Introduction
Esophageal cancer is one of the most lethal human
cancers that occur worldwide. It is the eighth most common cancer
in several European countries and its incidence is on the increase
in Western countries (1–3). Barrett’s esophagus, the only known
precursor to esophageal adenocarcinoma, is prevalent in Western
countries (1–3). In Barrett’s esophagus, a human
metaplastic condition is characterized by a posterior
intestinal-like phenotype in an anterior organ. Underlying it is a
mechanism of epigenetically regulated, developmentally critical
genes, such as the HOXB family (4). By contrast, the squamous cell
carcinoma of the esophagus is predominant in Asia, including Japan
(1–3).
A previous study suggested that the genetic and
epigenetic alterations, which constrain tumor suppressor genes and
activate oncogenes, are involved in the initiation, progression and
development of carcinogenesis in the esophagus, which is
asssociated with exposure to environmental carcinogens (5). Specifically, animal model analogies
of environmental carcinogenesis in humans indicated that
alterations in the expression of microRNAs, such as miR-31
and miR-21, characterized epithelial tumor progression in
the esophagus. The microRNAs were also detected in circulating
blood and were associated with inflammation of the esophagus
(6–8).
The most reliable markers currently available for
predicting cancer risk are findings of the degree of dysplasia in
endoscopic biopsies of the esophagus (9). Although epigenetic regulation is
eventually involved in tumor development in the esophagus, few
molecular biomarkers have been translated to widespread clinical
practice (9). Epigenetic studies
have shown that the aberrant DNA methylation of tumor suppressor
genes is involved in esophageal cancer, as well as in
adenocarcinoma, squamous cell carcinoma and Barrett’s esophagus. In
addition, several aberrantly methylated genes have been studied
with regard to early detection or as diagnostic markers and for
estimating prognosis or predicting responses to treatment (9).
In esophageal cancers, alterations in histone
modifications have also been identified. Histone deacetylase
inhibitors have been shown to enhance radiation responses through a
mechanism accompanied by an increase in the levels of γH2ax, an
indicator of double-strand breaks (DSBs), and a decrease in Rad51
expression, a DSB repair protein. This suggests that histone
deacetylase inhibitors are safe, promising radiosensitizers for
esophageal cancer radiotherapy (10). Nevertheless, the significance of
histone modifiers remains to be determined.
Through the use of H3K4 demethylase Jarid1b
(Kdm5b/Plu-1/Rbp2-h1) as a biomarker, a small subpopulation of
tumor-initiating melanoma cells was isolated. JARID1B
knockdown ultimately inhibited melanoma cell growth (11). In this study, we investigated the
effects of JARID1B knockdown on squamous cell carcinoma of
the esophagus using lentiviral transfer of small hairpin (sh) RNA
molecules for inhibition. Our findings are compatible with the
hypothesis that, similar to genetic alterations, epigenetic
aberrations including histone modifications significantly
contribute to tumor initiation and progression in gastrointestinal
cancers. This observation provides a rationale to study the
usefulness of JARID1B in the diagnosis and therapeutic
approaches to esophageal cancer.
Materials and methods
Cell culture and transfection
Human esophageal squamous cell carcinoma cell lines
(TE4 and TE8) were maintained at 37°C in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS). For shRNA-mediated
knockdown of endogenous JARID1B, lentiviruses were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The
cells were cultured in 12-well plates. After 24 h, cells were
infected with 20 μl/well of shRNA lentivirus particles in
the presence of 5 μg/ml polybrene (Sigma-Aldrich, St. Louis,
MO, USA). After another 24 h, the culture medium was removed and
replaced with 1 ml of complete medium without polybrene.
Subsequently, shRNA-infected cells were treated and selected with 2
μg/ml of puromycin (Sigma-Aldrich).
RNA extraction and real-time quantitative
polymerase chain reaction (PCR)
Total RNA was extracted from cells using Qiagen
RNeasy mini kits and was reverse-transcribed (RT) into cDNA using
High Capacity RNA to cDNA kits (Applied Biosystems, Carlsbad, CA,
USA). Samples were analyzed by real-time quantitative RT-PCR
(TaqMan Master Mix Kit, Applied Biosystems) to detect the
expression of the human genes JARID1B, SNAIL,
VIMENTIN and ACTB. The primers were used were:
JARID1B, forward: 5′-GCTTAATGGCAA AAGGCAAAC-3′ and reverse:
5′-CGGAGCTCATTCACT GTCAAC-3′; SNAIL, forward:
5′-GCTGCAGGACTCT AATCCAGA-3′ and reverse: 5′-ATCTCCGGAGGTGGG
ATG-3′; VIMENTIN, forward: 5′-AAAGTGTGGCTGCCAA GAAC-3′ and
reverse: 5′-AGCCTCAGAGAGGTCAGCAA-3′; ACTB, forward:
5′-AGAGCTACGAGCTGCCTGAC-3′ and reverse:
5′-CGTGGATGCCACAGGACT-3′.
Proliferation assay
Cell proliferation was determined with the WST-8
assay using Cell Counting kit-8 (Dojindo, Kumamoto, Japan), in
which 2,000 cells/well were placed in a 96-well plate. After 24,
48, 72 and 96 h, 10 μl of Cell Counting kit-8 solution
[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
monosodium salt] was added to each well and incubated for 1 h. Cell
viability was determined by reading the optical density in each
well at 450 nm.
Invasion assay
Cancer cell invasion was assessed using 24-well
BioCoat™ Matrigel Invasion Chambers (8 μm; Becton-Dickinson,
Franklin Lakes, NJ, USA) according to the manufacturer’s protocol.
Briefly, 5×104 cells were placed in the top chamber. The
bottom chamber contained 10% FBS as a chemoattractant. After 96-h
incubation, the non-invasive cells on the upper surface of the
membrane were removed with cotton swabs. The cells that adhered to
the lower surface of the membrane were fixed and stained using
Diff-Quick (Sysmex Internal Reagents Co., Ltd., Kobe, Japan) and
the number of cells was counted.
Animal experiments
A total of 102 or 103 cells
(JARID1B knockdown TE4 cells and control TE4 cells), mixed
with BD matrigel (Becton Dickinson) at a 1:1 ratio, were injected
subcutaneously into NOD/SCID mice. These mice were examined for up
to 10 weeks and sacrificed when the tumors reached a maximum
diameter of 15 mm. The animal studies were approved by the Animal
Experiments Committee of Osaka University (Suita, Japan).
Results
JARID1B knockdown suppresses esophageal
cancer cell growth
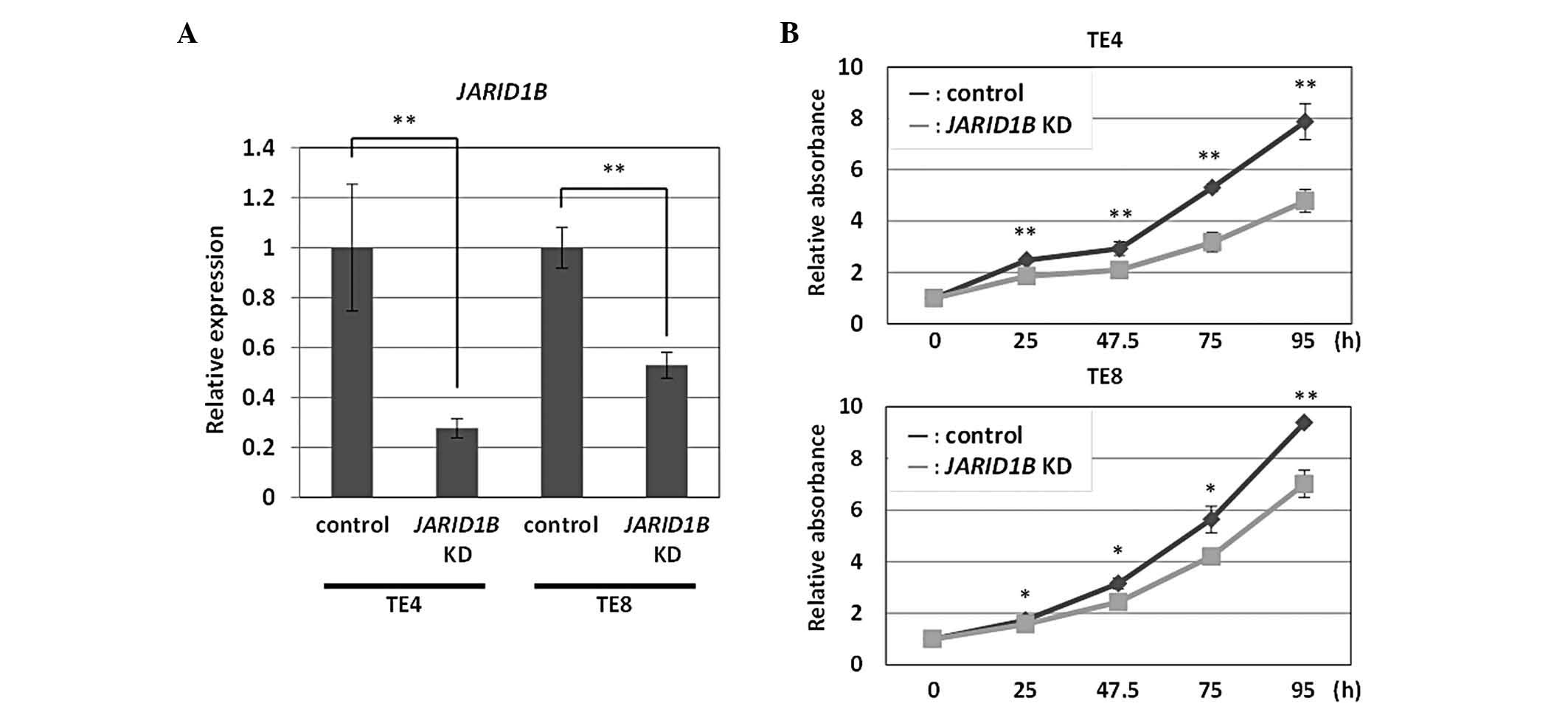
A lentiviral vector-mediated shRNA knockdown system
was developed for the efficient knockdown of JARID1B.
Following the introduction of shRNA, TE4 and TE8 esophageal
squamous cell carcinoma cells were grown in growth medium to select
transfectants. RNA was extracted from these cells and used for
quantitative RT-PCR analysis. The transfectants with the
JARID1B knockdown vector had reduced amounts of endogenous
JARID1B transcripts as compared with the control vector
transfectants for TE4 and TE8 cells (Fig. 1A). Based on cell counts,
JARID1B knockdown TE4 cells exhibited reduced cell growth
during the periods indicated in Fig.
1B. Similar results were obtained with JARID1B knockdown
TE8 cells (Fig. 1B). These results
indicated that JARID1B knockdown suppressed esophageal tumor
cell growth.
JARID1B knockdown suppresses esophageal
cancer cell invasion
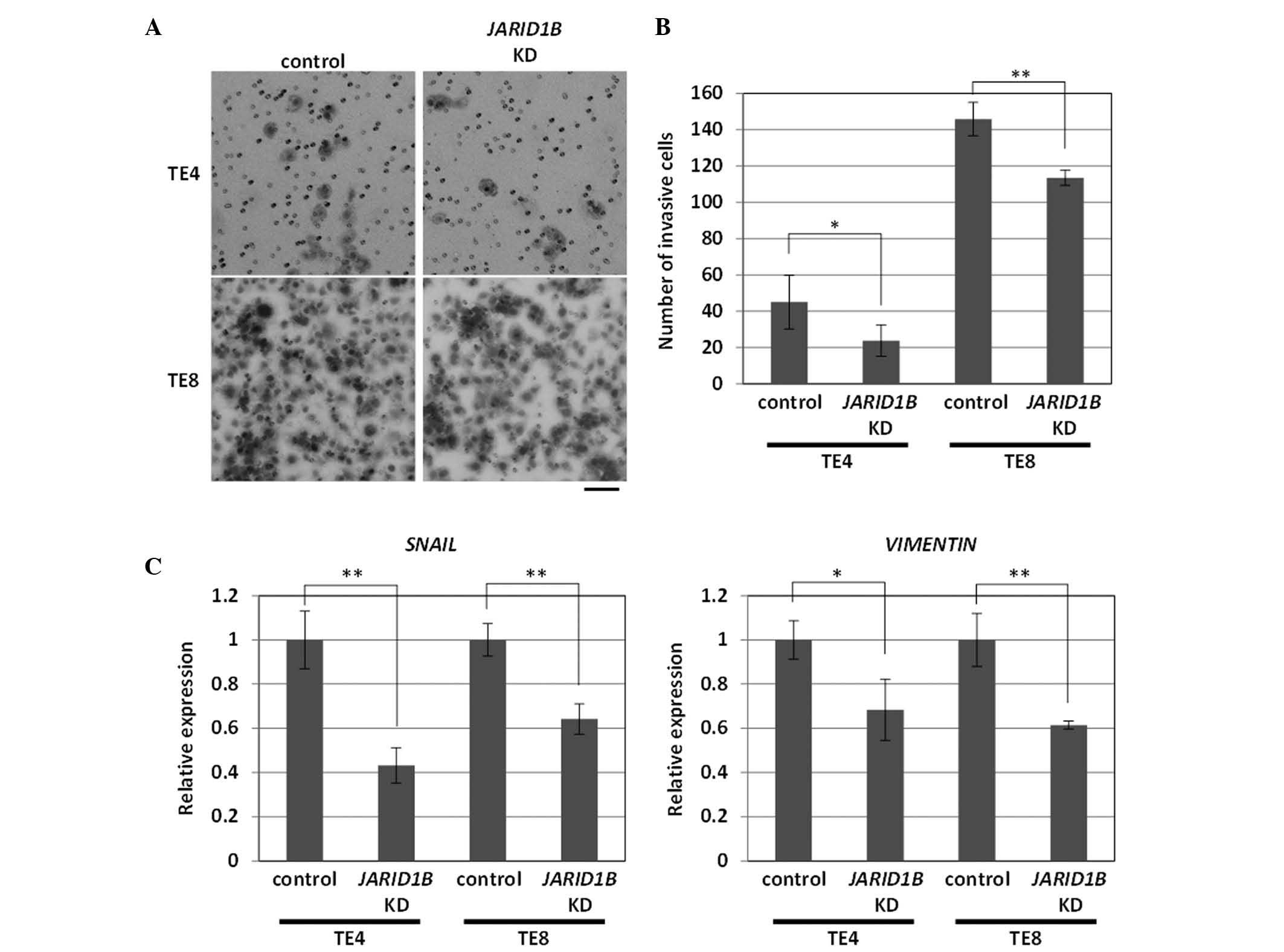
Cancer invasion and metastasis are frequently
associated with cancer heterogeneity and are important factors that
affect cancer management (12,13).
Thus, the control of cancer invasion is crucial. Concomitant with
the observed cell growth inhibition, the invasion ability of
JARID1B knockdown TE4 cells was significantly suppressed
(Fig. 2A). Similar results were
obtained with JARID1B knockdown TE8 cells, although total
cell invasion was more apparent than with TE4 cells (Fig. 2A).
To explore the possible underlying mechanisms, we
examined the expression of epithelial-mesenchymal transition (EMT)
genes, ES-like genes (SOX2, OCT3/4, KLF4 and
c-MYC) for which aggressive phenotypes have been suggested
(14) and tumor suppressor genes
(p21/Waf1/Cip1/Sdi1 and p16/INK4A). We found
reproducible results for the significant inhibition of EMT-related
genes, SNAIL and VIMENTIN, in JARID1B
knockdown TE4 and TE8 cells (Fig.
2B). Thus, these results indicated that JARID1B
knockdown reduced tumor cell growth and invasion via the induction
of a network of EMT-related genes.
JARID1B knockdown suppresses esophageal
cancer sphere formation
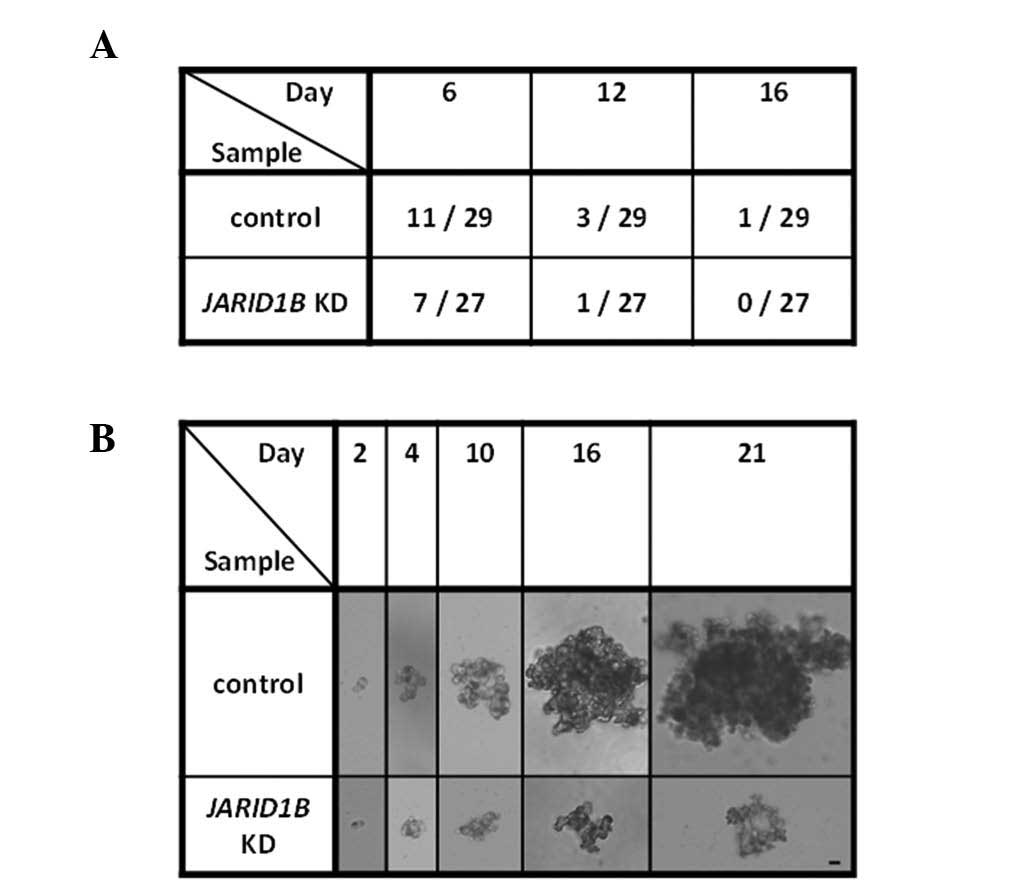
Concerning the heterogeneity of cells within tumors,
the involvement of cancer stem cells has been discussed with regard
to self-renewal and re-establishment of tumor tissues (15,16).
To assess the self-renewal of cancer cells, TE4 and TE8
JARID1B knockdown and control transfectant cells were used
in sphere formation assays. JARID1B knockdown resulted in
the inhibition of sphere formation as observed on days 6, 12 and 16
(representative data shown in Fig. 3A
and B). Thus, these results suggest that JARID1B
knockdown reduced the self-renewal activity of esophageal cancer
cells.
JARID1B knockdown suppresses esophageal
cancer tumorigenicity
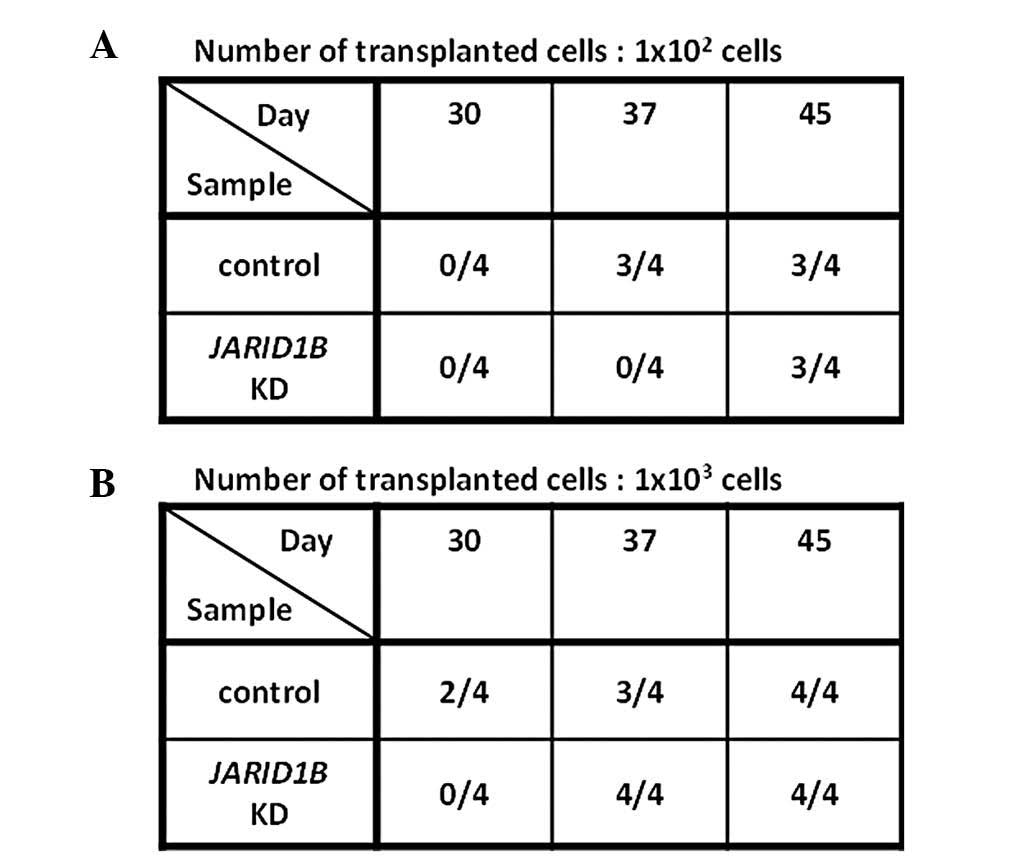
The effects of JARID1B knockdown in
vivo were examined by inoculating JARID1B knockdown TE4
and TE8 cells into immune-deficient NOD/SCID mice. When
102 JARID1B knockdown TE4 or TE8 cells were
inoculated subcutaneously into mice, tumorigenicity was reduced as
observed on days 30 and 37 (representative data shown in Fig. 4A).
However, our vector system used an antibiotics
selection system to enrich the transfectants and our in vivo
observations were made in the absence of antibiotics selection.
Thus, reversed clones that escaped from an initial treatment with
JARID1B knockdown may have developed after a long period of
time. Consistent with this possibility, observations on day 45
indicated that even initially-JARID1B knockdown
vector-treated cells exhibited tumorigenicity. This suggested that
some lentiviral-mediated JARID1B knockdown cells may have
lost the transgene, leading to the development of transgene-free
clones.
Similarly, inoculating 103 cells
initially showed reduced tumorigenicity on day 30, although tumor
growth was observed on day 45. These results indicated that,
although JARID1B inhibition may be a candidate molecular
target for cancer therapy, a continuous inhibition system would be
necessary to achieve eradication of therapy-resistant esophageal
cancer.
Discussion
In general, methylation and demethylation of
histones turns genes ‘off’ and ‘on’ either by loosening their
tails, which allows transcriptional factors to access DNA, or by
reversing this access. Dysregulation of these activities are
hallmarks of cancer through genetic and epigenetic alterations
(12,13).
It was recently observed that Jarid1a/b-mediated
demethylation of histone H3K4 contributed to silencing
retinoblastoma target genes in senescent cells, presumably through
closing the chromatin in which the silencing of retinoblastoma
trigger genes was involved (17).
Thus, distinct senescence-associated changes in
histone-modification patterns are consistent with a repressive
chromatin environment in the retinoblastoma tumor suppressor
pathway (17). The results of the
present study indicated that JARID1B knockdown (i.e.,
inhibition of H3K4 demethylation) resulted in the suppression of
tumor cell growth in vitro and in vivo. This suggests
that JARID1B is involved in regulating tumor cell growth in
the human esophagus and is in agreement with findings of a previous
report on melanoma (11).
Among retinoblastoma-mediated genes, tumor
suppressor p16/INK4A is well documented as being involved
with a senescence-associated phenotype (18). p16/INK4A-mediated senescence
occurs through the retinoblastoma-inhibiting action of
cyclin-dependent kinases and leads to G1 cell cycle arrest
(18) through the interplay
between their pathways and reactive oxygen species (ROS) (19). Our study indicated that exposure to
hydrogen peroxide (a typical inducer of ROS) did not result in any
apparent induction of a senescence-associated phenotype in
esophageal squamous cell carcinoma cells that lacked tumor
suppressor p16/INK4A in the retinoblastoma pathway. This
suggests a role for p16/INK4A in inducing a
senescence-associated phenotype with JARID1B inhibition
(data not shown).
Therapeutic approaches for esophageal cancer include
conventional treatments, such as surgical removal and
chemoradiation treatment as well as gene therapy strategies, such
as the introduction of the tumor suppressor p16/INK4A
(20), expression of IL-2,
IL-6 and GM-CSF gene products (21,22),
and the transduction of the herpes simplex virus-thymidine kinase
gene (23,24). To achieve continuous knockdown of
JARID1B, options include antisense oligonucleotides or low
molecular therapeutic pharmacology (25). As an example, a combination of
introducing the tumor suppressor gene p16/INK4A as gene
therapy with anti-JARID1B treatment potentially leads to the
efficient induction of a senescence-associated phenotype in
esophageal cancer. This combination therapy would be efficient for
eradicating therapy-resistant cancer cells, which survive after
conventional treatment such as surgery, chemotherapy and radiation
therapy.
Acknowledgements
This study was partly supported by a
grant from the Core Research for a Grant-in-Aid for Scientific
Research from the Ministry of Education, Culture, Sports, Science
and Technology (H.I., M.M.), a Grant-in-Aid for the 3rd
Comprehensive 10-year Strategy for Cancer Control Ministry of
Health, Labour and Welfare (H.I., M.M.), a grant from the Kobayashi
Cancer Research Foundation (H.I.) and a grant from the Princess
Takamatsu Cancer Research Fund, Japan (H.I.).
References
|
1.
|
Hansson LE, Sparen P and Nyren O:
Increasing incidence of both major histological types of esophageal
carcinomas among men in Sweden. Int J Cancer. 54:402–407. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Ozols R: Esophageal cancer. Curr Problems
Cancer. XVIII:191–246. 1994.
|
|
3.
|
Conteduca V, Sansonno D, Ingravallo G,
Marangi S, Russi S, Lauletta G and Dammacco F: Barrett’s esophagus
and esophageal cancer: An overview. Int J Oncol. 41:414–424.
2012.
|
|
4.
|
di Pietro M, Lao-Sirieix P, Boyle S,
Cassidy A, Castillo D, Saadi A, Eskeland R and Fitzgerald RC:
Evidence for a functional role of epigenetically regulated
midcluster HOXB genes in the development of Barrett esophagus. Proc
Natl Acad Sci USA. 109:9077–9082. 2012.PubMed/NCBI
|
|
5.
|
Coia LR: The esophagus. Moss’ Radiation
Oncology: Rationale, Techniques, Results. Cox J: Mosby-Year Book;
St. Louise: pp. 4091994
|
|
6.
|
Follis RH Jr: The pathology of zinc
deficiency. Zinc Metabolism. Prasad AS: Springfield; pp. 129–141.
1966
|
|
7.
|
Taccioli C, Chen H, Jiang Y, Liu XP, Huang
K, Smalley KJ, Farber JL, Croce CM and Fong LY: Dietary zinc
deficiency fuels esophageal cancer development by inducing a
distinct inflammatory signature. Oncogene. 31:4550–4558. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Alder H, Taccioli C, Chen H, Jiang Y,
Smalley KJ, Fadda P, Ozer HG, Huebner K, Farber JL, Croce CM and
Fong LY: Dysregulation of miR-31 and miR-21 induced by zinc
deficiency promotes esophageal cancer. Carcinogenesis.
33:1736–1744. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kaz AM and Grady WM: Epigenetic biomarkers
in esophageal cancer. Cancer Lett. Mar 7–2012.(Epub ahead of
print).
|
|
10.
|
Shoji M, Ninomiya I, Makino I, Kinoshita
J, Nakamura K, Oyama K, Nakagawara H, Fujita H, Tajima H, Takamura
H, Kitagawa H, Fushida S, Harada S, Fujimura T and Ohta T: Valproic
acid, a histone deacetylase inhibitor, enhances radiosensitivity in
esophageal squamous cell carcinoma. Int J Oncol. 40:2140–2146.
2012.PubMed/NCBI
|
|
11.
|
Roesch A, Fukunaga-Kalabis M, Schmidt EC,
Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T
and Herlyn M: A temporarily distinct subpopulation of slow-cycling
melanoma cells is required for continuous tumor growth. Cell.
141:583–594. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
13.
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell
LL, Polyak K, Brisken C, Yang J and Weinberg RA: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Dewi DL, Ishii H, Kano Y, Nishikawa S,
Haraguchi N, Sakai D, Satoh T, Doki Y and Mori M: Cancer stem cell
theory in gastrointestinal malignancies: recent progress and
upcoming challenges. J Gastroenterol. 46:1145–1157. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Chicas A, Kapoor A, Wang X, Aksoy O,
Evertts AG, Zhang MQ, Garcia BA, Bernstein E and Lowe SW: H3K4
demethylation by Jarid1a and Jarid1b contributes to
retinoblastoma-mediated gene silencing during cellular senescence.
Proc Natl Acad Sci USA. 109:8971–8976. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Rayess H, Wang MB and Srivatsan ES:
Cellular senescence and tumor suppressor gene p16. Int J Cancer.
130:1715–1725. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Vurusaner B, Poli G and Basaga H: Tumor
suppressor genes and ROS: complex networks of interactions. Free
Radic Biol Med. 52:7–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Schrump DS, Chen GA, Consuli U, Jin X and
Roth JA: Inhibition of esophageal cancer proliferation by
adenovirally mediated delivery of p16INK4. Cancer Gene Ther.
3:357–364. 1996.PubMed/NCBI
|
|
21.
|
Matsubara H, Tagawa M, Gunji Y, Takenaga
K, Sugaya M, Urashima T, Koide Y, Suzuki T, Asano T, Ochiai T,
Isono K, Kageyama H, Nakamura Y and Sakiyama S: Study of
irradiation effects on cytokine secretion from
retrovirally-transduced tumor cells: a model for tumor vaccination.
Anticancer Res. 16:645–650. 1996.PubMed/NCBI
|
|
22.
|
Matsubara H, Koide Y, Sugaya M, Gunji Y,
Asano T, Ochiai T, Takegana K, Sakiyama S and Tagawa M: Antitumor
response of genetically engineered IL-2 expression to human
esophageal carcinoma cells in mature T cell-defective condition.
Int J Oncol. 13:1217–1239. 1998.
|
|
23.
|
Miyauchi M, Shimada H, Kadomatsu K,
Muramatsu T, Matsubara S, Ikematsu S, Takenaga K, Asano T, Ochiai
T, Sakiyama S and Tagawa M: Frequent expression of midkine gene in
esophageal cancer suggests a potential usage of its promoter for
suicide gene therapy. Jpn J Cancer Res. 90:469–475. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Matsubara H, Kawamura K, Sugaya M, Koide
Y, Gunji Y, Takenaga K, Asano T, Ochiai T, Sakiyama S and Tagawa M:
Differential efficacy of suicide gene therapy by herpes simplex
virus-thymidine kinase gene reflects the status of p53 gene in
human esophageal cancer cells. Anticancer Res. 19:4157–4160.
1999.
|
|
25.
|
Yamamoto T, Nakatani M, Narukawa K and
Obika S: Antisense drug discovery and development. Future Med Chem.
3:339–365. 2011. View Article : Google Scholar
|