Introduction
Ewing Sarcoma (ES) is a bone and soft tissue tumor
of possible neuroectodermal or mesenchymal origin that afflicts
children and young adults. Approximately 20–25% of cases are
metastatic at diagnosis, and the survival rates are poor in the
advanced setting (1,2). ES is characterized by a frequent
characteristic cytogenetic translocation of the EWSR1 (22q12) and
FLI-1 (11q24) genes. The resulting fusion protein EWS-FLI-1 is
responsible for oncogene activation, inhibition of tumor
suppression, chromatin remodeling and epigenomic reprogramming
(3–5).
Standard multimodality treatment consists of
induction chemotherapy followed by local control with surgery
and/or radiotherapy and then consolidation chemotherapy with
multiple drugs. Active chemotherapy agents in the first-line
setting include doxorubicin, vincristine, cyclophosphamide,
etoposide and ifosfamide. Treatment efficacy appears to reach a
plateau after dose intensification and interval compression of all
active agents (6,7), and the development of resistance to
chemotherapy remains as the main cause of treatment failure
(8,9). Cooperative groups have examined
different chemotherapy combinations, dose-intensifying regimens
with bone marrow transplantation, metronomic therapy and other
synergistic mechanisms associated with the EWS fusion protein in
ES. However, these approaches have failed to improve survival rates
in clinical trials thus far (10,11).
Elucidating the molecular mechanisms underlying the development of
chemotherapy resistance in ES may help develop new agents with
improved synergy.
Doxorubicin belongs to the class of anthracyclines
and acts by topoisomerase II poisoning, creation of double-strand
DNA breaks (DSBs), and impairment of DNA repair and supercoiling,
leading to changes in epigenetic processes (12,13).
Mechanisms involved in resistance to doxorubicin include drug
efflux transporters, alterations in the ability of doxorubicin to
form DSBs, and alterations in downstream apoptosis signaling
triggered by DNA damage (14).
Vincristine, a natural alkaloid extracted from Vinca
rosea, interferes with microtubule formation and stability through
depolymerization, resulting in cell cycle arrest and apoptosis
(15). In addition to affecting
chromatin stability (16) by
interfering with DNA binding and histone eviction, vincristine also
affects topoisomerase IIa levels (17). Resistance to vinca alkaloids involves
overexpression of ATP-binding cassette (ABC) transporters such as
P-glycoprotein, alterations of β-tubulin (βII, βIII and βIV) and
multidrug-resistance proteins (18).
The aim of the present study was to investigate
whether the expression of genes associated with resistance to
chemotherapy in other tumor types is different in
chemotherapy-resistant ES cells. The literature was first
data-mined for genes associated with resistance to drugs used in ES
treatment, focusing on doxorubicin and vincristine, and then
SK-ES-1 cells resistant to these drugs were developed.
Subsequently, the expression of the selected genes was evaluated
with quantitative polymerase chain reaction (qPCR) analysis.
Materials and methods
Data mining and refining
To select drug resistance genes, the literature was
searched for studies investigating resistance to doxorubicin and
vincristine in all cancer types. Approximately 270 genes were
identified, but only 23 genes appropriately validated by
experimental methodologies (mutational and knockout gene analysis)
were selected. This database was further enriched with information
from online tools, such as DrugBank, Gene Ontology and UniProt.
Finally, a set of five genes (CCAR1, TUBA1A, POLDIP2, SMARCA4 and
SMARCB1) was selected based on pathways that are important for ES
development.
Cell culture
The standard Ewing sarcoma cell line SK-ES-1
(American Type Culture Collection, Manassas, VA, USA) was cultured
in RPMI-1640 medium (Gibco®; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% bovine serum, 4 mg/ml
gentamicin (Nova Farma, São Paulo, Brazil) and amphotericine B
(Fungizone®, Invitrogen; Thermo Fisher Scientific, Inc.).
Chemoresistance induction
For chemotherapy resistance induction, the cells
underwent treatment for 72 h, with increasing drug concentration
every 2 weeks, for a period of ≥10 weeks. The concentrations were
10, 20, 30, 40 and 50 mM for doxorubicin (Libbs, São Paulo, Brazil)
and 0.5, 1, 2, 3 and 4 nM for vincristine (Pfizer, Inc., New York,
NY, USA). Untreated cells served as control (9).
Cell proliferation
SK-ES-1 cells were seeded at a density of 2.104
cells per well in 48-well plates. After 24 h, the cells were
treated with vincristine and doxorubicin individually, including
control. The concentrations for this treatment were 10, 30 and 50
nM for doxorubicin and 1, 3 and 5 nM for vincristine. The medium
was removed after 72 h of treatment; the cells were washed with
phosphate-buffered saline, detached with 0.25% trypsin solution (no
EDTA; Invitrogen; Thermo Fisher Scientific) and then counted with
the trypan blue exclusion method in a hemocytometer, as previously
described (19). The mean of three
experiments for each dose was utilized for calculations.
RNA extraction and cDNA synthesis
RNA was extracted using TRIzol®
(Invitrogen; Thermo Fisher Scientific) according to the
manufacturer's protocol. cDNA was synthesized using the reverse
transcription (RT)-PCR kit SuperScriptTM III First-Strand Synthesis
SuperMix (Invitrogen; Thermo Fisher Scientific).
qPCR
qPCR was performed using the AB 7500 system (Thermo
Fisher Scientific Inc.), with positive and negative controls.
Reactions were prepared with KiCqStart® qPCR Ready mix™
(Sigma-Aldrich; Merck KGaA, St. Louis, MO, USA), using a 0.5-µl
sample cDNA. Expression levels were evaluated using the 2-ΔΔCq
method, with GAPDH used as the housekeeping gene. The primers used
for CCAR1, TUBA1A, POLDIP2, SMARCA4 and SMARCB1 are listed in
Table I.
 | Table I.Quantitative polymerase chain reaction
primer sequences. |
Table I.
Quantitative polymerase chain reaction
primer sequences.
| Gene | Primer sequence | Product |
|---|
| SMARCB1 | F:
5′-TCCGTATGTTCCGAGGTTCT-3′ | 154 |
|
| R:
5′-CTGGTGGCTAGAGTCGTGTA-3′ |
|
| SMARCA4 | F:
5′-GCTCCGAGGTCTGATAGTGA-3′ | 133 |
|
| R:
5′-CGCTGTCTGGATCTGGAATC-3′ |
|
| CCAR1 | F:
5′-AGAGTTCGACGTGTTGTTCC-3′ | 90 |
|
| R:
5′-GCGCCTTAGTTCCATCATGT-3′ |
|
| TUBA1A | F:
5′-TTGTTCACTGGTACGTTGGG-3′ | 105 |
|
| R:
5′-AATCCACACCAACCTCCTCA-3′ |
|
| POLDIP2 | F:
5′-TTCCAGTATAGCAGCCACGT-3′ | 97 |
|
| R:
5′-GAACATCAAAGTGGGAGCCA-3′ |
|
| GAPDH | F:
5′-CAAGATCATCAGCAATGCCTCC-3′ | 103 |
|
| R:
5′-GACTGTGGTCATGAGTCCTTCC-3′ |
|
Statistical analysis
Statistical differences were analyzed using one-way
analysis of variance, with the Sidak correction method for multiple
comparison tests. Experiments were conducted three times and in
triplicates. All statistical analyses were performed using SPSS
16.0 for Windows. The differences were considered statistically
significant when P-values were <0.05.
Results
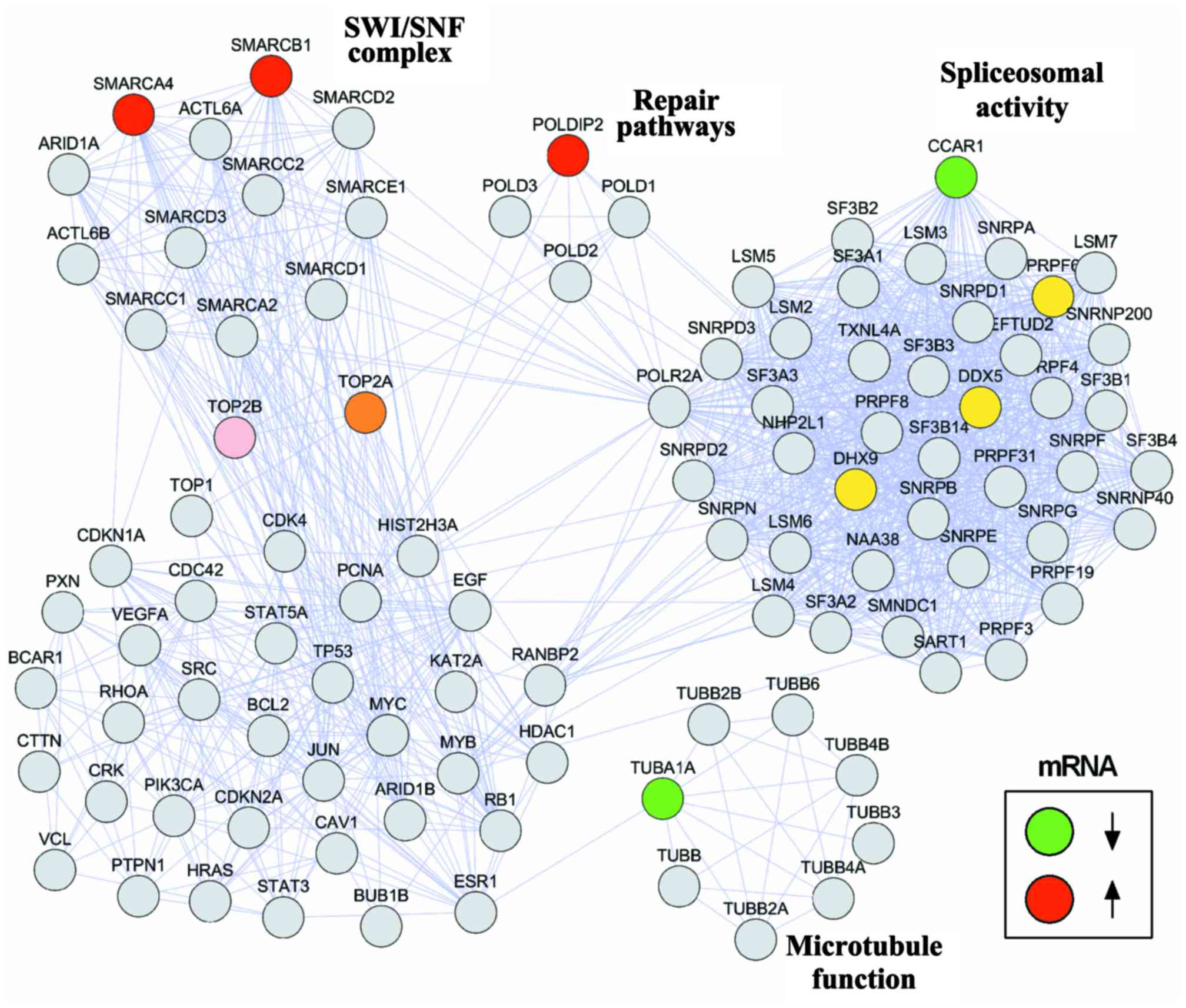
Protein network integration
Protein interaction networks are crucial for
understanding the biological cellular processes. We designed a
protein network to visualize the interactions between the selected
genes and molecular pathways associated with ES. A set of genes
associated with resistance to doxorubicin and vincristine was
manually curated. The open-source software programs Cytoscape
version 3.6.0 and String version 10.5 were used to build the
network. The interactions between the investigated genes (CCAR1,
TUBA1A, POLDIP2, SMARCA4 and SMARCB1) and DNA-topoisomerase II
(TOP2A), a target of both doxorubicin and vincristine, and genes
directly binding to EWS-FLI1 fusion protein, including those
associated with spliceosomal activity that is crucial to ES
pathogenesis, are shown in Fig.
1.
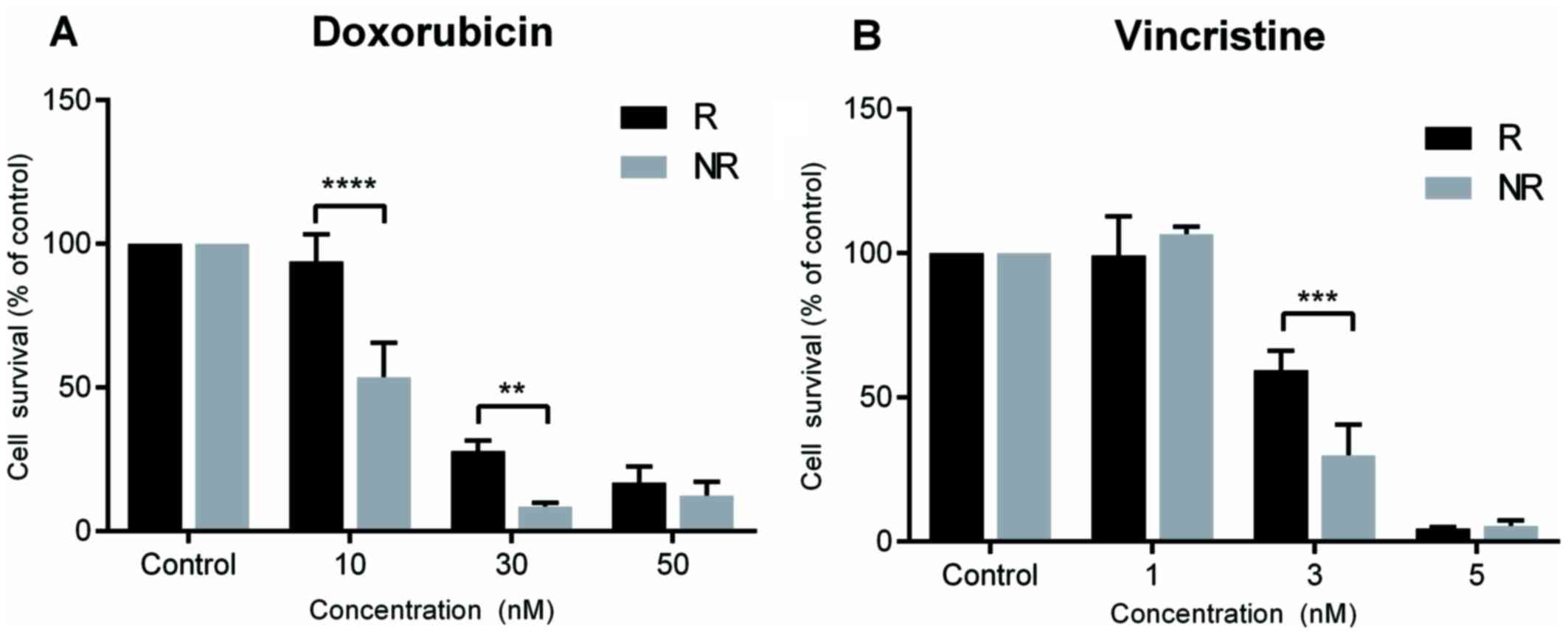
Induction of chemotherapy
resistance
Chemotherapy resistance was successfully induced for
both doxorubicin and vincristine in SK-ES-1 cells. Doxorubicin
resistance was evidenced at concentrations of 10 nM (P<0.0001
compared with controls) and 30 nM (P<0.005 compared with
controls), and vincristine resistance at a concentration of 3 nM
(P<0.0005 compared with controls; Fig. 2).
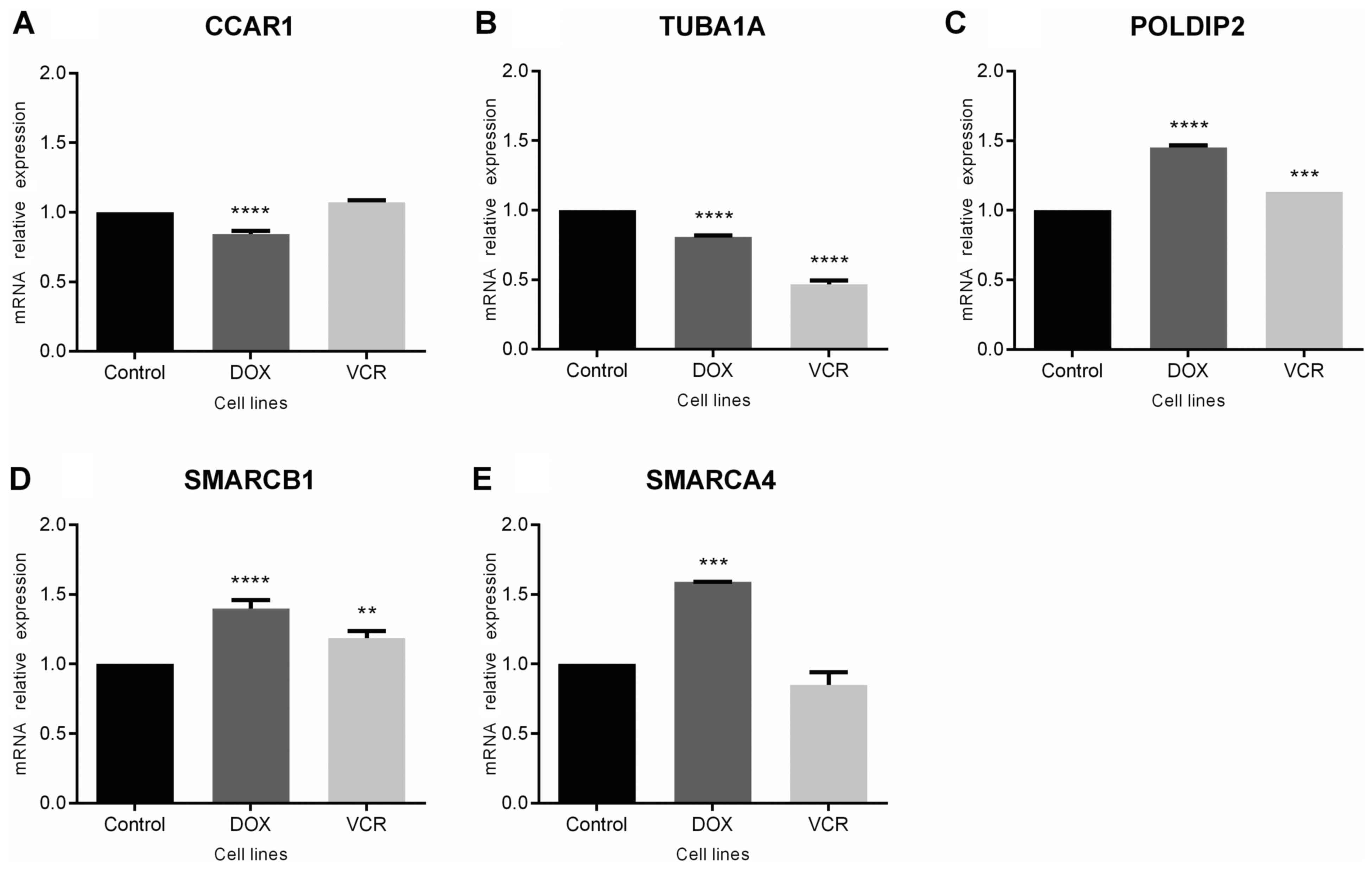
Gene expression
Changes in expression for the selected genes in
chemoresistant ES cells were analyzed. The results revealed a
modest, but statistically significant downregulation of CCAR1
(P<0.0001 compared with control cells; Fig. 3A) and TUBA1A (P<0.0001 compared
with controls) in doxorubicin-resistant cells, with decreased
expression of TUBA1A also observed in vincristine-resistant cells
(P<0.0001 compared with controls; Fig. 3B). By contrast, POLDIP2 was
upregulated in cells resistant to either drug (P<0.0001 for
doxorubicin-resistant and P<0.0005 for vincristine-resistant
cells; Fig. 3C). For SMARCB1 and
SMARCA4, gene expression was upregulated in cells resistant to
doxorubicin (P<0.0001 and P<0.0005, respectively), whereas
changes were observed in vincristine-resistant cells for SMARCB1
only (P<0.0005; Fig. 3D and
E).
Discussion
Resistance to chemotherapy remains the main reason
for treatment failure in patients with ES. In the present study, a
group of genes was selected (TUBA1A, POLDIP2, SMARCA4, SMARCB1 and
CCAR1), known in other tumors to be associated with resistance to
drugs commonly used in ES, and the gene expression in
chemotherapy-resistant ES cells was evaluated. It was demonstrated
that the TUBA1A expression levels were lower in cells resistant to
either doxorubicin or vincristine, when compared with the
non-resistant cells. The TUBA1A gene encodes the α-tubulin protein,
which belongs to the tubulin family of proteins that form and
organize microtubules that are required for cell division and
movement. Given that vincristine is a microtubule-depolymerizing
agent, resistance to this agent may develop from changes in tubulin
levels (20). Doxorubicin may
interfere in TUBA1A levels, with lower gene expression observed in
resistant MCF-7 breast cancer cells (21).
Upregulation of POLDIP2 in the
chemotherapy-resistant ES cells was also observed. POLDIP2 encodes
a protein implicated in the activity of translesional polymerases.
The translesion synthesis polymerase and primer extension
activities of PrimPol play a role in DNA damage tolerance (22); its involvement in repair processes
may explain the increases in cell sensitivity to oxidative stress
when POLDIP2 is silenced (23). In
addition, POLDIP2 plays an important role in DNA replication/repair
and regulation of reactive oxygen species and participates in
cytoskeletal reorganization as well as key pathways in cancer,
including those involved in autophagy and cell cycle regulation
(24). POLDIP2 is also involved in
vascular integrity, smooth cell migration and adhesion (25). These processes are important for
tumor development and survival, and its upregulation in
drug-resistant cells may contribute to increased tumor
aggressiveness.
Both SMARCB1 and SMARCA4 are part of the SWI/SNF
chromatin-remodeling complex, which recruits TOP2A to DNA and leads
to the formation of DSBs and cell death. Loss of the SWI/SNF
complex results in drug resistance, including DSBs and repair
pathways (26). Knockdown of SMARCA4
and SMARCB1 leads to increased chemotherapy resistance (27). Our results revealed higher expression
levels of SMARCA4 and SMARCB1 in doxorubicin-resistant ES cells,
suggesting a different landscape in ES. The oncogenic EWS-FLI1
fusion induces chromatin-remodeling patterns, stimulating or
repressing enhancers and establishing a modified oncogenic
regulatory and interaction network that may explain divergences in
drug resistance mechanisms among different tumors (4).
The results of the present study are consistent with
previous evidence indicating that loss of SMARCB1 is found in a
small percentage of ES patients, and this may interfere favorably
with outcome. It is possible that the combination of EWSR1
translocation and SMARCB1 loss increases susceptibility of tumor
cells to treatment (28).
CCAR1 is a biphasic regulator of cell growth and
apoptosis, and plays an important role in tumorigenesis in gastric
cancer (29) and hepatocellular
carcinoma (30). CCAR1 can target
gene activation by estrogen and glucocorticoid receptors in breast
cancer cells (31), and androgen
receptors in prostate cancer (32).
EWS-FLI1 alters mRNA splicing in ES cells, giving rise to several
protein isoforms implicated in oncogenesis (33), and CCAR1 is associated with
spliceosomal activity. An unexpected decrease in CCAR1 expression
levels was observed in doxorubicin-resistant cells. Further
experiments are required to elucidate how this gene is associated
with chemotherapy resistance and alternative splicing in ES.
Although the genes selected in the present study have different
functions and are involved in diverse pathways, our integration
network reveals a possible connection among these different
mechanisms. Therefore, repair pathways, SWI/SNF chromatin
remodeling, microtubule rearrangements and spliceosomal activity
may be interacting to maintain chemoresistance mechanisms in
ES.
In summary, the present findings provide early
evidence revealing novel changes in gene expression associated with
chemotherapy resistance in ES cells. Gene knockout assays,
characterization of resistance pathways and the use of tumor
samples from patients are among the next steps required to confirm
and extend these findings, and validate this set of genes as
possible targets to counteract therapy resistance in ES.
Acknowledgements
Not applicable.
Funding
This research was supported by PRONON/Ministry of
Health, Brazil (grant no. 25000.162.034/2014-21 to C.B.F); the
Children's Cancer Institute (ICI); the National Council for
Scientific and Technological Development (CNPq; grant no.
303276/2013-4 to R.R.) and the Clinical Hospital institutional
research fund (FIPE/HCPA - 150419).
Availability of data and materials
The materials included in the manuscript will be
made freely available to any researchers who wish to use them for
non-commercial purposes, while preserving any necessary
confidentiality and anonymity.
Authors' contributions
LH, MS, CAS, DBO and CBF made substantial
contributions to the design of the present study; LH, MS, CAS and
DBO performed the experiments; LH, MS, and CBF analysed the data;
LH, MS, CAS, DBO, LJG, ALB, ATB, RR and CBF critically revised the
manuscript for important intellectual content, wrote and reviewed
the manuscript; LH, MS, ATB, RR and CBF wrote the manuscript. All
authors critically revised the manuscript and all authors have read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Balamuth NJ and Womer RB: Ewing's sarcoma.
Lancet Oncol. 11:184–192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gaspar N, Hawkins DS, Dirksen U, Lewis IJ,
Ferrari S, Le Deley MC, Kovar H, Grimer R, Whelan J, Claude L, et
al: Ewing sarcoma: Current management and future approaches through
collaboration. J Clin Oncol. 33:3036–3046. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lawlor ER and Thiele CJ: Epigenetic
changes in pediatric solid tumors: Promising new targets. Clin
Cancer Res. 18:2768–2779. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Riggi N, Knoechel B, Gillespie SM,
Rheinbay E, Boulay G, Suvà ML, Rossetti NE, Boonseng WE, Oksuz O,
Cook EB, et al: EWS-FLI1 utilizes divergent chromatin remodeling
mechanisms to directly activate or repress enhancer elements in
Ewing sarcoma. Cancer Cell. 26:668–681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sheffield NC, Pierron G, Klughammer J,
Datlinger P, Schönegger A, Schuster M, Hadler J, Surdez D,
Guillemot D, Lapouble E, et al: DNA methylation heterogeneity
defines a disease spectrum in Ewing sarcoma. Nat Med. 23:386–395.
2017. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Womer RB, West DC, Krailo MD, Dickman PS,
Pawel BR, Grier HE, Marcus K, Sailer S, Healey JH, Dormans JP, et
al: Randomized controlled trial of interval-compressed chemotherapy
for the treatment of localized Ewing sarcoma: A report from the
Children's Oncology Group. J Clin Oncol. 30:4148–4154. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grier HE, Krailo MD, Tarbell NJ, Link MP,
Fryer CJ, Pritchard DJ, Gebhardt MC, Dickman PS, Perlman EJ, Meyers
PA, et al: Addition of ifosfamide and etoposide to standard
chemotherapy for Ewing's sarcoma and primitive neuroectodermal
tumor of bone. N Engl J Med. 348:694–701. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ahmed AA, Zia H and Wagner L: Therapy
resistance mechanisms in Ewing's sarcoma family tumors. Cancer
Chemother Pharmacol. 73:657–663. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Heinen TE, Dos Santos RP, da Rocha A, Dos
Santos MP, Lopez PL, Filho Silva MA, Souza BK, Rivero LF, Becker
RG, Gregianin LJ, et al: Trk inhibition reduces cell proliferation
and potentiates the effects of chemotherapeutic agents in Ewing
sarcoma. Oncotarget. 7:34860–34880. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anderson PM, Bielack SS, Gorlick RG,
Skubitz K, Daw NC, Herzog CE, Monge OR, Lassaletta A, Boldrini E,
Pápai Z, et al: A phase II study of clinical activity of SCH 717454
(robatumumab) in patients with relapsed osteosarcoma and Ewing
sarcoma. Pediatr Blood Cancer. 63:1761–1770. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wagner LM, Fouladi M, Ahmed A, Krailo MD,
Weigel B, DuBois SG, Doyle LA, Chen H and Blaney SM: Phase II study
of cixutumumab in combination with temsirolimus in pediatric
patients and young adults with recurrent or refractory sarcoma: A
report from the Children's Oncology Group. Pediatr Blood Cancer.
62:440–444. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pang B, Qiao X, Janssen L, Velds A,
Groothuis T, Kerkhoven R, Nieuwland M, Ovaa H, Rottenberg S, van
Tellingen O, et al: Drug-induced histone eviction from open
chromatin contributes to the chemotherapeutic effects of
doxorubicin. Nat Commun. 4:19082013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang F, Teves SS, Kemp CJ and Henikoff S:
Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys
Acta. 1845:84–89. 2014.PubMed/NCBI
|
|
14
|
Cox J and Weinman S: Mechanisms of
doxorubicin resistance in hepatocellular carcinoma. Hepat Oncol.
3:57–59. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kavallaris M: Microtubules and resistance
to tubulin-binding agents. Nat Rev Cancer. 10:194–204. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mohammadgholi A, Rabbani-Chadegani A and
Fallah S: Mechanism of the interaction of plant alkaloid
vincristine with DNA and chromatin: Spectroscopic study. DNA Cell
Biol. 32:228–235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Skladanowski A, Côme MG, Sabisz M,
Escargueil AE and Larsen AK: Down-regulation of DNA topoisomerase
IIalpha leads to prolonged cell cycle transit in G2 and early M
phases and increased survival to microtubule-interacting agents.
Mol Pharmacol. 68:625–634. 2005.PubMed/NCBI
|
|
18
|
Zhang Y, Yang SH and Guo XL: New insights
into Vinca alkaloids resistance mechanism and circumvention in lung
cancer. Biomed Pharmacother. 96:659–666. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Flores DG, de Farias CB, Leites J, de
Oliveira MS, Lima RC, Tamajusuku AS, Di Leone LP, Meurer L,
Brunetto AL, Schwartsmann G, et al: Gastrin-releasing peptide
receptors regulate proliferation of C6 Glioma cells through a
phosphatidylinositol 3-kinase-dependent mechanism. Curr Neurovasc
Res. 5:99–105. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huzil JT, Chen K, Kurgan L and Tuszynski
JA: The roles of beta-tubulin mutations and isotype expression in
acquired drug resistance. Cancer Inform. 3:159–181. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Villeneuve DJ, Hembruff SL, Veitch Z,
Cecchetto M, Dew WA and Parissenti AM: cDNA microarray analysis of
isogenic paclitaxel- and doxorubicin-resistant breast tumor cell
lines reveals distinct drug-specific genetic signatures of
resistance. Breast Cancer Res Treat. 96:17–39. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guilliam TA, Bailey LJ, Brissett NC and
Doherty AJ: PolDIP2 interacts with human PrimPol and enhances its
DNA polymerase activities. Nucleic Acids Res. 44:3317–3329. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maga G, Crespan E, Markkanen E, Imhof R,
Furrer A, Villani G, Hübscher U and van Loon B: DNA polymerase
δ-interacting protein 2 is a processivity factor for DNA polymerase
λ during 8-oxo-7,8-dihydroguanine bypass. Proc Natl Acad Sci USA.
110:18850–18855. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brown DI, Lassègue B, Lee M, Zafari R,
Long JS, Saavedra HI and Griendling KK: Poldip2 knockout results in
perinatal lethality, reduced cellular growth and increased
autophagy of mouse embryonic fibroblasts. PLoS One. 9:e966572014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sutliff RL, Hilenski LL, Amanso AM,
Parastatidis I, Dikalova AE, Hansen L, Datla SR, Long JS, El-Ali
AM, Joseph G, et al: Polymerase delta interacting protein 2
sustains vascular structure and function. Arterioscler Thromb Vasc
Biol. 33:2154–2161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wijdeven RH, Pang B, van der Zanden SY,
Qiao X, Blomen V, Hoogstraat M, Lips EH, Janssen L, Wessels L,
Brummelkamp TR, et al: Genome-wide identification and
characterization of novel factors conferring resistance to
topoisomerase II poisons in cancer. Cancer Res. 75:4176–4187. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dubey R, Lebensohn AM, Bahrami-Nejad Z,
Marceau C, Champion M, Gevaert O, Sikic BI, Carette JE and Rohatgi
R: Chromatin-remodeling complex SWI/SNF controls multidrug
resistance by transcriptionally regulating the drug efflux pump
ABCB1. Cancer Res. 76:5810–5821. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jahromi MS, Putnam AR, Druzgal C, Wright
J, Spraker-Perlman H, Kinsey M, Zhou H, Boucher KM, Randall RL,
Jones KB, et al: Molecular inversion probe analysis detects novel
copy number alterations in Ewing sarcoma. Cancer Genet.
205:391–404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang TS, Wei KL, Lu CK, Chen YH, Cheng
YT, Tung SY, Wu CS and Chiang MK: Inhibition of CCAR1, a
coactivator of beta-catenin, suppresses the proliferation and
migration of gastric cancer cells. Int J Mol Sci. 18:E4602017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ha SY, Kim JH, Yang JW, Kim J, Kim B and
Park CK: The overexpression of CCAR1 in hepatocellular carcinoma
associates with poor prognosis. Cancer Res Treat. 48:1065–1073.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim JH, Yang CK, Heo K, Roeder RG, An W
and Stallcup MR: CCAR1, a key regulator of mediator complex
recruitment to nuclear receptor transcription complexes. Mol Cell.
31:510–519. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Seo WY, Jeong BC, Yu EJ, Kim HJ, Kim SH,
Lim JE, Kwon GY, Lee HM and Kim JH: CCAR1 promotes chromatin
loading of androgen receptor (AR) transcription complex by
stabilizing the association between AR and GATA2. Nucleic Acids
Res. 41:8526–8536. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Selvanathan SP, Graham GT, Erkizan HV,
Dirksen U, Natarajan TG, Dakic A, Yu S, Liu X, Paulsen MT, Ljungman
ME, et al: Oncogenic fusion protein EWS-FLI1 is a network hub that
regulates alternative splicing. Proc Natl Acad Sci USA.
112:E1307–E1316. 2015. View Article : Google Scholar : PubMed/NCBI
|