297–315.
2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Pal MM: Glutamate: The master
neurotransmitter and its implications in chronic stress and mood
disorders. Front Hum Neurosci. 15(722323)2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Pochwat B, Nowak G and Szewczyk B: An
update on NMDA antagonists in depression. Expert Rev Neurother.
19:1055–1067. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Li Y: Modern epigenetics methods in
biological research. Methods. 187:104–113. 2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sun L, Zhang H and Gao P: Metabolic
reprogramming and epigenetic modifications on the path to cancer.
Protein Cell. 13:877–919. 2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Gougousis S, Petanidis S, Poutoglidis A,
Tsetsos N, Vrochidis P, Skoumpas I, Argyriou N, Katopodi T and
Domvri K: Epigenetic editing and tumor-dependent immunosuppressive
signaling in head and neck malignancies. Oncol Lett.
23(196)2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Dawson MA and Kouzarides T: Cancer
epigenetics: From mechanism to therapy. Cell. 150:12–27.
2012.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Ling C and Rönn T: Epigenetics in human
obesity and type 2 diabetes. Cell Metab. 29:1028–1044.
2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Surace AEA and Hedrich CM: The role of
epigenetics in autoimmune/inflammatory disease. Front Immunol.
10(1525)2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Menke A, Klengel T and Binder EB:
Epigenetics, depression and antidepressant treatment. Curr Pharm
Des. 18:5879–5889. 2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Fardi M, Solali S and Hagh MF: Epigenetic
mechanisms as a new approach in cancer treatment: An updated
review. Genes Dis. 5:304–311. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Singh KP, Miaskowski C, Dhruva AA, Flowers
E and Kober KM: Mechanisms and measurement of changes in gene
expression. Biol Res Nurs. 20:369–382. 2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Corbett AH: Post-transcriptional
regulation of gene expression and human disease. Curr Opin Cell
Biol. 52:96–104. 2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Landini A, Trbojević-Akmačić I, Navarro P,
Tsepilov YA, Sharapov SZ, Vučković F, Polašek O, Hayward C,
Petrović T, Vilaj M, et al: Genetic regulation of
post-translational modification of two distinct proteins. Nat
Commun. 13(1586)2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Li X, Zhao Q, Wei W, Lin Q, Magnan C,
Emami MR, Wearick-Silva LE, Viola TW, Marshall PR, Yin J, et al:
The DNA modification N6-methyl-2'-deoxyadenosine (m6dA) drives
activity-induced gene expression and is required for fear
extinction. Nat Neurosci. 22:534–544. 2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Kiselev IS, Kulakova OG, Boyko AN and
Favorova OO: DNA methylation as an epigenetic mechanism in the
development of multiple sclerosis. Acta Naturae. 13:45–57.
2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Dhar GA, Saha S, Mitra P and Chaudhuri RN:
DNA methylation and regulation of gene expression: Guardian of our
health. Nucleus (Calcutta). 64:259–270. 2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Lee YS: Are we studying non-coding RNAs
correctly? Lessons from nc886. Int J Mol Sci.
23(4251)2022.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Diamantopoulos MA, Tsiakanikas P and
Scorilas A: Non-coding RNAs: The riddle of the transcriptome and
their perspectives in cancer. Ann Transl Med. 6(241)2018.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Kumar S, Gonzalez EA, Rameshwar P and
Etchegaray JP: Non-Coding RNAs as mediators of epigenetic changes
in malignancies. Cancers (Basel). 12(3657)2020.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Padda IS, Mahtani AU and Parmar M: Small
interfering RNA (siRNA) based therapy. StatPearls. Treasure island
(FL): StatPearls Publishing Copyright ©. 2022, StatPearls
Publishing LLC.; 2022.
|
|
62
|
Zhang X, Wang W, Zhu W, Dong J, Cheng Y,
Yin Z and Shen F: Mechanisms and functions of long non-coding RNAs
at multiple regulatory levels. Int J Mol Sci.
20(5573)2019.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Chen JJ, Stermer D and Tanny JC: Decoding
histone ubiquitylation. Front Cell Devel Biol.
10(968398)2022.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Miller JL and Grant PA: The role of DNA
methylation and histone modifications in transcriptional regulation
in humans. Subcell Biochem. 61:289–317. 2013.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Zhang Y, Sun Z, Jia J, Du T, Zhang N, Tang
Y, Fang Y and Fang D: Overview of histone modification. Adv Exp Med
Biol. 1283:1–16. 2021.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Alhamwe BA, Khalaila R, Wolf J, von Bülow
V, Harb H, Alhamdan F, Hii CS, Prescott SL, Ferrante A, Renz H, et
al: Histone modifications and their role in epigenetics of atopy
and allergic diseases. Allergy Asthma Clin Immunol.
14(39)2018.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Barnes CE, English DM and Cowley SM:
Acetylation & Co: An expanding repertoire of histone acylations
regulates chromatin and transcription. Essays Biochem. 63:97–107.
2019.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Sekiguchi M and Matsushita N: DNA damage
response regulation by histone ubiquitination. Int J Mol Sci.
23(8187)2022.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Wang J, Qiu Z and Wu Y: Ubiquitin
regulation: The histone modifying Enzyme's story. Cells.
7(118)2018.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Penner-Goeke S and Binder EB: Epigenetics
and depression. Dialogues Clin Neurosci. 21:397–405.
2019.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Menke A and Binder EB: Epigenetic
alterations in depression and antidepressant treatment. Dialogues
Clin Neurosci. 16:395–404. 2014.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Wankerl M, Miller R, Kirschbaum C, Hennig
J, Stalder T and Alexander N: Effects of genetic and early
environmental risk factors for depression on serotonin transporter
expression and methylation profiles. Transl Psychiatry.
4(e402)2014.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Lee JS, Jaini PA and Papa F: An epigenetic
perspective on lifestyle medicine for depression: Implications for
primary care practice. Am J Lifestyle Med. 16:76–88.
2022.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Meng L, Bai X and Zheng Y, Chen D and
Zheng Y: Altered expression of norepinephrine transporter
participate in hypertension and depression through regulated TNF-α
and IL-6. Clin Exp Hypertens. 42:181–189. 2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Xu Q, Jiang M, Gu S, Wang F and Yuan B:
Early life stress induced DNA methylation of monoamine oxidases
leads to depressive-like behavior. Front Cell Dev Biol.
8(582247)2020.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Humphreys KL, Moore SR, Davis EG, MacIsaac
JL, Lin DTS, Kobor MS and Gotlib IH: DNA methylation of HPA-axis
genes and the onset of major depressive disorder in adolescent
girls: A prospective analysis. Transl Psychiatry.
9(245)2019.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Murgatroyd C, Patchev AV, Wu Y, Micale V,
Bockmühl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OFX and
Spengler D: Dynamic DNA methylation programs persistent adverse
effects of early-life stress. Nat Neurosci. 12:1559–1566.
2009.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Duan Z and Lu J: DNA methyltransferases in
depression: An update. Front Psychiatry. 11(538683)2020.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Crawford B, Craig Z, Mansell G, White I,
Smith A, Spaull S, Imm J, Hannon E, Wood A, Yaghootkar H, et al:
DNA methylation and inflammation marker profiles associated with a
history of depression. Hum Mol Genet. 27:2840–2850. 2018.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Ryan J, Pilkington L, Neuhaus K, Ritchie
K, Ancelin ML and Saffery R: Investigating the epigenetic profile
of the inflammatory gene IL-6 in late-life depression. BMC
Psychiatry. 17(354)2017.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Peña CJ and Nestler EJ: Progress in
epigenetics of depression. Prog Mol Biol Transl Sci. 157:41–66.
2018.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Park HS, Kim J, Ahn SH and Ryu HY:
Epigenetic targeting of histone deacetylases in diagnostics and
treatment of depression. Int J Mol Sci. 22(5398)2021.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Wu MS, Li XJ, Liu CY, Xu Q, Huang JQ, Gu S
and Chen JX: Effects of histone modification in major depressive
disorder. Curr Neuropharmacol. 20:1261–1277. 2022.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Rey R, Chauvet-Gelinier JC, Suaud-Chagny
MF, Ragot S, Bonin B, d'Amato T and Teyssier JR: Distinct
expression pattern of epigenetic machinery genes in blood
leucocytes and brain cortex of depressive patients. Mol Neurobiol.
56:4697–4707. 2019.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Policarpo R, Sierksma A, De Strooper B and
d'Ydewalle C: From junk to function: LncRNAs in CNS health and
disease. Front Mol Neurosci. 14(714768)2021.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Lin R and Turecki G: Noncoding RNAs in
depression. Adv Exp Med Biol. 978:197–210. 2017.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Shi Y, Wang Q, Song R, Kong Y and Zhang Z:
Non-coding RNAs in depression: Promising diagnostic and therapeutic
biomarkers. EBioMedicine. 71(103569)2021.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Yoshino Y and Dwivedi Y: Non-coding RNAs
in psychiatric disorders and suicidal behavior. Front Psychiatry.
11(543893)2020.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Wu Y, Rong W, Jiang Q, Wang R and Huang H:
Downregulation of lncRNA GAS5 alleviates hippocampal neuronal
damage in mice with depression-like behaviors via modulation of
MicroRNA-26a/EGR1 axis. J Stroke Cerebrovasc Dis.
30(105550)2021.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Zhou Y and Chen B: GAS5-mediated
regulation of cell signaling (Review). Mol Med Rep. 22:3049–3056.
2020.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Webb LM, Phillips KE, Ho MC, Veldic M and
Blacker CJ: The relationship between DNA methylation and
antidepressant medications: A systematic review. Int J Mol Sci.
21(826)2020.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Czarny P, Białek K, Ziółkowska S,
Strycharz J, Barszczewska G and Sliwinski T: The importance of
epigenetics in diagnostics and treatment of major depressive
disorder. J Person Med. 11(167)2021.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Kanherkar RR, Getachew B, Ben-Sheetrit J,
Varma S, Heinbockel T, Tizabi Y and Csoka AB: The effect of
citalopram on genome-wide DNA methylation of human cells. Int J
Genomics. 2018(8929057)2018.PubMed/NCBI View Article : Google Scholar
|