Introduction
Kimura's disease (KD) was first reported in China by
Kim in 1937 as eosinophilic hyperplastic lymphogranuloma (1). Until 1948, the disease was widely
recognized in Japan and systematically described by Professor
Kimura as ‘unusual granulations combined with hyperplastic changes
in lymphoid tissue’ (2,3). KD is a chronic disease and its etiology
has not been fully elucidated to date. Patients suffering from this
condition mainly present with solitary or multiple painless masses
in the maxillofacial and other regions, which often recur after
treatment (4,5). Clinically, KD is always accompanied by
enlarged regional lymph nodes, eosinophilia and markedly elevated
serum IgE levels (2,5). This disease has been most commonly
diagnosed in middle-aged individuals in the Far East, for example
China and Japan (6–8). Thus far, only 200 cases have been
reported worldwide. We herein report a rare case of KD in a
48-year-old man and review the relevant literature to help improve
the level of clinicians' knowledge regarding the diagnosis and
treatment of this disease.
Case report
A 48-year-old man was admitted to the Hainan Branch
of Chinese People's Liberation Army General Hospital (Sanya, China)
in December 2017, with recurrent masses in the right parotid gland
and neck region for ~15 years. The patient was in good overall
condition and had no other complaints, such as pain, swelling,
local numbness or xerostomia. There were no reported allergies or
other systemic manifestations. The patient had undergone surgical
resection of the masses twice (1997 and 2000), but the masses
recurred both times after ~6 months. Both postoperative
pathological diagnoses were lymphadenitis.
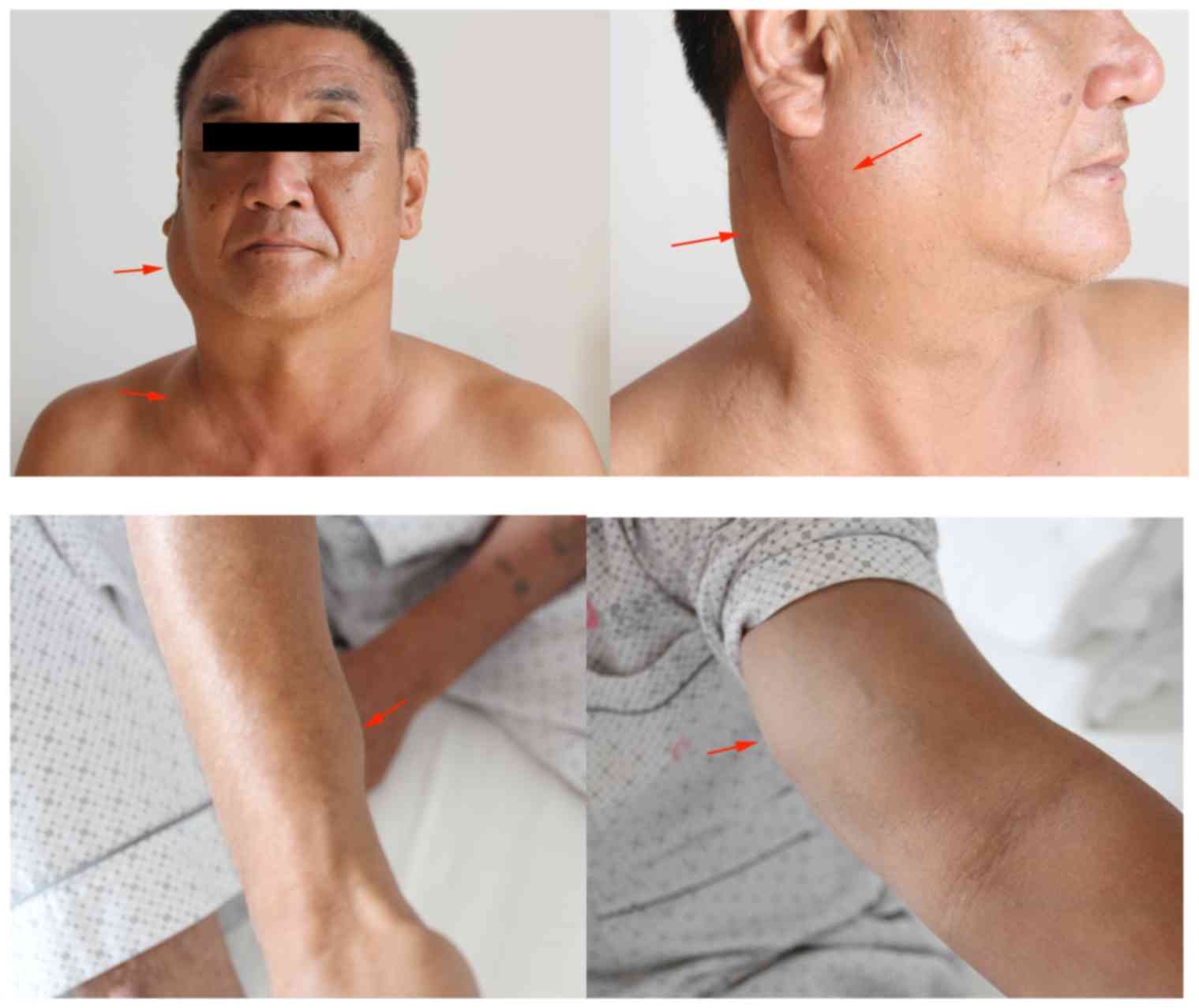
Physical examination revealed maxillofacial
asymmetry. Hard, mobile masses were identified in the region of the
right parotid gland and neck, arranged longitudinally. On
palpation, the borders of the masses were clear, without adhesions
to the surrounding tissue; the total size was 15×10×3 cm. Masses
were also identified in the left supraclavicular fossa, the right
forearm near the wrist joint and the left medial upper arm, sized
5×5×2, 4×3×2 and 4×3×1 cm, respectively (Fig. 1). In addition, multiple enlarged
lymph nodes were palpated in the submandibular area and the neck
region.
Reviewing the results of the laboratory tests, with
an increased eosinophil percentage (0.55%; normal, 0.01–0.05%),
markedly elevated serum IgE levels (27,100 IU/ml; normal, 0–100
IU/ml) and leukocyte count (21.78×109/l; normal,
3.5×109/l), and decreased neutrophil percentage (0.28%;
normal, 0.50–0.70%). The results of other investigations, including
routine urinalysis, liver and kidney function tests, were within
normal limits. Chest radiography was also normal.
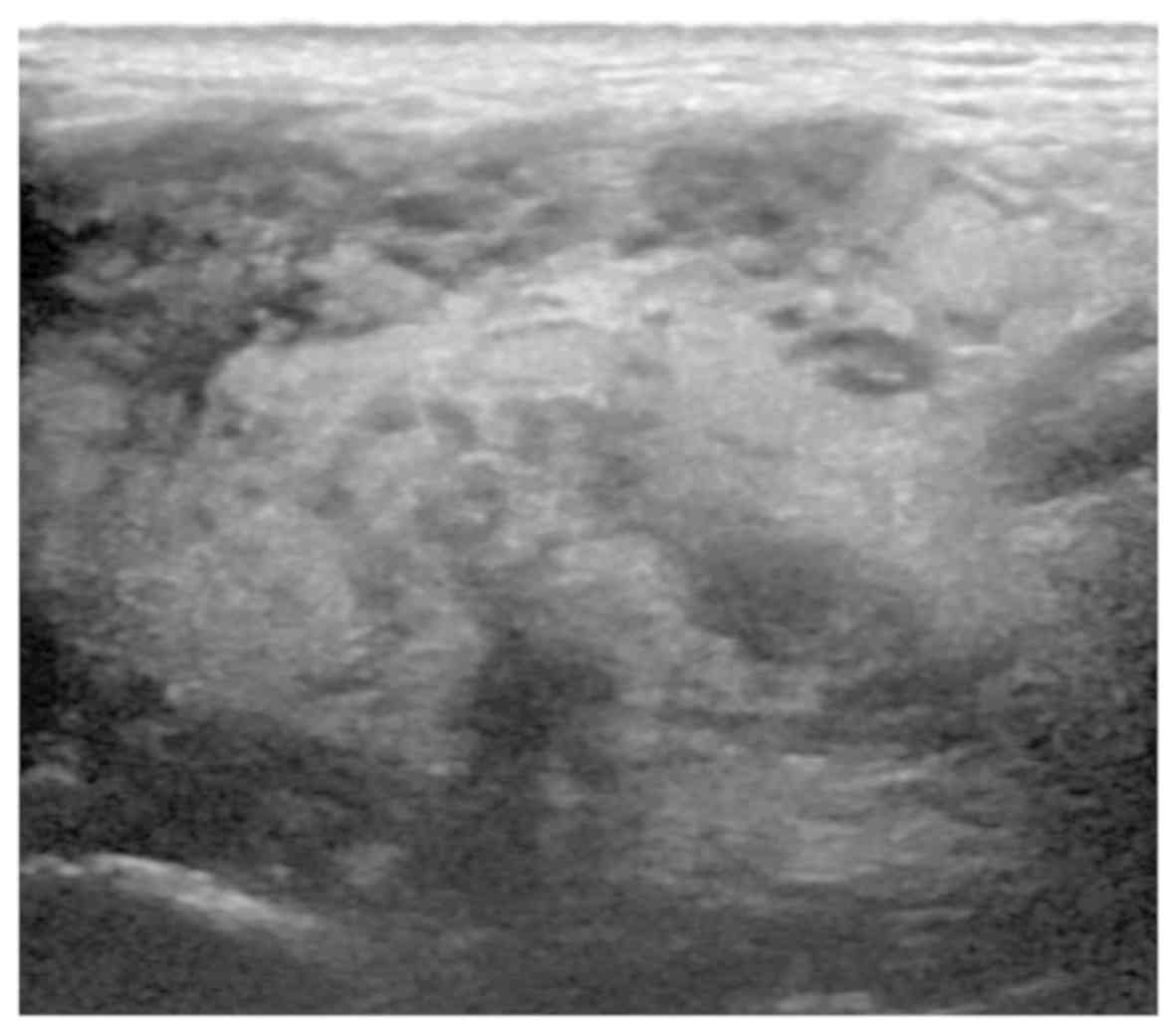
Ultrasound examination of the right parotid gland
region revealed uneven echogenicity, consistent with a high
likelihood of a benign tumor (Fig.
2); a low-echogenicity nodule was identified in the right
submaxillary region, which was considered to be a reactive
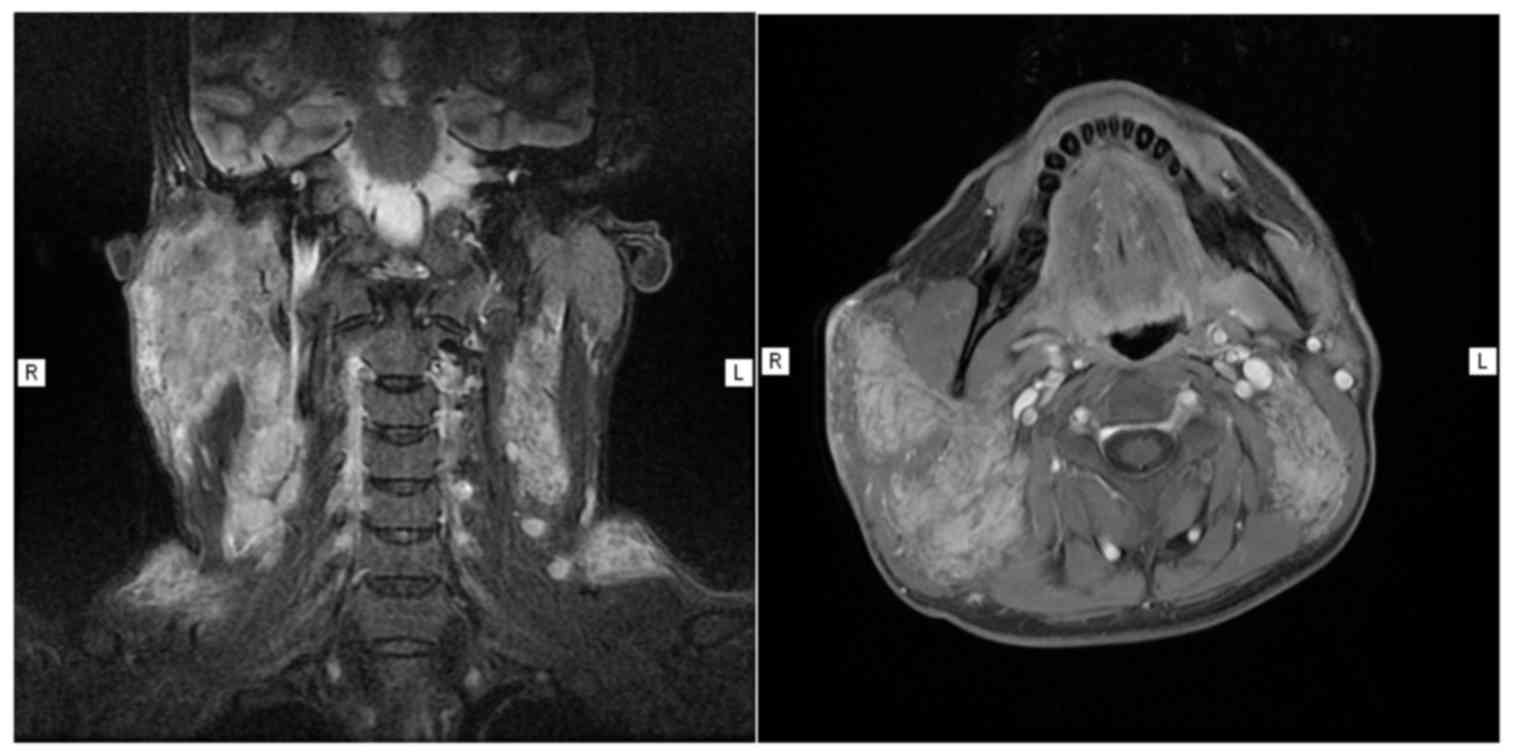
hyperplastic lymph node. On magnetic resonance imaging (MRI)
examination, a widespread abnormal signal was detected in the right
parotid gland and submaxillary region, and a contrast-enhanced scan
revealed inhomogeneous enhancement; the right side of the
sternocleidomastoid muscle and the parotid gland were partially
involved, with half of the lesions wrapped around the carotid
sheath (Fig. 3); multiple enlarged
lymph nodes were also identified displaying obvious
enhancement.
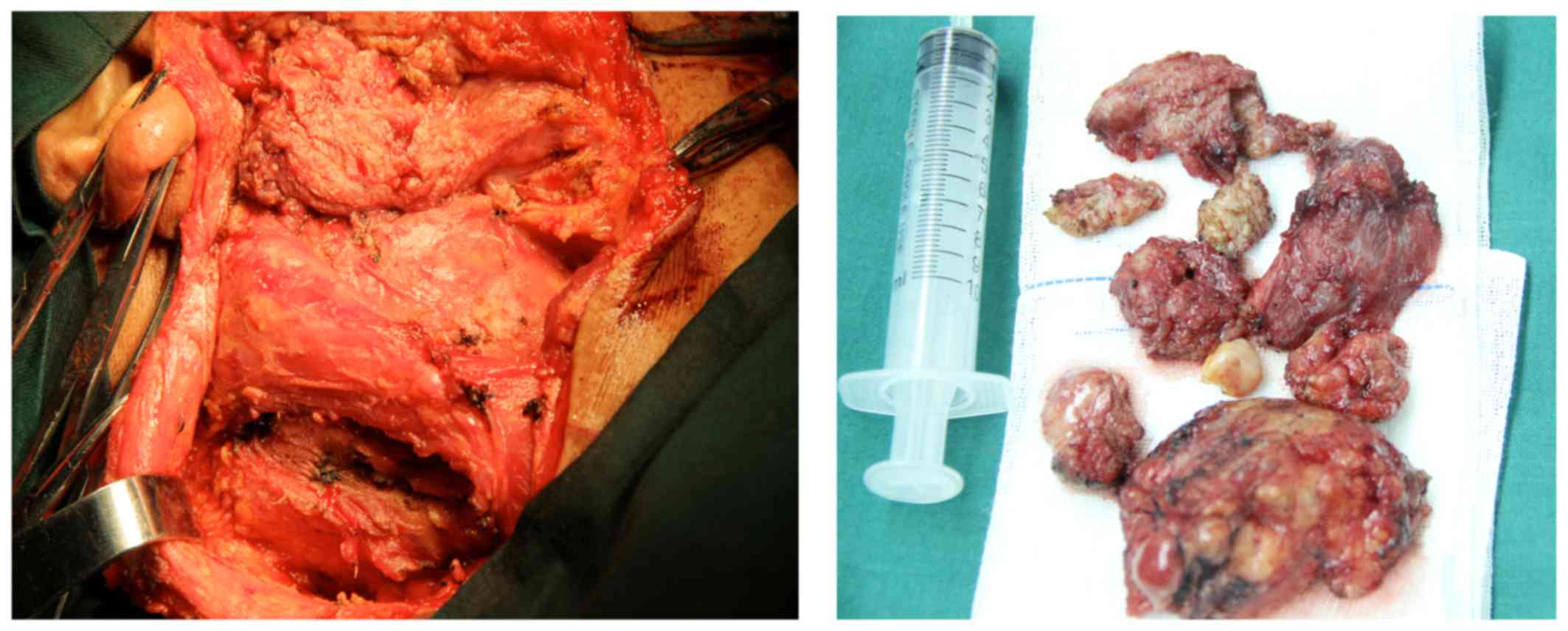
A detailed surgical plan was formulated and,
following a full preoperative preparation, the right parotid gland
and neck masses were resected under general anesthesia, together
with the right superficial parotid lobe (Fig. 4).
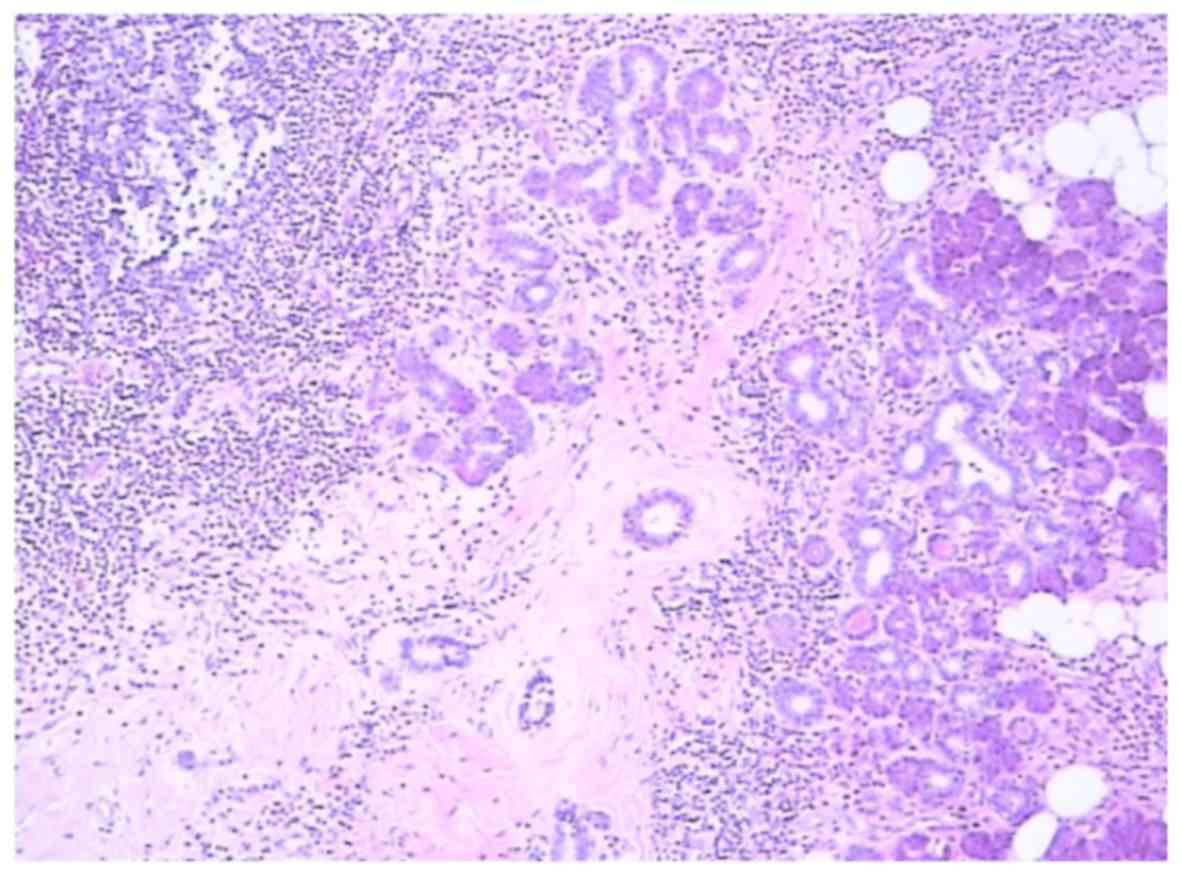
Postoperative pathological examination established
the diagnosis of eosinophilic hyperplastic lymphogranuloma, also
referred to as KD (Fig. 5), with
lesions involving the parotid gland and lymph node tissue.
The patient was administered 25 mg prednisone twice
daily for 2 weeks after the definitive diagnosis. On repeat blood
tests, the eosinophil count returned to normal levels, and the
masses of the left supraclavicular fossa, right forearm and left
medial upper arm notably decreased in size. Therefore, the initial
treatment was successful in controlling the disease. Next, the
patient will be advised to undergo radiotherapy (total dose, 20–50
Gy; 1.8–2.0 Gy per day, five days a week, over the course of 2–5
weeks).
Discussion
KD is a rare condition, with <300 reported cases
to date. Based on these cases, KD is rarely observed in Europe and
America (9,10), but frequently occurs in East Asian
and Southeast Asian populations (11–14),
with China and Japan being the most common. The pathogeny of KD
remains unknown. The minimum age reported is 5 years, but it mostly
occurs in middle-aged men (15–17).
Women only account for a small proportion of the patients, with a
male:female ratio of ~3:1 (18).
Clinically, KD has a specific predilection for the head, neck
(5,10) and limbs, with the lesions most
commonly developing in the soft tissue, whereas maxillofacial
salivary glands and lymph nodes may also be affected. Patients with
KD disease often have a long course, manifesting solitary or
multiple soft tissue masses. KD is usually associated with
peripheral blood eosinophilia and markedly elevated IgE levels.
The etiology of KD remains unknown (4,5,10,13). It
has been confirmed that there is no correlation with tuberculosis,
bacterial, fungal or viral infections, poisoning or syphilis
(19). Most scholars tend to
consider KD as an IgE-mediated type I allergy and inflammatory
disease (20), and this hypothesis
is supported by the increased eosinophil count and IgE levels in
the peripheral blood (21,22). Googe et al reported increased
IgE levels in KD patients with renal involvement, which supports
the theory that KD is an immune response (23); moreover, clinical application of
steroid therapy appears to be effective, further supporting this
theory. In recent years, Ohta et al measured Th1/Th2 and
Tc1/Tc2 lymphocyte subsets by flow cytometry in KD patients, and
interestingly observed that Th2 and Tc1 cell numbers increased
significantly in patients with KD compared with the healthy control
group; therefore, they reported that Th2 and Tc1 cells appear to
play an important role in the pathogenesis of KD (24). Other studies demonstrated that the
release of cytokines in patients with KD can increase the
permeability of the glomerular basement membrane, causing
proteinuria, which may ultimately cause renal damage (25,26).
Chim et al considered a clonal T-cell lymphoproliferative
disorder as the possible cause of KD; however, this result was from
a single case study, and the etiological analysis remains
speculative (27).
KD patients are typically men in their 30s and the
highest incidence of this disease worldwide is observed in the
Asian population. Although Asian and non-Asian patients have
certain characteristics in common, it has been reported that
non-Asian patients do not generally exhibit involvement of the
salivary glands (10); by contrast,
in Asian patients KD has a predilection for pre-auricular, parotid
and submandibular salivary glands, the neck and the maxillofacial
region. The majority of the patients present with firm, painless,
single or multiple subcutaneous masses, progressively increasing in
size. Mass size may vary greatly, ranging from 1 to 7 cm (28). In addition, multiple regional lymph
nodes are involved, manifesting as a single or multiple fused
nodules. Although KD mostly involves the maxillofacial region,
involvement of other sites has also been reported, including the
axilla, trunk, kidney, arm, nerves, orbit, spermatic cord and groin
(6,29). In the case presented herein, the
patient had a right parotid swelling, along with multiple
supraclavicular fossa and arm masses for 15 years. The skin
overlying the KD lesions was not inflamed, although mild itching
was occasionally reported. With disease progression, skin
pigmentation, thickening, local erosion and ulceration may develop,
sometimes affecting the whole body in severe cases. Some scholars
report that renal involvement is a common systemic manifestation
that may affect up to 60% of the patients (5,25,30,31).
In addition, the disease may be complicated by asthma, fatigue,
fever, allergic rhinitis, atopic dermatitis, urticaria and other
allergic symptoms (20). KD is
generally benign and self-limited, with a prolonged course. On
long-term follow-up, malignant transformation has not been
reported. The overall prognosis is good, but recurrence is
common.
KD patients almost always have marked eosinophilia
and elevated serum IgE levels. Some studies report that blood
eosinophil counts are closely associated with the size of the
lesion, i.e., the bigger the lesion, the higher the eosinophil
count (32,33). Physical examination along with US, CT
and MRI may help determine the characteristics, boundaries and
blood supply of the mass, as well as the presence of lymph node
involvement. On US, the mass may exhibit heterogeneous or
homogeneous echogenicity (34),
occasionally displaying increased vascularity. On CT scans, the
lesions are strongly enhanced, reflecting their vascular nature;
lymphadenopathy is reported among the typical findings. On MRI, the
lesion exhibits intermediate to high signal intensity on
T1-weighted images and hyperintense signals on T2-weighted images
(35,36). Therefore, imaging may be a useful way
of demonstrating the morphology and extent of the lesion, as well
as its association with adjacent structures (8).
There are currently no uniform diagnostic criteria
for KD. The following characteristics should raise suspicion of KD:
i) Young male, with head and/or neck painless mass; ii) local
enlarged lymph nodes; iii) long history; iv) parts of body other
than the head and neck displaying multiple painless masses,
accompanied by pruritus of the overlying skin; v) increased blood
eosinophil count and serum IgE level (a high reference value is
needed to make a correct diagnosis); and vi) CT and MRI showing a
wide range of lesions and multiple enlarged lymph nodes. Based on
these clinical manifestations and blood test results, the diagnosis
of KD may be preliminarily considered, but the final diagnosis
relies on pathological examination. The histopathological
characteristics of KD include follicular hyperplasia, eosinophilic
infiltrates and proliferation of postcapillary venules (2,10,12,23).
Clinically, the differential diagnosis of KD may
include angiolymphoid hyperplasia with eosinophilia (ALHE),
Hodgkin's lymphoma, angioimmunoblastic T-cell lymphoma, Langerhans
cell histiocytosis, florid follicular hyperplasia, Castleman's
disease, dermatopathic lymphadenopathy, sinonasal eosinophilic
angiocentric fibrosis, drug reactions and parasitic infections. In
the literature, the most common misdiagnosis is ALHE (10,37);
however, patients with ALHE have normal IgE levels and no kidney
damage (3,9,38,39).
In conclusion, the prognosis for KD is quite
favorable, but may recur frequently in the original location
(4), with a recurrence rate of up to
25% (40), posing a major challenge
to the physician and patient. A standard treatment for KD has not
yet been established. The goal of treatment currently is to
maintain appearance and functionality, while preventing recurrence
and long-term sequelae, including nephritis and myocarditis.
Although surgery is the most widely used treatment
method and it can help reach a definite diagnosis (32,33,35,41,42),
other options include radiotherapy, systemic corticosteroids,
cytotoxic agents, cyclosporin and pentoxifylline (32,33,42).
Oral corticosteroids are usually recommended in cases of
symptomatic nephrotic syndrome (26)
and, in order to prevent relapse and reduce the long-term side
effects of steroid therapy, postoperative radiation therapy may be
used (low-dose local irradiation, ~25–30 Gy) (43,44).
Reportedly, the recurrence rate appears to be lower if two
treatment modalities are combined (41). Recently, anti-IgE therapy has been
introduced (45), and the size of
lesion and peripheral blood eosinophil count of KD patients were
reported to decrease following anti-IgE therapy.
In conclusion, KD is a chronic disorder of unknown
etiology. In clinical practice, KD is easily misdiagnosed as other
inflammatory lesions or benign tumors of the head and neck, so more
thorough diagnostic workup is required. Only through a careful
medical history and comprehensive clinical examination, combined
with laboratory, imaging and histopathological examinations, can a
definitive diagnosis be reached and individualized treatment
administered.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Hainan Key
Research and Development Program (grant no. ZDYF2016170) and the
Clinical Research Support Fund of PLA General Hospital (grant no.
2017FC-WJFWZX-22).
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient provided consent for publication of the
case details and associated images.
Availability of data and materials
Not applicable.
Authors' contributions
XL took care of the patient, wrote the medical
records and wrote the manuscript. JW and HL collected relevant
medical records and assisted with writing. MZ was responsible for
surgical treatment and paper revision. All the authors have read
and approved the final version of this manuscript.
Competing interests
The authors declare that they have no competing
interests to disclose.
References
|
1
|
Kim HT: Eosinophilic hyperplastic
lymphogranuloma, comparison with Mikulicz's disease. Chin Med J.
23:6991937.
|
|
2
|
Kung IT, Gibson JB and Bannatyne PM:
Kimura's disease: A clinico-pathological study of 21 cases and its
distinction from angiolymphoid hyperplasia with eosinophilia.
Pathology. 16:39–44. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kimura T and Yoshimura S: Unusual
granuloma combined with hyperplastic changes in lymphatic tissues.
Trans Soc Path Jpn. 13:179–180. 1948.
|
|
4
|
Abuel-Haija M and Hurford MT: Kimura
disease. Arch Pathol Lab Med. 131:650–651. 2007.PubMed/NCBI
|
|
5
|
Kuo TT, Shih LY and Chan HL: Kimura's
disease. Involvement of regional lymph nodes and distinction from
angiolymphoid hyperplasia with eosinophilia. Am J Surg Pathol.
12:843–854. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takeishi M, Makino Y, Nishioka H, Miyawaki
T and Kurihara K: Kimura disease: Diagnostic imaging findings and
surgical treatment. J Craniofac Surg. 18:1062–1067. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iwai H, Nakae K, Ikeda K, Ogura M,
Miyamoto M, Omae M, Kaneko T and Yamashita T: Kimura disease:
Diagnosis and prognostic factors. Otolaryngol Head Neck Surg.
137:306–311. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park SW, Kim HJ, Sung KJ, Lee JH and Park
IS: Kimura disease: CT and MR imaging findings. AJNR Am J
Neuroradiol. 33:784–788. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iguchi Y, Inoue T, Shimono M, Yamamura T,
Shigematsu T and Takahashi S: Kimura's disease and its relation to
angiolymphoid hyperplasia with eosinophilia: Report of three cases
and review of the literature. J Oral Pathol. 15:132–137. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen H, Thompson LD, Aguilera NS and
Abbondanzo SL: Kimura disease: A clinicopathologic study of 21
cases. Am J Surg Pathol. 28:505–513. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gumbs MA, Pai NB, Saraiya RJ, Rubinstein
J, Vythilingam L and Choi YJ: Kimura's disease: A case report and
literature review. J Surg. 70:190–193. 1999.
|
|
12
|
Hui PK, Chan JK, Ng CS, Kung IT and Gwi E:
Lymphadenopathy of Kimura's disease. Am J Surg Pathol. 13:177–186.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Urabe A, Tsuneyoshi M and Enjoji M:
Epithelioid hemangioma versus Kimura's disease. A comparative
clinicopathologic study. Am J Surg Pathol. 11:758–766. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu X, Fu J, Fang Y and Liang L: Kimura
disease in children: A case report and a summary of the literature
in Chinese. J Pediatr Hematol Oncol. 33:306–311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Francis IC, Kappagoda MB, Smith J and
Kneale K: Kimura's disease of the orbit. Ophthal Plast Reconstr
Surg. 4:235–239. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seregard S: Angiolymphoid hyperplasia with
eosinophilia should not be confused with Kimura's disease. Acta
Ophthalmol Scand. 79:91–93. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Takagi K, Harada T and Ishikawa E:
Kimura's disease (eosinophilic lymphfolliculoid granuloma). Nihon
Rinsho. 51:785–788. 1993.(In Japanese). PubMed/NCBI
|
|
18
|
Tseng CF, Lin HC, Huang SC and Su CY:
Kimura's disease presenting as bilateral parotid masses. Eur Arch
Otorhinolaryngol. 262:8–10. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Asadi AK: Angiolymphoid hyperplasia with
eosinophilia. Dermatol Online J. 8:102002.PubMed/NCBI
|
|
20
|
Kimura Y, Pawankar R, Aoki M, Niimi Y and
Kawana S: Mast cells and T cells in Kimura's disease express
increased levels of interleukin-4, interleukin-5, eotaxin and
RANTES. Clin Exp Allergy. 32:1787–1793. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oguz KK, Ozturk A and Cila A: Magnetic
resonance imaging findings in Kimura's disease. Neuroradiology.
46:855–858. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choi JA, Lee GK, Kong KY, Hong SH, Suh JS,
Ahn JM, Lee YJ, Cho KH, Park JG, Choi JY, et al: Imaging findings
of Kimura's disease in the soft tissue of the upper extremity. AJR
Am J Roentgenol. 184:193–199. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Googe PB, Harris NL and Mihm MC Jr:
Kimura's disease and angiolymphoid hyperplasia with eosinophilia:
Two distinct histopathological entities. J Cutan Pathol.
14:263–271. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ohta N, Fukase S, Suzuki Y, Ito T,
Yoshitake H and Aoyagi M: Increase of Th2 and Tc1 cells in patients
with Kimura's disease. Auris Nasus Larynx. 38:77–82. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rajpoot DK, Pahl M and Clark J: Nephrotic
syndrome associated with Kimura disease. Pediatr Nephrol.
14:486–488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Uysal IO, Eryilmaz MA, Salk I and
Abasiyanik F: Kimura disease in the parotid gland. J Craniofac
Surg. 22:337–338. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chim CS, Fung A, Shek TW, Liang R, Ho WK
and Kwong YL: Analysis of clonality in Kimura's disease. Am J Surg
Pathol. 26:1083–1086. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dik VK, van de Wiel BA and Vasmel WL:
Kimura's disease of the parotid glands and multiple cervical lymph
nodes. Neth J Med. 68:175–177. 2010.PubMed/NCBI
|
|
29
|
Gopinathan A and Tan TY: Kimura's disease:
Imaging patterns on computed tomography. Clin Radiol. 64:994–999.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chan TM, Chan PC, Chan KW and Cheng IK:
IgM nephropathy in a patient with Kimura's disease. Nephron.
58:489–490. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Akosa AB, Sherif A and Maidment CG:
Kimura's disease and membranous nephropathy. Nephron. 58:472–474.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun QF, Xu DZ, Pan SH, Ding JG, Xue ZQ,
Miao CS, Cao GJ and Jin DJ: Kimura disease: Review of the
literature. Intern Med J. 38:668–672. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sakamoto M, Komura A and Nishimura S:
Hematoserological analysis of Kimura's disease for optimal
treatment. Otolaryngol Head Neck Surg. 132:159–160. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ragu R, Eng JY and Azlina AR: Kimura's
disease of the parotid: A complete clinical-radiological-pathology
report. Med J Malaysia. 69:199–201. 2014.PubMed/NCBI
|
|
35
|
Som PM and Biller HF: Kimura disease
involving parotid gland and cervical nodes: CT and MR findings. J
Comput Assist Tomogr. 16:320–322. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takahashi S, Ueda J, Furukawa T, Tsuda M,
Nishimura M, Orita H, Tsujimura T and Araki Y: Kimura disease: CT
and MR findings. AJNR Am J Neuroradiol. 17:382–385. 1996.PubMed/NCBI
|
|
37
|
Buggage RR, Spraul CW, Wojno TH and
Grossniklaus HE: Kimura disease of the orbit and ocular adnexa.
Surv Ophthalmol. 44:79–91. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Katagiri K, Itami S, Hatano Y, Yamaguchi T
and Takayasu S: In vivo expression of IL-4, IL-5, IL-13 and
IFN-gamma mRNAs in peripheral blood mononuclear cells and effect of
cyclosporin A in a patient with Kimura's disease. Br J Dermatol.
137:972–977. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kimura T and Yoshimura S: E. I: Unusual
granuloma combined with hyperplastic changes in lymphatic tissues.
Trans Soc Path Jpn. 37:1791948.
|
|
40
|
Allen PW, Ramakrishna B and MacCormac LB:
The histiocytoid hemangiomas and other controversies. Pathol Annu.
27:51–87. 1992.PubMed/NCBI
|
|
41
|
Hiwatashi A, Hasuo K, Shiina T, Ohga S,
Hishiki Y, Fujii K and Ishitoya J: Kimura's disease with bilateral
auricular masses. AJNR Am J Neuroradiol. 20:1976–1978.
1999.PubMed/NCBI
|
|
42
|
Shetty AK, Beaty MW, McGuirt WF Jr, Woods
CR and Givner LB: Kimura's disease: A diagnostic challenge.
Pediatrics. 110:e392002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Itami J, Arimizu N, Miyoshi T, Ogata H and
Miura K: Radiation therapy in Kimura's disease. Acta Oncol.
28:511–514. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kim GE, Kim WC, Yang WI, Kim SK, Oh WY,
Suh HS, Hahn JS and Park CS: Radiation treatment in patients with
recurrent Kimura's disease. Int J Radiat Oncol Biol Phys.
38:607–612. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nonaka M, Sakitani E and Yoshihara T:
Anti-IgE therapy to Kimura's disease: A pilot study. Auris Nasus
Larynx. 41:384–388. 2014. View Article : Google Scholar : PubMed/NCBI
|