Introduction
Primary hepatic angiosarcoma (PHA) is an aggressive
malignant neoplasm arising from the endothelium of blood vessels
within the liver (1). PHA is rarely
encountered in Western countries (2–4), where it
reportedly accounts for 0.6% (1) to
2% (4) of all primary liver
tumours.
Due to its aggressive growth patterns and indistinct
clinical characteristics, the diagnosis of PHA is often made
post-mortem (1,4). Approximately 1 in 5 patients are
diagnosed in vivo when they present with capsular rupture
and intraperitoneal bleeding (2,4,5); these tumours, however, tend to be
locally advanced when diagnosed ante-mortem (4).
This is the case report of a 60-year old female
patient who presented with a ruptured hepatic tumour
histopathologically diagnosed as PHA and a review of the
clinicopathological characteristics of PHAs.
Case report
A 60-year old woman with no known chronic medical
conditions experienced vague upper abdominal pain for 6 weeks prior
to presentation. The patient reported no weight loss, anorexia,
gastrointestinal symptoms or history of trauma. The physical
examination revealed no abnormalities.
The blood tests revealed a haemoglobin count of
10,000/dl. The electrolyte levels and the renal and liver function
tests were normal. An endoscopic evaluation excluded the presence
of gastrointestinal lesions. However, the abdominal ultrasound
revealed the presence of free intraperitoneal fluid and a tumour in
segment III of the liver. Contrast-enhanced computed tomography
scans of the chest, abdomen and pelvis confirmed the presence of
perihepatic fluid and the presence of a heterogenous 4-cm tumour in
segment III of the liver. The remaining segments of the liver were
normal, with no evidence of metastatic disease.
The patient was put under general anaesthesia and
underwent laparoscopic liver resection. There was no evidence of
metastatic disease in the peritoneal cavity and the tumour was
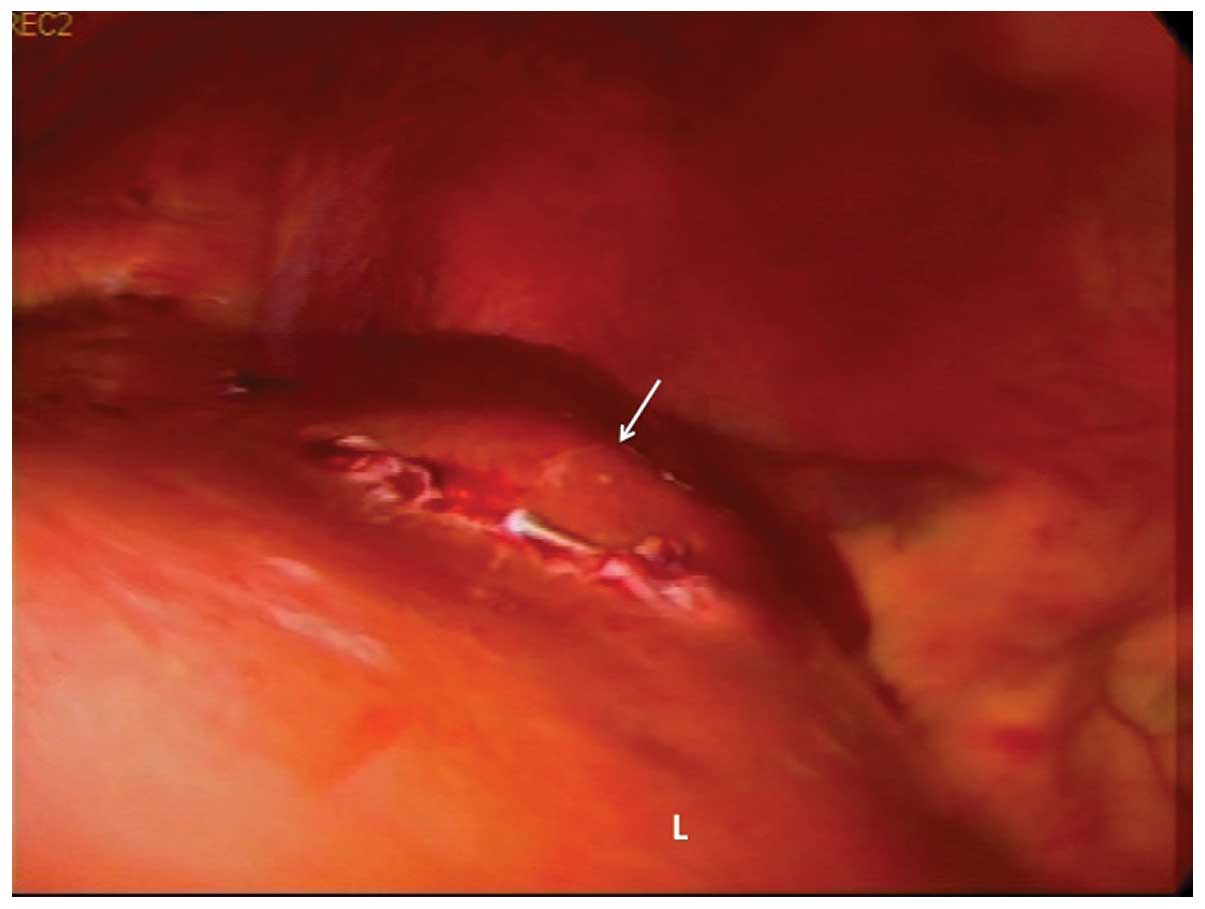
readily identified by its bosselated surface (Fig. 1). Laparoscopic ultrasonography was
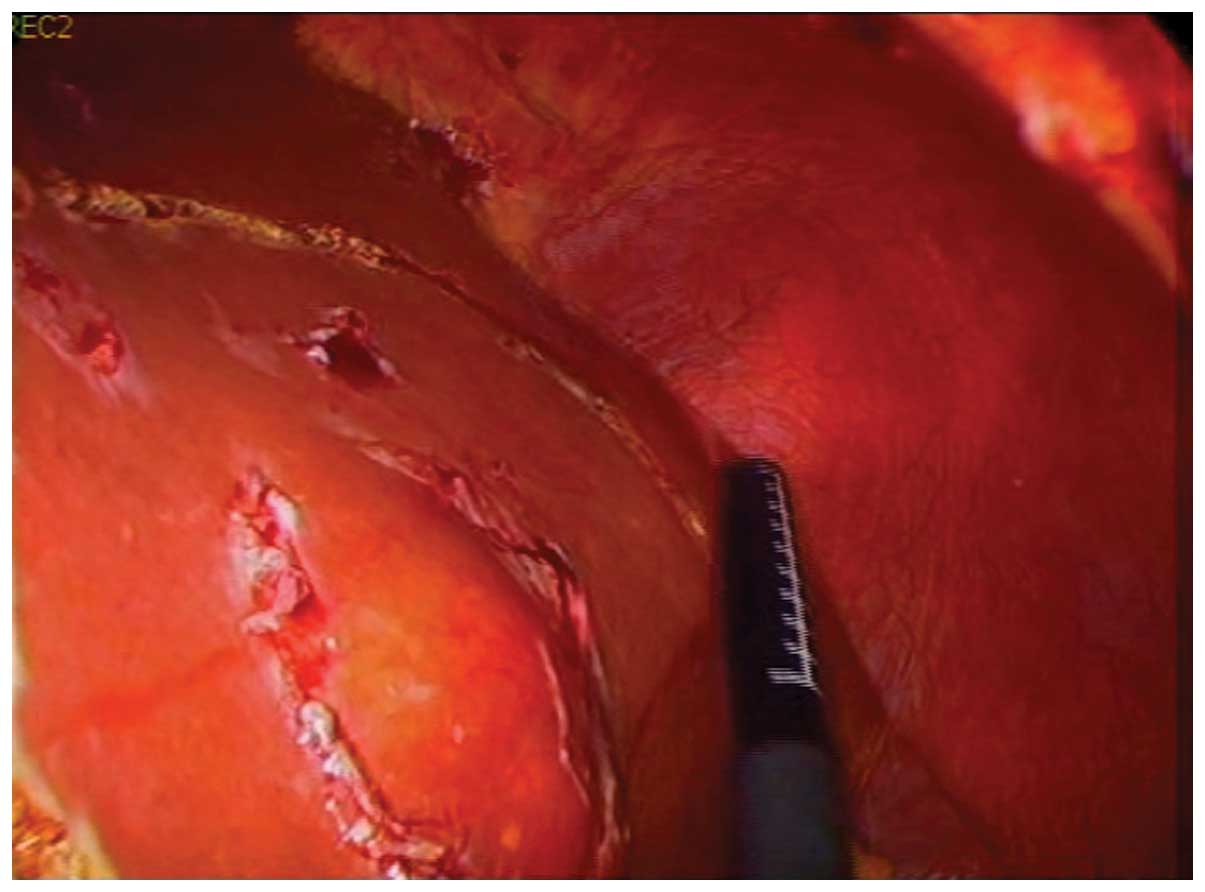
used to achieve clear resection margins (Fig. 2). A laparoscopic left lateral
sectionectomy was completed uneventfully.
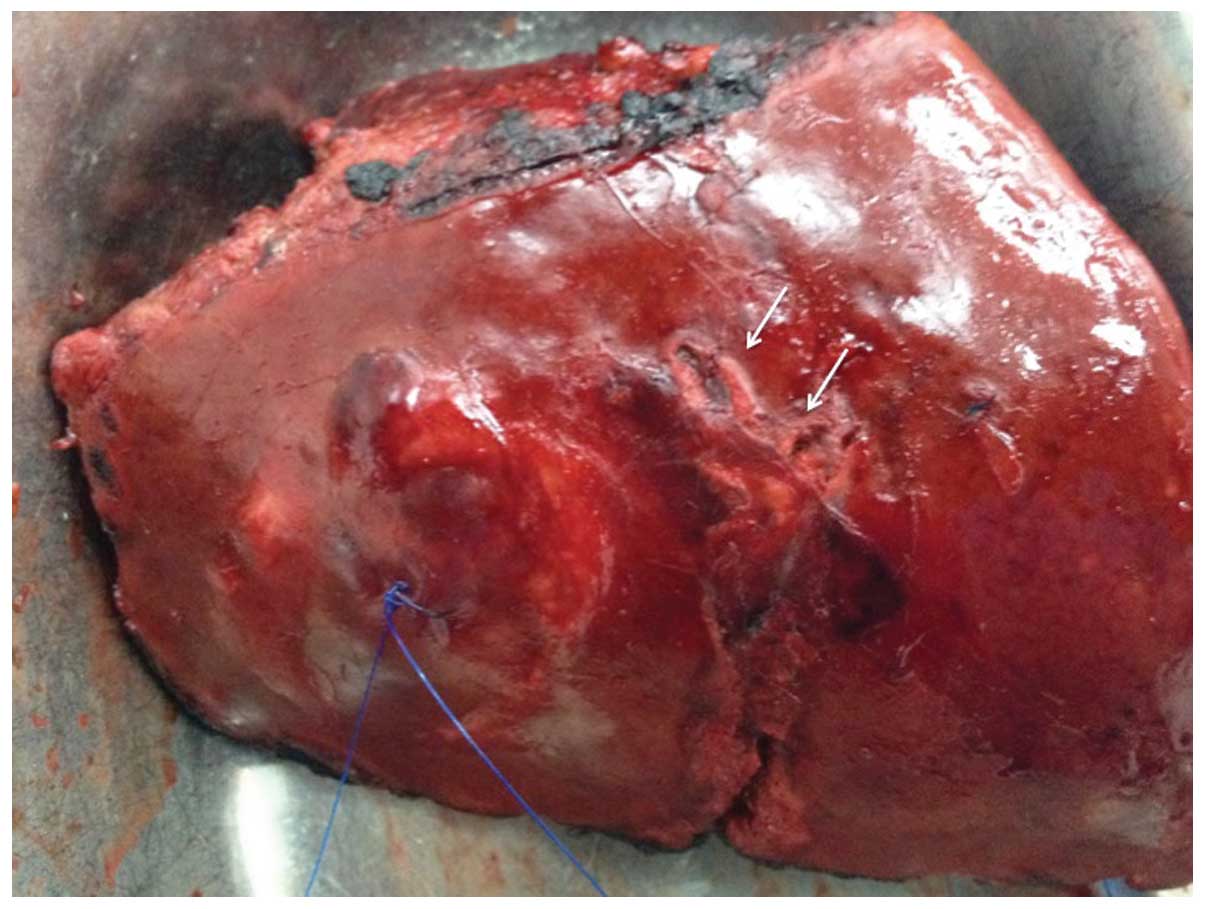
The pathological examination confirmed the presence
of a tumour sized 3×4×2.5 cm, with a focal point at which Glisson's
capsule was breached (Fig. 3). The
resection margins were clear of tumour for >2 cm.
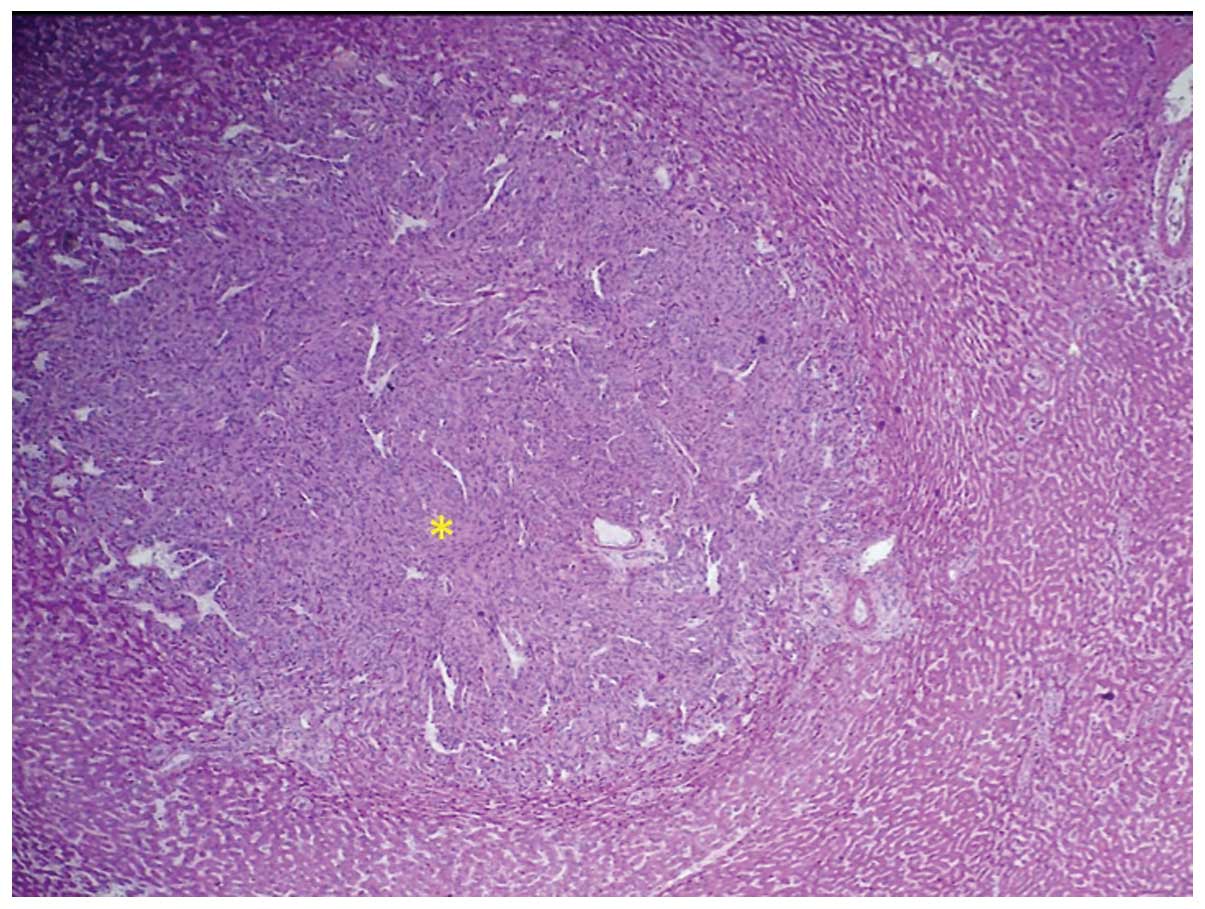
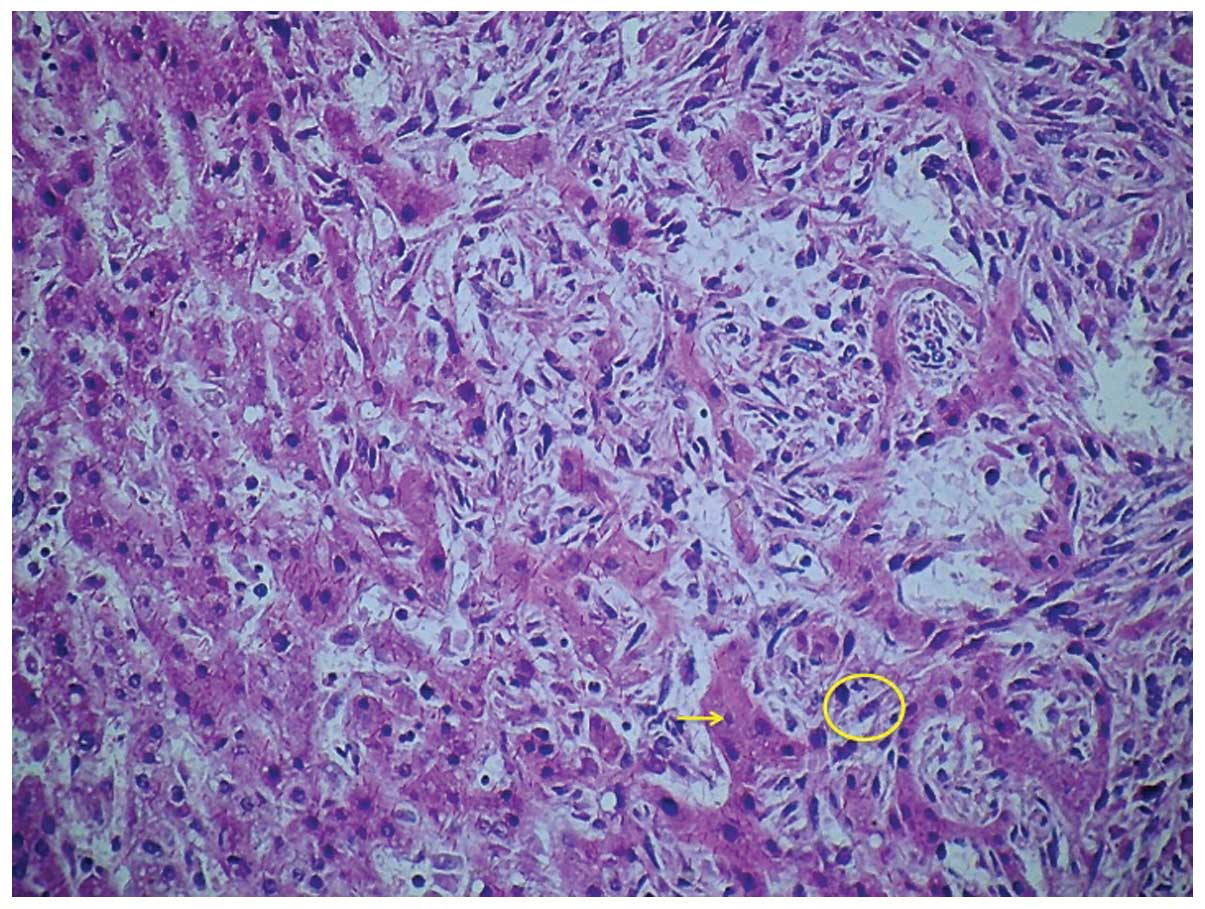
Histologically, the tumour was composed of sheets of poorly
differentiated neoplastic cells (Fig.
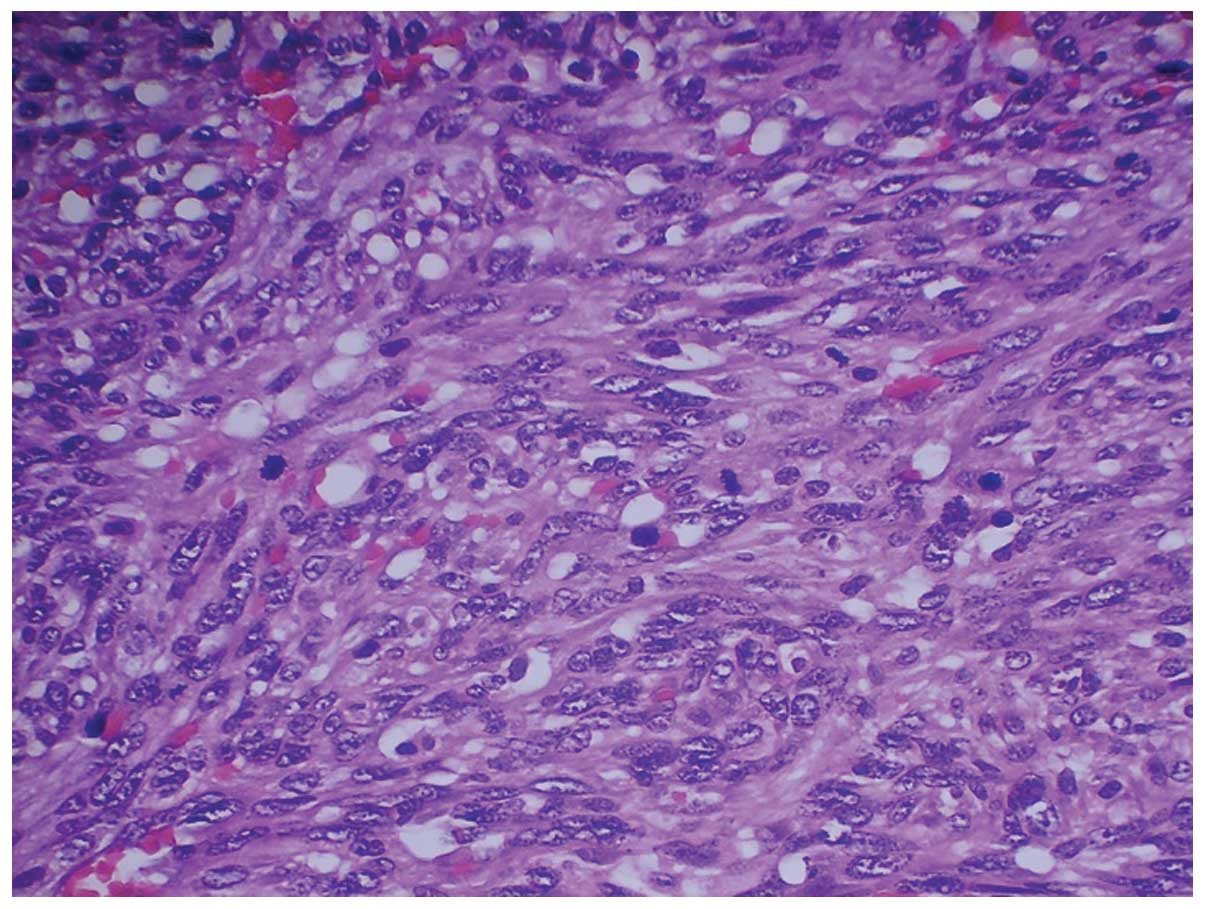
4). The malignant cells were spindle-shaped and possessed
hyperchromatic nuclei, indistinct cell borders and scant cytoplasm
(Fig. 5). Significant abnormal
mitotic activity was observed and bizarre multinucleated giant
cells were present, particularly at the periphery of the lesion
(Fig. 6). The tumour
characteristically grew along the hepatic sinusoids and around
residual hyperplastic hepatocytes, which were otherwise normal.
There were scattered islands of haematopoietic cells and large
areas of thrombosis and infarction, with accompanying tumour
necrosis. The histological appearance indicated a high-grade
pleomorphic sarcoma consistent with PHA. The diagnosis was verified
by immunohistochemistry, which was positive for CD31 and factor
VIII (FVIII)-R antigen, but negative for desmin, keratin, S-100
protein and discovered on GIST-1.
Discussion
Angiosarcomas are aggressive malignant neoplasms
originating from the endothelium of blood vessels (2). The liver is the fifth most common site
of origin for angiosarcomas (2–3). Although
it accounts for 0.6% (1) to 2%
(4) of primary liver tumours in
Western countries, PHA is a rare occurrence (1–4). The three
largest published series are audits from national sarcoma
registries, with a limited number of cases, namely 6 cases over 15
years in China, 26 cases over 19 years in Taiwan (6) and 32 cases over 16 years in Britain
(7).
In the largest reported series of 32 cases from the
British Hepatic Angiosarcoma Register, Baxter (7) reported the annual incidence of PHA to be
0.16 cases per million persons. An annual incidence as high as 0.26
per million was reported in an audit from the United States by
Vianna et al (8). There has
been no prior report on PHA from the Anglophone Caribbean, a region
consisting of 17 English-speaking Caribbean countries with a
cumulative population of 6.5 million (9). We commenced a hepatobiliary registry to
record all primary and metastatic liver tumours encountered in the
three main referral centres for the Anglophone Caribbean in
January, 2011 (9). This was the first
case encountered in the registry, allowing us to estimate the
annual incidence as 0.05 per million population in the Anglophone
Caribbean, which is comparable to the incidence in Western
countries (7,8).
Our patient was a 60-year old woman; this is the
usual age at which a PHA is diagnosed (4), although they are more common in men
(4,5,7). The
reported male:female ratio ranges from 1.9:1 in Taiwan (7) to as high as 4:1 in Britain (8).
We were unable to identify an aetiological factor in
our patient. Early studies suggested that environmental exposure to
Thorotrast (4,7), vinyl (7),
polyvinyl chloride-manufacturing materials (7), arsenic, pesticides and radium (4) were risk factors for PHA; however,
exposure to these agents has become uncommon in modern practice. It
was not surprising, therefore, that neither of these environmental
risk factors were present in our case.
PHAs are often recognized only at autopsy (4,10), as the
diagnosis is often missed in vivo when patients report
vague, non-specific symptoms and the tumour exhibits aggressive
growth patterns (2,4,10,11). When the diagnosis is made ante-mortem,
PHAs are usually identified at an advanced stage, when therapeutic
options are limited (4,12,13). In
our case, the diagnosis was made relatively early in the absence of
metastatic or locally advanced disease.
The patient presented after the tumour had ruptured.
Intraperitoneal haemorrhage following tumour rupture appears to be
a relatively common method of acute presentation (2,4,5,14,15), usually accompanied by a high mortality
risk due to exsanguination (2,4,15). Survivors usually present with a herald
bleed, as in our case. This presentation accounts for 22% (4) to 27% (10)
of the cases diagnosed ante-mortem.
The histological characteristics observed in this
case were typical of PHA. Normal hepatocytes were observed, with
infiltrating islands of neoplastic endothelial cells extending
along sinusoidal vascular channels (2,4). The
individual neoplastic cells are often anaplastic or spindle-shaped,
with pale cytoplasm, pleomorphic nuclei and indistinct borders
(4). The presence of bizzare giant
cells and epithelioid cell patterns, as seen in this case, are also
characteristic (2). The cells stained
positive for CD31 and FVIII, which are characteristic of PHA
(2,4).
Although not present in this case, PHA cells may also stain
positive for CD34 (2,4) and vimentin (2).
PHAs are aggressive tumours. Without treatment, the
median survival is reported to be only 6 months from the time of
diagnosis (2,4,10,16). As the response to systemic
chemotherapy and radiotherapy is unpredictable (2,17), it is
crucial for surgeons to achieve complete resection with clear
margins. However, even following complete (R0) resection, long-term
survival is poor, with existing 5-year survival rate estimates of
3% (2,4,10,16).
In conclusion, PHAs are rare tumours, with an
estimated annual incidence of 0.05 per million in the Caribbean.
Intraperitoneal haemorrhage from tumour rupture appears to be a
common method of presentation. Patients with herald bleeds should
be urgently treated, prior to the development of exsanguinating
haemorrhage, by hepatobiliary surgeons performing aggressive
resection to ensure clear margins.
References
|
1
|
Molina E and Hernandez A: Clinical
manifestations of primary hepatic angiosarcoma. Dig Dis Sci.
48:677–682. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chien CY, Hwang CC, Yeh CN, Chen HY, Wu
JT, Cheung CS, Lin CL, Yen CL, Wang WY and Chiang KC: Liver
angiosarcoma, a rare liver malignancy, presented with
intraabdominal bleeding due to rupture - a case report. World J
Surg Oncol. 10:232012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Young RJ, Brown NJ, Reed MW, Hughes D and
Woll PJ: Angiosarcoma. Lancet Oncol. 11:983–991. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang NC, Wann SR, Chang HT, Lin SL, Wang
JS and Guo HR: Arsenic, vinyl chloride, viral hepatitis, and
hepatic angiosarcoma: A hospital-based study and review of
literature in Taiwan. BMC Gastroenterol. 11:1422011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ho S-Y, Tsai C-C, Tsai Y-C and Guo H-R:
Hepatic angiosarcoma presenting as hepatic rupture in a patient
with long-term ingestion of arsenic. J Formos Med Assoc.
103:374–379. 2004.PubMed/NCBI
|
|
6
|
Wang TH, Pan ZG and Ren ZG: Rare primary
liver malignant tumor. Fudan Univ J Med Sci. 36:221–224. 2009.
|
|
7
|
Baxter PJ: The British hepatic
angiosarcoma register. Environ Health Perspect. 41:115–116. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vianna NJ, Brady JA and Cardamone AT:
Epidemiology of angiosarcoma of liver in New York State. N Y State
J Med. 81:895–899. 1981.PubMed/NCBI
|
|
9
|
Cawich SO, Johnson PB, Shah S, et al:
Overcoming obstacles to establish a multidisciplinary team approach
to hepatobiliary diseases: a working model in a Caribbean setting.
J Multidiscip Healthc. 7:227–230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Locker GY, Doroshow JH, Zwelling LA and
Chabner BA: The clinical features of hepatic angiosarcoma: A report
of four cases and a review of the English literature. Medicine
(Baltimore). 58:48–64. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Timaran CH, Grandas OH and Bell JL:
Hepatic angiosarcoma: Long-term survival after complete surgical
removal. Am Surg. 66:1153–1157. 2000.PubMed/NCBI
|
|
12
|
Poggio JL, Nagorney DM, Nascimento AG,
Rowland C, Kay P, Young RM and Donohue JH: Surgical treatment of
adult primary hepatic sarcoma. Br J Surg. 87:1500–1505. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jaffe BM, Donegan WL, Watson F and Spratt
JS Jr: Factors influencing survival in patients with untreated
hepatic metastases. Surg Gynecol Obstet. 127:1–11. 1968.PubMed/NCBI
|
|
14
|
Tsai CC, Hsieh JF, Han SJ and Mo LR:
Hemoperitoneum secondary to biopsy of the hepatic angiosarcoma.
Chin J Radiol. 24:37–40. 1999.
|
|
15
|
Lee SW, Song CY, Gi YH, Kang SB, Kim YS,
Nam SW, Lee DS and Kim JO: Hepatic angiosarcoma manifested as
recurrent hemoperitoneum. World J Gastroenterol. 14:2935–2938.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Falk H, Herbert JT, Edmonds L, Heath CW
Jr, Thomas LB and Popper H: Review of four cases of childhood
hepatic angiosarcoma - elevated environmental arsenic exposure in
one case. Cancer. 47:382–391. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Almogy G, Lieberman S, Gips M, Pappo O,
Edden Y, Jurim O, Simon Slasky B, Uzieli B and Eid A: Clinical
outcomes of surgical resections for primary liver sarcoma in
adults: Results from a single centre. Eur J Surg Oncol. 30:421–427.
2004. View Article : Google Scholar : PubMed/NCBI
|