Introduction
Germ cell tumors (GCTs) are the most common
malignancies in males aged 15–35 years, whereas only 2–5% of these
tumors arise in extragonadal sites (1). The most common extragonadal
localization is the mediastinum, followed by the retroperitoneum,
pineal gland and suprasellar region (2,3). GCTs
comprise a variety of histologically different types that carry
different prognoses. The presence of yolk sac elements is
associated with a dismal prognosis (4–6) and is
found in 30–40% of GCTs (1,7). Yet pure yolk sac tumors (YSTs) in adult
males are rare. We herein report a case of an extragonadal YST with
hepatoid differentiation (hepYST), primary localized in the brain
and lung.
Case report
A 41-year old man was admitted to our emergency
department with a generalized seizure. No motor or sensory symptoms
were present, but there was retrograde amnesia and altered mental
status. The physical examination was unremarkable. The patient's
past medical history included orchidopexy in childhood.
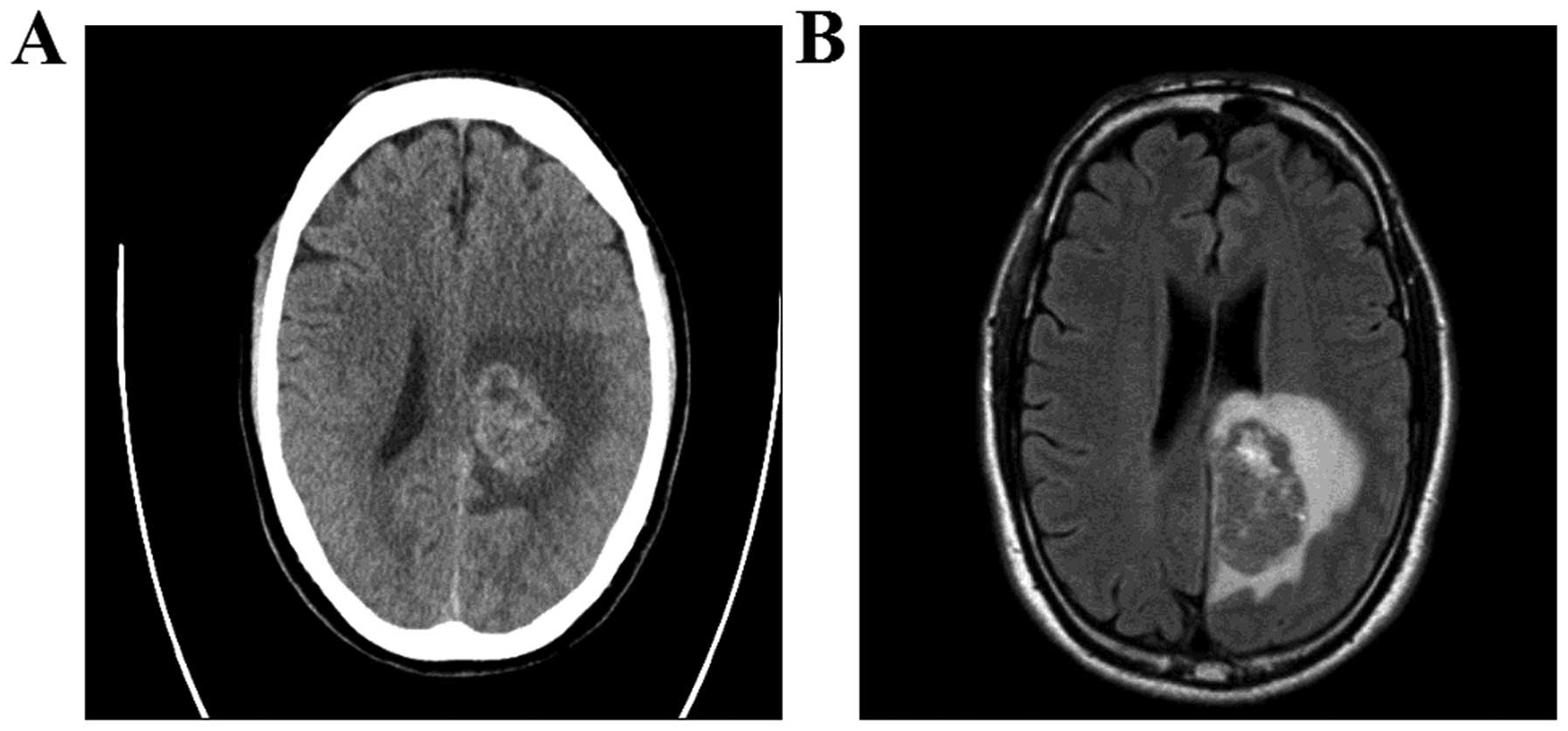
A head computed tomography (CT) and magnetic
resonance imaging (MRI) examination revealed a large mass (5×3 cm)
in the left occipital lobe with associated edema (Fig. 1). The patient underwent total tumor
removal via left occipital craniotomy. The postoperative course was
complicated by severe transitional hemiparesis.
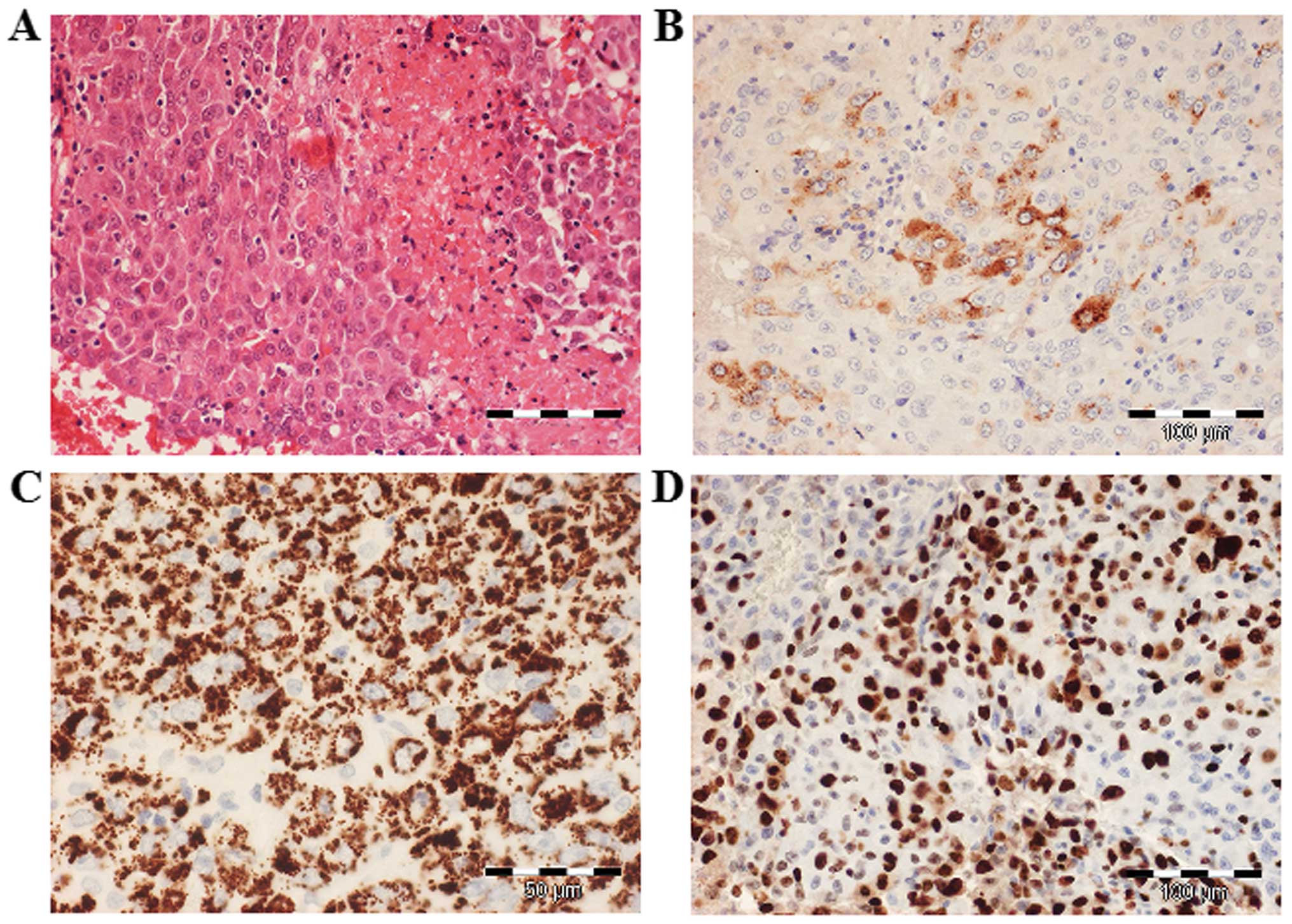
The histopathological examination of the resected
tumor revealed large, eosinophilic cells with round, centrally
located nuclei, arranged in cords or trabeculae. The tumor cells
focally contained PAS-positive intracytoplasmic and
extracytoplasmic hyaline bodies. Immunohistochemically, the tumor
cells were diffusely positive for Hep Par-1, glypican-3 and
cytokeratin (CK) 8; α-fetoprotein (AFP) was focally expressed and
Ki-67 staining revealed 80% positive tumor cell nuclei, indicating
a very high proliferation activity (Fig.
2). By contrast, the tumor cells were negative for placental
alkaline phosphatase, octamer-binding transcription factor 3/4,
CK20, CD30 and c-kit. The immunohistochemical findings were thus
consistent with malignant epithelial GCT with a hepatoid
pattern.
The serum lactate dehydrogenase (LDH) and AFP levels
were markedly elevated (Table I).
The serum human chorionic gonadotropin level was within the normal
range. Chest CT revealed a lesion sized 2×1 cm in the upper lobe of
the right lung. No other lesions were identified using testicular
ultrasound, abdominal CT, liver MRI and gastroscopy.
 | Table I.Tumor marker levels during the course
of the disease. |
Table I.
Tumor marker levels during the course
of the disease.
| Treatment | AFP
(ng/ml)a | LDH
(µmol/sec/l)b |
|---|
|
Preoperative/naïve | n/a | 7.53 |
| Postoperative | 265 | 3.40 |
| After 1 PEI | 230 | 3.48 |
| After 2 PEI | 410 | 4.53 |
| After 1 TI | 332 | 12.76 |
| After 2 TI | 262 | 3.37 |
| After 1 CE +
autoTx | 614 | 4.08 |
| After radiation and
lung resection | 397 | 6.43 |
| After 2 CE +
autoTx | 559 | 8.62 |
Taking all laboratory findings into account, the
diagnosis of an extragonadal hepYST was considered to be
likely.
Initially, the patient underwent two courses of
cisplatin, etoposide and ifosfamide (PEI regimen). However,
subsequent restaging revealed an increase in the serum AFP and LDH
levels, as well as an increase in the size of the intracranial
lesion; the lung lesion remained stable. Given the refractory
disease and unfavorable prognostic characteristics (8), a decision was made to treat the patient
with a high-dose salvage chemotherapy protocol according to
Kondagunta et al [two courses of paclitaxel and ifosfamide
(TI regimen) followed by three courses of high-dose carboplatin and
etoposide (CE regimen) plus peripheral blood stem cell support]
(9). However, after the first
high-dose chemotherapy course, the patient developed hemiparesis.
The serum AFP and LDH levels increased and the head CT revealed
marked tumor progression. In order to achieve a fast tumor mass
reduction, cranial irradiation and surgical removal of the lung
tumor were performed. The histopathological examination was
consistent with hepYST. High-dose chemotherapy (two remaining
high-dose CE courses) was resumed as ultima ratio for
disease control.
Unfortunately, despite therapy, subsequent restaging
showed an increase in tumor markers, as well as new and multiple
lung and liver metastases. Palliative therapy with oral etoposide
was administered and the patient succumbed to refractory disease 10
months after the initial diagnosis.
Discussion
YSTs are rare, aggressive tumors, occurring mainly
in young adults, with a peak incidence at 21 years (10,11).
YSTs arise most commonly in the gonads, but extragonadal sites of
origin are reported in 24% of the cases (11). The most common localization of YST is
the anterior mediastinum, followed by the retroperitoneum and
cranium (2,3); exceedingly rare sites, such as the
lungs, pancreas, kidney and spinal cord, have also been reported
(12–15). Regarding the intracranial
manifestations, YSTs are typically located midline in the pineal
region or the suprasellar region (3). One case of mixed GCT with extensive
yolk sac elements outside the midline (in the frontal lobe) in an
adult was also reported (16).
Intracranial YSTs present a unique entity. They may
disseminate along the neuroaxis, at the time of diagnosis or early
during the course of the disease. However, to the best of our
knowledge, a case of intracranial YST with concomitant extracranial
manifestation has not been reported thus far. In the case presented
herein, the tumor was located in the occipital lobe and lungs. Due
to its atypical location and the radiological findings (absence of
necrosis, hemorrhage, cysts) (17),
the suspected preoperative diagnosis was primary brain tumor rather
than GCT. However, the histological examination did not support
this diagnosis. Immunohistochemically, the tumor cells expressed
Hep Par-1, glypican 3 and AFP; this immunoprofile is compatible
with hepatocellular carcinoma (HCC), hepatoid adenocarcinoma (HAC),
as well as hepYST (Table II)
(10). Hep Par-1 is expressed in
normal and neoplastic hepatocytes and has been used to confirm
hepatoid differentiation. The degree of staining may correlate with
hepatoid differentiation, and a strong Hep Par-1 positivity favors
HCC diagnosis, but is found only in a minority of hepYST (18). Glypican 3 and AFP are also quite
specific for hepatocellular differentiation and are typically
expressed in HCC, HAC and hepYST (18).
| Table II.Characteristics of immunohistochemical
staining of hepatocellular carcinoma, hepatoid adenocarcinoma and
hepatoid yolk sac tumor. |
Table II.
Characteristics of immunohistochemical
staining of hepatocellular carcinoma, hepatoid adenocarcinoma and
hepatoid yolk sac tumor.
| Staining | HCC | HAC | hepYST |
|---|
| AFP | + | + | + |
| pCEA | + | + | + |
|
| Canalicular | Diffuse
membranous |
|
|
| pattern | or canalicular
patterns |
|
| Glypican 3 | + | ++ | + |
| SALL4 | + | + | ++ |
|
| (Cut-off: 7% in 25%
of cells) |
| (Cut-off: 100% in 25%
of cells) |
| Hep Par1 | + | + | − |
| CK7/CK20 | −/− | + or −/+ or − | − |
The clinical characteristics of our patient made the
diagnosis of HCC and HAC highly unlikely, as both those tumors
predominantly affect older males (19,20).
Brain metastases at the time of diagnosis are extremely rare
(<1% in HCC and not reported in HAC) (19,20).
Most importantly, the absence of a primary tumor in the liver or
other gastrointestinal organs on initial presentation strongly
argued against HCC or HAC. Based on all the characteristics
mentioned and discussed above, hepYST was the most likely
diagnosis.
hepYSTs are exceedingly rare, with only 32 reported
cases in the English medical literature to date (10). The majority of these tumors arise
from the ovary, whereas only single cases with an extraovarian
origin are described. In adult males, cases of sole mediastinal and
testicular hepYSTs are reported (21,22).
The treatment of YSTs may be challenging. Standard
care is similar to that for other types of non-germinoma GCT and
includes platinum-based chemotherapy protocols, followed by
surgical resection of the residual tumor or radiation (23). The most widely adopted regimens are
PEI and bleomycin, etoposide and cisplatin (2,24). In
our case, due to tumor location in the brain and lung, PEI was
selected. Unfortunately, the patient had already experienced
progression during therapy. Subsequent salvage high-dose
chemotherapy, including tumor resection and radiation, did not
achieve disease control and the patient succumbed to the disease 10
months after diagnosis. Similar survival rates were reported by
Moran et al (21). In a case
series of 4 patients with mediastinal hepYST, 3 patients succumbed
to the disease within 1 year after the initial diagnosis. A large
series with 788 YST patients reported sustainably better survival
rates: The 5-year overall survival was 55% (54% for mediastinal and
60% for retroperitoneal YSTs) (11).
Unfortunately, survival rates according to histological YST
subtypes are not available, which poses the question whether
histological YST subtype, i.e., hepatoid differentiation, affects
survival. Goebel et al (Proc ASCO 22: abs. 1842, 2003)
reported an association of hepYST with a higher risk of relapse
following initial treatment, as supported by our case.
In conclusion, hepYST is a rare differential
diagnosis in the spectrum of brain tumors, including metastases,
with poor prognosis. Due to the rarity of these tumors, clear
therapeutic recommendations and survival data are currently not
available. Thus, treatment of hepYST is considered to be an
interdisciplinary challenge.
Acknowledgements
We would like to thank Dr Guido Reifenberger,
Department of Neuropathology, Heinrich Heine University,
Düsseldorf, Germany and Dr Ivo Leuschner, Department of Pediatric
Pathology, Kiel Pediatric Tumor Registry, University Hospital of
Schleswig-Holstein, Kiel, Germany, for the contribution to the
discussion of the histological findings.
References
|
1
|
Stang A, Trabert B, Wentzensen N, Cook MB,
Rusner C, Oosterhuis JW and McGlynn KA: Gonadal and extragonadal
germ cell tumors in the United States, 1973–2007. Int J Androl.
35:616–625. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Albany C and Einhorn LH: Extragonadal germ
cell tumors: Clinical presentation and management. Curr Opin Oncol.
25:261–265. 2013.PubMed/NCBI
|
|
3
|
Schmoll HJ: Extragonadal germ cell tumors.
Ann Oncol. 13:(Suppl 4). 265–272. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McKenney JK, Heerema-McKenney A and Rouse
RV: Extragonadal germ cell tumors: A review with emphasis on
pathologic features, clinical prognostic variables, and
differential diagnostic considerations. Adv Anat Pathol. 14:69–92.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Toner GC, Geller NL, Lin SY and Bosl GJ:
Extragonadal and poor risk nonseminomatous germ cell tumors.
Survival and prognostic features. Cancer. 67:2049–2057. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rodney AJ, Tannir NM, Siefker-Radtke AO,
Liu P, Walsh GL, Millikan RE, Swisher SG, Tu SM and Pagliaro LC:
Survival outcomes for men with mediastinal germ-cell tumors: The
university of Texas M. D. Anderson cancer center experience. Urol
Oncol. 30:879–885. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Talerman A: Endodermal sinus (yolk sac)
tumor elements in testicular germ-cell tumors in adults: Comparison
of prospective and retrospective studies. Cancer. 46:1213–1217.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Motzer RJ, Geller NL, Tan CC, Herr H,
Morse M, Fair W, Sheinfeld J, Sogani P, Russo P and Bosl GJ:
Salvage chemotherapy for patients with germ cell tumors: The
memorial Sloan-Kettering cancer center experience (1979–1989).
Cancer. 67:1305–1310. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kondagunta GV, Bacik J, Sheinfeld J,
Bajorin D, Bains M, Reich L, Deluca J, Budnick A, Ishill N,
Mazumdar M, et al: Paclitaxel plus ifosfamide followed by high-dose
carboplatin plus etoposide in previously treated germ cell tumors.
J Clin Oncol. 25:85–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rittiluechai K, Wilcox R, Lisle J, Everett
E, Wallace HJ III and Verschraegen CF: Prognosis of hepatoid yolk
sac tumor in women: What's up, Doc? Eur J Obstet Gynecol Reprod
Biol. 175:25–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shah JP, Kumar S, Bryant CS, Ali-Fehmi R,
Malone JM Jr, Deppe G and Morris RT: A population-based analysis of
788 cases of yolk sac tumors: A comparison of males and females.
Int J Cancer. 123:2671–2675. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pelosi G, Petrella F, Sandri MT, Spaggiari
L, Galetta D and Viale G: A primary pure yolk sac tumor of the lung
exhibiting CDX-2 immunoreactivity and increased serum levels of
alkaline phosphatase intestinal isoenzyme. Int J Surg Pathol.
14:247–251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang B, Gao S, Chen Y and Wu Y: Primary
yolk sac tumor arising in the pancreas with hepatic metastasis: A
case report. Korean J Radiol. 11:472–475. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kumar Y, Bhatia A, Kumar V and Vaiphei K:
Intrarenal pure yolk sac tumor: An extremely rare entity. Int J
Surg Pathol. 15:204–206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guzel A, Tatli M, Belen D and Seckin H:
Spinal cord compression of primary extragonadal giant yolk sac
tumor. Spinal Cord. 45:254–257. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takahashi T, Ishikawa E, Masuda Y,
Yamamoto T, Sato T, Shibuya M and Matsumura A: Mixed germ cell
tumor with extensive yolk sac tumor elements in the frontal lobe of
an adult. Case Rep Surg. 2012:4737902012.PubMed/NCBI
|
|
17
|
Ueno T, Tanaka YO, Nagata M, Tsunoda H,
Anno I, Ishikawa S, Kawai K and Itai Y: Spectrum of germ cell
tumors: From head to toe. Radiographics. 24:387–404. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fan Z, van de Rijn M, Montgomery K and
Rouse RV: Hep par 1 antibody stain for the differential diagnosis
of hepatocellular carcinoma: 676 tumors tested using tissue
microarrays and conventional tissue sections. Mod Pathol.
16:137–144. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Su JS, Chen YT, Wang RC, Wu CY, Lee SW and
Lee TY: Clinicopathological characteristics in the differential
diagnosis of hepatoid adenocarcinoma: A literature review. World J
Gastroenterol. 19:321–327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi HJ, Cho BC, Sohn JH, Shin SJ, Kim SH,
Kim JH and Yoo NC: Brain metastases from hepatocellular carcinoma:
Prognostic factors and outcome: Brain metastasis from HCC. J
Neurooncol. 91:307–313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moran CA and Suster S: Hepatoid yolk sac
tumors of the mediastinum: A clinicopathologic and
immunohistochemical study of four cases. Am J Surg Pathol.
21:1210–1214. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Horie Y and Kato M: Hepatoid variant of
yolk sac tumor of the testis. Pathol Int. 50:754–558. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schmoll HJ, Souchon R, Krege S, Albers P,
Beyer J, Kollmannsberger C, Fossa SD, Skakkebaek NE, de Wit R,
Fizazi K, et al: European consensus on diagnosis and treatment of
germ cell cancer: A report of the European germ cell cancer
consensus group (EGCCCG). Ann Oncol. 15:1377–1399. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Packer RJ, Cohen BH and Cooney K:
Intracranial germ cell tumors. Oncologist. 5:312–320.
2000.PubMed/NCBI
|