Introduction
Primary central nervous system (CNS) lymphoma
(PCNSL) is the second most common malignant brain tumor, accounting
for ∼2.9% of the primary brain tumors in Japan (1). Age, performance status (PS), lactate
dehydrogenase (LDH) serum levels, cerebrospinal fluid protein
concentrations and the involvement of deep structures of the brain
are independent predictors of survival (2). The 2-year overall survival (OS) rate
varies from 24 to 85% in proportion to the prognostic score based
on the aforementioned risk factors (2). Although the majority of PCNSLs are
diffuse large B-cell lymphomas (DLBCLs), primary T-cell lymphoma of
the CNS accounts for only 2–8.5% of PCNSLs (3,4).
However, the survival of patients with primary T-cell lymphoma is
similar to that of patients with B-cell PCNSL (5).
Anaplastic large-cell lymphoma (ALCL) is an uncommon
type of T-cell lymphoma first reported by Stein et al
(6). It is characterized by large
pleomorphic CD30 (Ki-1)-expressing lymphoid blasts containing
horseshoe-shaped nuclei (7).
Currently, the fourth edition of the WHO classification of Tumors
of Haematopoietic Lymphoid Tissues, published in 2008, divides
systemic ALCLs into two entities: anaplastic lymphoma kinase
(ALK)-positive and ALK-negative (8). In total, 60–85% of systemic ALCLs are
ALK-positive lymphomas that exhibit the characteristic t(2;5)
(p23;q35) translocation that produces the ALK protein (7). This type is associated with a younger
age of onset and a more favorable prognosis (9–11).
Patients with ALK-positive ALCL are usually younger and have a
better prognosis compared to those with ALK-negative ALCL. The
5-year OS rates are 80 and 40% in ALK-positive and -negative
patients, respectively (7).
Although ALCL frequently involves lymph nodes and
occasionally involves extranodal sites, such as the skin, soft
tissues, bone, bone marrow, liver, lungs and gastrointestinal
tract, it rarely occurs in the CNS (9,12)
and ALCL of the CNS is limited to case reports. A few case reviews
suggested that although ALK positivity and younger age appear to be
favorable prognostic factors, this disease is generally much more
aggressive compared to systemic ALCL or PCNSL (13,14).
However, the precise prognosis and standard treatment have not yet
been determined due to the small number of reported cases.
In this study, we reviewed previously published
cases of ALCL and discussed the therapeutic management of ALCL of
the CNS. We also report a case study of a patient with ALK-positive
brain ALCL who underwent successful treatment with high-dose
methotrexate (HD-MTX) alone and has not exhibited recurrence for
>5 years.
Materials and methods
A search was conducted using PubMed for published
studies in English on cases of immunocompetent patients with ALCL
of the brain. The keywords used were ‘anaplastic large-cell
lymphoma’, ‘ALK’ and ‘primary central nervous system lymphoma’.
Information regarding age, gender, location, ALK positivity,
treatment and clinical course were collected from each study.
In addition to the 13 cases reviewed by George et
al (13), an additional 13
reported cases (12–33) were selected and a total of 27 cases
were summarized, including those of the present study (Table I). The survival time of 2 patients
(cases 10 and 22) was not described, 13 patients had succumbed to
the disease and 12 patients exhibited no evidence of disease at the
time the studies were published.
 | Table IReported cases with ALCL. |
Table I
Reported cases with ALCL.
| No. | ALK | Age | Gender | Location | Marker | Lesion | RT | CT regimens | Survival | Authors (Refs.) |
|---|
| 1 | + | 4.5 | F | Multifocal brain,
brain stem, spinal cord, intra-axial and meningeal | Null-cell | M | + | CHOP | NED at 6.1 years | Havlioglu et
al (15) |
| 2 | + | 10 | F | Parietal lobe
abutting against falx | T-cell | S | + | CHOP, MTX | Dead at 6 months from
CT | Buxton et al
(16) |
| 3 | + | 13 | M | Frontal,
parietal | T-cell | M | − | CHOP, MTX | Dead shortly after
CT | Abdulkader et
al (12) |
| 4 | + | 29 | M | Frontotemporal
(macular) | T-cell | M | + | MTX | NED at 13 months | Ponzoni et al
(17) |
| 5 | + | 17 | M | Parietal dura | T-cell | S | + | - | NED at 4.8 years | George et al
(13) |
| 6 | + | 18 | F | Temporal dura | T-cell | M | + | CHOP, MTX | NED at 5.2
years | George et al
(13) |
| 7 | + | 9 | M | Bilateral
frontal | T-cell | M | + | MTX | NED at 26
months | Ozkaynak (33) |
| 8 | + | 17 | M | Frontoparietal
eroding skull | T-cell | M | + | CHOP | Dead at 1
month | Rupani et al
(18) |
| 9 | + | 39 | M |
Occipitoparietal | T-cell | S | + | MTX | NED at 9
months | Cooper et al
(19) |
| 10 | + | 4 | M | Pineal region,
LMM | T-cell | LMM | + | CHOP | NED after CT | Karikari et
al (20) |
| 11 | + | 20 | M | Region of the
sylvian fissure | T cell | S | + | CHOP | NED at 8 years | Vivekanandan et
al (22) |
| 12 | + | 38 | M |
Parietooccipital | T-cell | S | + | MTX | NED at 15
months | Carmichael
(23) |
| 13 | + | 20 | M | Frontal | T-cell | S | − | MTX | NED at 5 years | Present case |
| 14 | − | 22 | F | Dura cerebellum,
temporal, 4 additional lesions | T-cell | M | − | - | Dead at 11
days | George et al
(13) |
| 15 | − | 46 | F |
Parietooccipital | T-cell | S | + | - | NED at 25
months | Chuang et al
(24) |
| 16 | − | 50 | M | Parietal, 2
additional supratentorial, dura | Null-cell | M | + | - | Dead at 2
months | George et al
(13) |
| 17 | − | 63 | M | Four
frontoparietal, dura, brain | T-cell | M | + | - | Dead at 11
weeks | Paulus et al
(25) |
| 18 | − | 66 | F | Temporal | T-cell | S | − | - | Dead at 4 days | Nuckols et
al (26) |
| 19 | − | 79 | M |
Parietoocipital | T-cell | S | − | - | Dead at 4
months | Kodama et al
(14) |
| 20 | − | 82 | M | Posterior fossa
lesion attaching to the tentorium | T-cell | S | − | - | Dead at 6
weeks | Gonzales (27) |
| 21 | − | 75 | M | Bilateral
hemisphere mimicking lymphomatosis cerebri | T-cell | M | + | - | Dead at 8
months | Sugino et al
(28) |
| 22 | − | 65 | M | Temporal | T-cell | S | + | MTX | NED when 2 courses
of CT | Colen et al
(29) |
| 23 | Unknown | 12 | F | Occipital | Null-cell | S | + | Unknown | Dead at 4
months | Bergmann and Edel
(30) |
| 24 | Unknown | 20 | M | Parietal | T-cell | S | + | Others | Dead at 24
months | Feldges et
al (31) |
| 25 | Unknown | 63 | M | Frontal,
parietal | T-cell | M | + | - | Dead at 3
months | Goldbrunner et
al (32) |
| 26 | Unknown | 6 | F | Unknown | T-cell | Unknown | - | MTX | NED at 79
months | Abla et al
(34) |
| 27 | Unknown | 7 | M | Unknown | T-cell | Unknown | + | MTX | NED at 98
months | Abla et al
(34) |
The OS for 25 patients was calculated by the
Kaplan-Meier survival method. Statistical analyses were performed
using JMP software version 9 (SAS Institute Inc., Cary, NC,
USA).
Results
Patient characteristics
Details of the 27 reported cases are provided in
Table II. The male:female ratio
was 19:8. The median age of the patients was 20 years (range,
4.0–82).
| Table IISummary of ALK lymphoma of reported
cases. |
Table II
Summary of ALK lymphoma of reported
cases.
| Variable | Total | ALK-positive | ALK-negative |
|---|
| No. | 27 | 13 | 9 |
| Median age
(range) | 20.0
(4.0–82.0) | 17.0
(4.0–39.0) | 65.0
(22.0–82.0) |
| Gender (M:F) | 19:8 | 10:3 | 6:3 |
| Lesions | | | |
| Single | 13 | 6 | 5 |
| Multiple | 12 | 7 | 4 |
| Unknown | 2 | 0 | 0 |
| Initial
therapy | | | |
| RT alone | 6 | 1 | 4 |
| RT+CT | 14 | 10 | 1 |
| CT alone | 3 | 2 | 0 |
| None | 4 | 0 | 4 |
| CT regimens | | | |
| MTX-based | 8 | 5 | 1 |
| CHOP-based | 4 | 4 | 0 |
|
MTX+CHOP-based | 3 | 3 | 0 |
| Others | 1 | 0 | 0 |
| Unknown | 1 | 0 | 0 |
Of the 27 patients, 13 (48.1%) were ALK-positive, 9
(33.3%) were ALK-negative and the ALK status was not determined in
the remaining 5 patients (18.5%). Twenty-four patients expressed
T-cell antigens and 3 patients had null-cell tumors (cases 1, 16
and 23). There was a difference in the age distribution between the
ALK-positive and -negative groups. The ALK-positive tumors occurred
in patients aged <40 years. Moreover, 10 out of the 13
ALK-positive patients (76.9%) were aged ≤20 years and the median
age of these 13 patients was 17 years (range, 4.0–39 years).
Conversely, ALK-negative tumors occurred in older patients. Out of
the 9 ALK-negative patients, 8 (88.9%) were >40 years of age and
the median age of these 9 patients was 65 years (range, 22–82
years). No ALK-negative patient was <20 years of age.
A single lesion was observed in 13 (48.1%) and
multiple lesions in 12 patients (44.4%), including 1 case of
leptomeningeal metastasis (case 10).
The postoperative treatment was chemoradiotherapy
for 14 (51.9%), chemotherapy alone for 3 (11.1%), radiotherapy
alone for 6 (22.2%) and supportive care alone for 4 patients
(14.8%). In total, 17 patients (63.0%) received some form of
chemotherapy. The chemotherapy regimens of these 17 patients were
as follows: MTX-based agents for 8 (47.1%), cyclophosphamide +
doxorubicin + vincristine + prednisolone (CHOP)-based agents for 4
(23.5%), both MTX- and CHOP-based agents for 3 (17.6%), other
agents for 1 (5.9%) and unknown for 1 patient (5.9%).
Clinical courses and OS
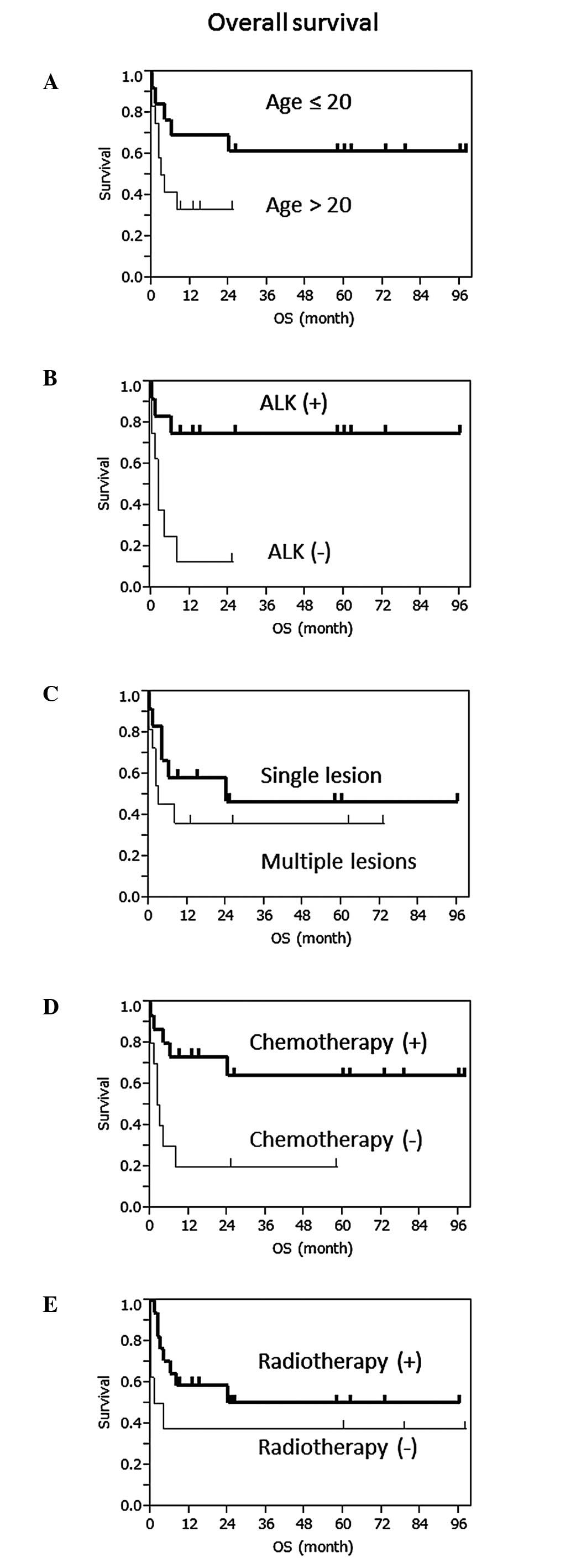
The patients aged <40 years (n=17) exhibited a
longer OS compared to those aged >40 years (n=8) and the median
OS of the groups was not reached (NR) and 2.5 months, respectively
(P<0.01, log-rank test; Fig.
1A), which demonstrated younger age as a good prognostic
factor. The median OS of patients aged ≤20 (n=13) and >20 (n=12)
years was NR and 3.5 months, respectively (P=0.089).
In total, 10 out of the 13 ALK-positive patients
(76.9%) exhibited no evidence of disease at the time of case report
publication. At least 4 out of the 13 ALK-positive patients
(30.7%), including our patient, exhibited no evidence of disease
for >5 years after the initial diagnosis, whereas 6 out of the 9
ALK-negative patients (66.7%) succumbed to the disease within 4
months after diagnosis. The median OS of each group was NR and 2
months, respectively (P<0.01; Fig.
1B). ALK was significantly correlated with the prognosis, with
a 5-year OS of 75.0 and <12.5% in patients with ALK-positive
(n=12) and -negative ALCL (n=8), respectively.
There was no difference in the OS patients with a
single lesion (n=12) and those with multiple lesions (n=11; P=0.36;
Fig. 1C).
Chemotherapy improved patient survival, as the
median OS with (n=10) or without (n=15) chemotherapy was NR and 2.5
months, respectively (P=0.01; Fig.
1D). Chemoradiotherapy tended to improve OS compared to
radiotherapy alone (P=0.08); however, there was no difference in OS
between patients who had received chemoradiotherapy and those who
had received chemotherapy alone (P=0.73, Fig. 1E). Three patients treated with
chemotherapy alone had no evidence of disease until the time when
their cases were reported and 2 patients them exhibited no
recurrence for >5 years.
Case presentation
We have treated 90 cases of histologically proven
PCNSL from 2000 to the present at our department, including 88
cases of DLBCL and 2 cases of T-cell lymphoma, including 1 ALCL. A
20-year-old immunocompetent man with no other significant medical
history was hospitalized with generalized seizures. He did not have
any neurological deficits and physical examination on admission
revealed no abnormal findings. Complete blood counts and serum
chemistries, including LDH and soluble interleukin-2 receptor
levels, were normal. Magnetic resonance imaging (MRI) with
gadolinium diethylenetriamine pentaacetic acid (Gd-DTPA) revealed a
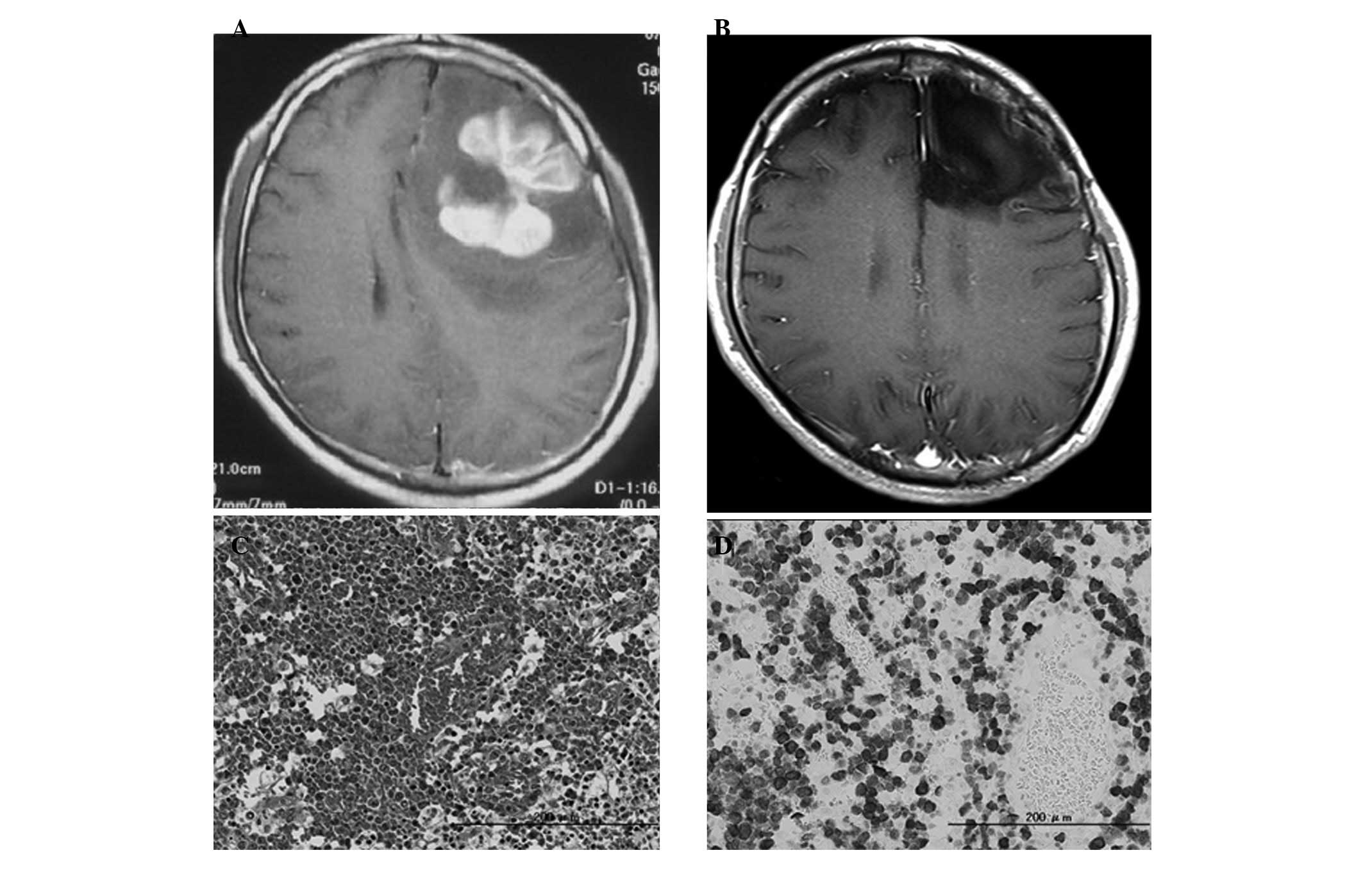
well-enhanced mass lesion in the left frontal lobe (Fig. 2A), with a surrounding
high-intensity lesion on T2-weighted images, indicating significant
edema. Whole-body CT scans with contrast medium revealed no
abnormality. Based on the suspicion of high-grade glioma, awake
brain surgery and gross total removal of the tumor were performed.
No residual tumor was identified on postoperative MRI with Gd-DTPA.
Pathological findings revealed the proliferation of large, atypical
lymphocytes containing scattered horseshoe-shaped nuclei on
hematoxylin and eosin (H&E)-stained slides (Fig. 2C). Immunohistochemistry revealed
that the tumor cells were positive for CD3 and ALK-1 (Fig. 2D), but negative for CD20. This
patient was finally diagnosed with ALK-1-positive ALCL. Bone marrow
examination findings were normal.
Treatment was immediately initiated with intravenous
HD-MTX (3.5 g/m2). The patient completed 3 courses of
HD-MTX, one every 2 weeks. Cerebral MRI and systemic positron
emission tomography with fluorodeoxyglucose (FDG-PET) revealed no
abnormal findings and radiotherapy was not performed. He had
regular MRI examinations every 3 months until 3 years after the
diagnosis and every 6 months thereafter. There has been no
recurrence of the disease for 5 years (Fig. 2B) and the patient has exhibited no
neurological deficits.
Discussion
We demonstrated that ALK-positive ALCL in the CNS
presents at a younger age, has a good prognosis and is sensitive to
chemoradiotherapy in the analysis of reported cases.
Shenkier et al (5) reported a retrospective analysis of
patients with T-cell PCNSL diagnosed between 1983 and 2003 at 12
institutions in 7 countries. In that report, among 25 cases of
T-cell PCNSL in which the pathology report was reviewed, 3 cases
(12%) exhibited the characteristics of ALCL. Thus, ALCL is a rare
disease of the brain, for which the literature is limited to case
reports. We collected twice as many cases as the largest reviews
(13) and although our review was
based on incomplete data from the literature, our findings may be
of value in understanding this rare disease.
In general, ALCL of the CNS has been recognized to
be significantly more aggressive compared to systemic ALCL or PCNSL
(13,14). However, our data demonstrate that
ALK-positive ALCL has a good prognosis, similar to systemic ALCL
(10,11). Only 3 ALK-positive patients were
reported to have succumbed to the disease. Tuberculosis or other
infectious diseases of the CNS were suspected in these 3 patients
(cases 2, 3 and 8) at the initial presentation and they were
treated with antituberculosis medications or antibiotics. Delayed
diagnosis may result in a worse outcome compared to that observed
in other ALK-positive patients. PCNSL is rare in adolescence
(34) and the radiological
findings of primary ALCL in the CNS often mimic those of infectious
or immunological disease. However, an early diagnosis and the
timely initiation of the appropriate treatment is critical.
There is no standard treatment for primary ALCL of
the CNS. Our findings demonstrate the importance of
chemoradiotherapy for ALCL. Of the 17 patients treated with
chemotherapy, 11 received MTX-based agents. Moreover, 9 of these 11
patients exhibited no evidence of tumor at the time of case report
publication, although 2 patients died shortly after treatment. As
it is known that the standard chemotherapy regimen for PCNSL is MTX
and that CHOP has not been proven to be effective against PCNSL
(35,36), MTX-based regimens are recommended
for ALCL of the CNS. Our patient was treated with 3 courses of
HD-MTX alone and he has exhibited no recurrence for >5 years.
Abla et al (21) reported
10 pediatric cases of PCNSL, including 2 ALCLs treated with
chemotherapy alone and the majority of the children achieved
long-term remissions. Radiotherapy may decrease cognitive function
and it is possible that ALK-positive patients, particularly younger
patients, may need to start treatment with chemotherapy alone.
Our review also demonstrated that ALK-negative ALCL
exhibits a poor prognosis and is very often fatal. The majority of
ALK-negative patients were treated with radiotherapy or supportive
care, due to their older age or poor PS. As ALK-negative ALCL tends
to occur in older individuals, similar to PCNSL and DLBCL,
chemoradiotherapy including HD-MTX should be initiated earlier.
In conclusion, our findings indicate that the
prognosis of ALCL of the CNS is correlated with ALK positivity and
patient age of <40 years. Chemoradiotherapy with MTX is
recommended as the standard treatment for ALCL. However, additional
multicenter studies including large numbers of cases are
required.
Abbreviations:
|
CNS
|
central nervous system
|
|
PCNSL
|
primary central nervous system
lymphoma
|
|
ALCL
|
anaplastic large-cell lymphoma
|
|
ALK
|
anaplastic lymphoma kinase
|
|
OS
|
overall survival
|
|
PS
|
performance status
|
|
LDH
|
lactate dehydrogenase
|
|
DLBCL
|
diffuse large B-cell lymphoma
|
|
HD-MTX
|
high-dose methotrexate
|
|
CHOP
|
cyclophosphamide + doxorubicin +
vincristine + prednisolone
|
|
NR
|
not reached
|
|
H&E
|
hematoxylin and eosin
|
|
MRI
|
magnetic resonance imaging
|
|
FDG-PET
|
positron emission tomography with
fluorodeoxyglucose
|
Acknowledgements
This study was supported by National
Cancer Center Research and Development Fund no. 55
References
|
1.
|
The committee of Brain Tumor Registry of
Japan: Report of brain tumor registry of Japan (1984–2000) 12th
edition. Neurol Med Chir (Tokyo). 49(Suppl): 1–101. 2009.
|
|
2.
|
Ferreri AJ, Blay JY, Reni M, et al:
Prognostic scoring system for primary CNS lymphomas: the
International Extranodal Lymphoma Study Group experience. J Clin
Oncol. 21:266–272. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Hayabuchi N, Shibamoto Y and Onizuka Y:
Primary central nervous system lymphoma in Japan: a nationwide
survey. Int J Radiat Oncol Biol Phys. 44:265–272. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Ferreri AJ, Reni M, Pasini F, et al: A
multicenter study of treatment of primary CNS lymphoma. Neurology.
58:1513–1520. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Shenkier TN, Blay JY, O’Neill BP, et al:
Primary CNS lymphoma of T-cell origin: a descriptive analysis from
the international primary CNS lymphoma collaborative group. J Clin
Oncol. 23:2233–2239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Stein H, Mason DY, Gerdes J, et al: The
expression of the Hodgkin’s disease associated antigen Ki-1 in
reactive and neoplastic lymphoid tissue: evidence that
Reed-Sternberg cells and histiocytic malignancies are derived from
activated lymphoid cells. Blood. 66:848–858. 1985.
|
|
7.
|
Jaffe ES, Harris NL, Stein H and Vardiman
JW: Anaplastic large cell lymphoma. Pathology and Genetics: Tumors
of Haematopoietic and Lymphoid Tissues. Jaffe ES, Harris NL, Stein
H, Vardiman JW, et al: IARC Press; pp. 230–235. Lyon: 2001
|
|
8.
|
Swerdlow S, Campo E, Harris N, et al:
Anaplastic large cell lymphoma WHO classification of Tumours of
Haematopoietic and Lymphoid Tissues. 4th edition. IARC Press; pp.
312–319. Lyon; 2008
|
|
9.
|
Stein H, Foss HD, Durkop H, et al: CD30(+)
anaplastic large cell lymphoma: a review of its histopathologic,
genetic, and clinical features. Blood. 96:3681–3695. 2000.
|
|
10.
|
Suzuki R, Kagami Y, Takeuchi K, et al:
Prognostic significance of CD56 expression for ALK-positive and
ALK-negative anaplastic large-cell lymphoma of T/null cell
phenotype. Blood. 96:2993–3000. 2000.PubMed/NCBI
|
|
11.
|
Falini B, Pileri S, Zinzani PL, et al:
ALK+ lymphoma: clinico-pathological findings and outcome. Blood.
93:2697–2706. 1999.
|
|
12.
|
Abdulkader I, Cameselle-Teijeiro J, Fraga
M, Rodriguez-Nunez A, Allut AG and Forteza J: Primary anaplastic
large cell lymphoma of the central nervous system. Hum Pathol.
30:978–981. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
George DH, Scheithauer BW, Aker FV, et al:
Primary anaplastic large cell lymphoma of the central nervous
system: prognostic effect of ALK-1 expression. Am J Surg Pathol.
27:487–493. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Kodama K, Hokama M, Kawaguchi K, Tanaka Y
and Hongo K: Primary ALK-1-negative anaplastic large cell lymphoma
of the brain: case report and review of the literature.
Neuropathology. 29:166–171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Havlioglu N, Manepalli A, Galindo L,
Sotelo-Avila C and Grosso L: Primary Ki-1 (anaplastic large cell)
lymphoma of the brain and spinal cord. Am J Clin Pathol.
103:496–499. 1995.PubMed/NCBI
|
|
16.
|
Buxton N, Punt J and Hewitt M: Primary
Ki-1-positive T-cell lymphoma of the brain in a child. Pediatr
Neurosurg. 29:250–252. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Ponzoni M, Terreni MR, Ciceri F, et al:
Primary brain CD30+ ALK1+ anaplastic large cell lymphoma
(‘ALKoma’): the first case with a combination of ‘not common’
variants. Ann Oncol. 13:1827–1832. 2002.
|
|
18.
|
Rupani A, Modi C, Desai S and Rege J:
Primary anaplastic large cell lymphoma of central nervous system -
a case report. J Postgrad Med. 51:326–327. 2005.PubMed/NCBI
|
|
19.
|
Cooper PB, Auerbach A, Aguilera NS, et al:
Rare primary CNS anaplastic large cell lymphoma in an
immunocompetent adult: a clinical-pathologic case report and review
case of the literature. Clin Neuropathol. 25:232–236.
2006.PubMed/NCBI
|
|
20.
|
Karikari IO, Thomas KK, Lagoo A, Cummings
TJ and George TM: Primary cerebral ALK-1-positive anaplastic large
cell lymphoma in a child. Case report and literature review.
Pediatr Neurosurg. 43:516–521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Abla O, Sandlund JT, Sung L, et al: A case
series of pediatric primary central nervous system lymphoma:
favorable outcome without cranial irradiation. Pediatr Blood
Cancer. 47:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Vivekanandan S, Dickinson P, Bessell E and
O’Connor S: An unusual case of primary anaplastic large cell
central nervous system lymphoma: an 8-year success story. BMJ Case
Rep. Feb 24–2011.(Epub ahead of print). View Article : Google Scholar
|
|
23.
|
Carmichael MG: Central nervous system
anaplastic large cell lymphoma in an adult: successful treatment
with a combination of radiation and chemotherapy. Mil Med.
172:673–675. 2007.PubMed/NCBI
|
|
24.
|
Chuang SS, Huang W, Lin CN, et al: Primary
cerebral anaplastic large cell lymphoma containing abundant
reactive histiocytes and eosinophils. A case report and literature
review. Pathol Res Pract. 197:647–652. 2001.
|
|
25.
|
Paulus W, Ott MM, Strik H, Keil V and
Muller-Hermelink HK: Large cell anaplastic (KI-1) brain lymphoma of
T-cell genotype. Hum Pathol. 25:1253–1256. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Nuckols JD, Liu K, Burchette JL, McLendon
RE and Traweek ST: Primary central nervous system lymphomas: a
30-year experience at a single institution. Mod Pathol.
12:1167–1173. 1999.PubMed/NCBI
|
|
27.
|
Gonzales M: Primary meningeal anaplastic
large cell lymphoma. Pathology. 35:451–452. 2003.PubMed/NCBI
|
|
28.
|
Sugino T, Mikami T, Akiyama Y, Wanibuchi
M, Hasegawa T and Mikuni N: Primary central nervous system
anaplastic large-cell lymphoma mimicking lymphomatosis cerebri.
Brain Tumor Pathol. Mar 18–2012.(Epub ahead of print).
|
|
29.
|
Colen CB, Rayes M, Kupsky WJ and
Guthikonda M: Synchronous meningioma and anaplastic large cell
lymphoma. Neuropathology. 30:260–266. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Bergmann M and Edel G: Primary
intracerebral non-Hodgkin’s lymphoma. Pathologe. 12:246–253.
1991.(In German).
|
|
31.
|
Feldges A, Gerhard L, Reinhardt V and
Budach V: Primary cerebral anaplastic T-cell-lymphoma (type Ki-1):
review and case report. Clin Neuropathol. 11:55–59. 1992.PubMed/NCBI
|
|
32.
|
Goldbrunner R, Warmuth-Metz M, Tonn JC,
Vince GH and Roosen K: Primary Ki-1-positive T-cell lymphoma of the
brain - an aggressive subtype of lymphoma: case report and review
of the literature. Surg Neurol. 46:37–41. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Ozkaynak MF: Favorable outcome of primary
CNS anaplastic large cell lymphoma in an immunocompetent patient. J
Pediatr Hematol Oncol. 31:128–130. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Abla O and Weitzman S: Primary central
nervous system lymphoma in children. Neurosurg Focus. 21:E82006.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
O’Neill BP, O’Fallon JR, Earle JD, Colgan
JP, Brown LD and Krigel RL: Primary central nervous system
non-Hodgkin’s lymphoma: survival advantages with combined initial
therapy? Int J Radiat Oncol Biol Phys. 33:663–673. 1995.
|
|
36.
|
Schultz C, Scott C, Sherman W, et al:
Preirradiation chemotherapy with cyclophosphamide, doxorubicin,
vincristine, and dexamethasone for primary CNS lymphomas: initial
report of radiation therapy oncology group protocol 88-06. J Clin
Oncol. 14:556–564. 1996.
|