Introduction
Hairy cell leukemia (HCL) is a very rare, indolent
disease that typically occurs in middle-aged adults; the disease
was first recognized by the World Health Organization in
2008(1). Its most common symptoms
include fatigue and left upper quadrant pain, with most patients
presenting with splenomegaly. The diagnosis is typically based on
the presence of hairy cells with a ‘fried-egg’ appearance in bone
marrow biopsy samples. Hairy cells are small- to medium-sized
mature B lymphoid cells with oval nuclei and pale blue cytoplasm
(2).
The HCL immunophenotypic profile is characterized by
the expression of CD19, CD20, CD22, and CD200. Cyclin D1 is
expressed in approximately 50-70% of cases, whereas CD5 is weakly
expressed in only approximately 0-2% (1,3). The
BRAF V600E gene mutation is found in >97% of HCL cases (4), and is associated with poor prognosis
(2). As its clinical symptoms are
not obvious, HCL must be differentiated from splenic diffuse red
pulp lymphoma, mantle cell lymphoma (MCL), and other B-cell
lymphomas (2). A combination of
clinical, morphological, immunohistochemical, and molecular
features are required for the diagnosis of HCL.
In the present study, we report a rare case of HCL
with lymphadenopathy in multiple nodes and a distinctive
immunophenotype characterized by CD5 and cyclin D1 expression in
the lymph nodes.
Case report
Patient and clinical data
A 48-year-old woman was admitted to our hospital
with a 2-year history of pancytopenia and splenomegaly. She had
worked in an oil refinery and was often exposed to ammonia,
hydrogen sulfide, and other gases for many years. Peripheral blood
analysis showed low neutrophil counts (2.0x109/l),
hemoglobin levels (98 g/l), and platelet counts
(59x109/l) (Table I).
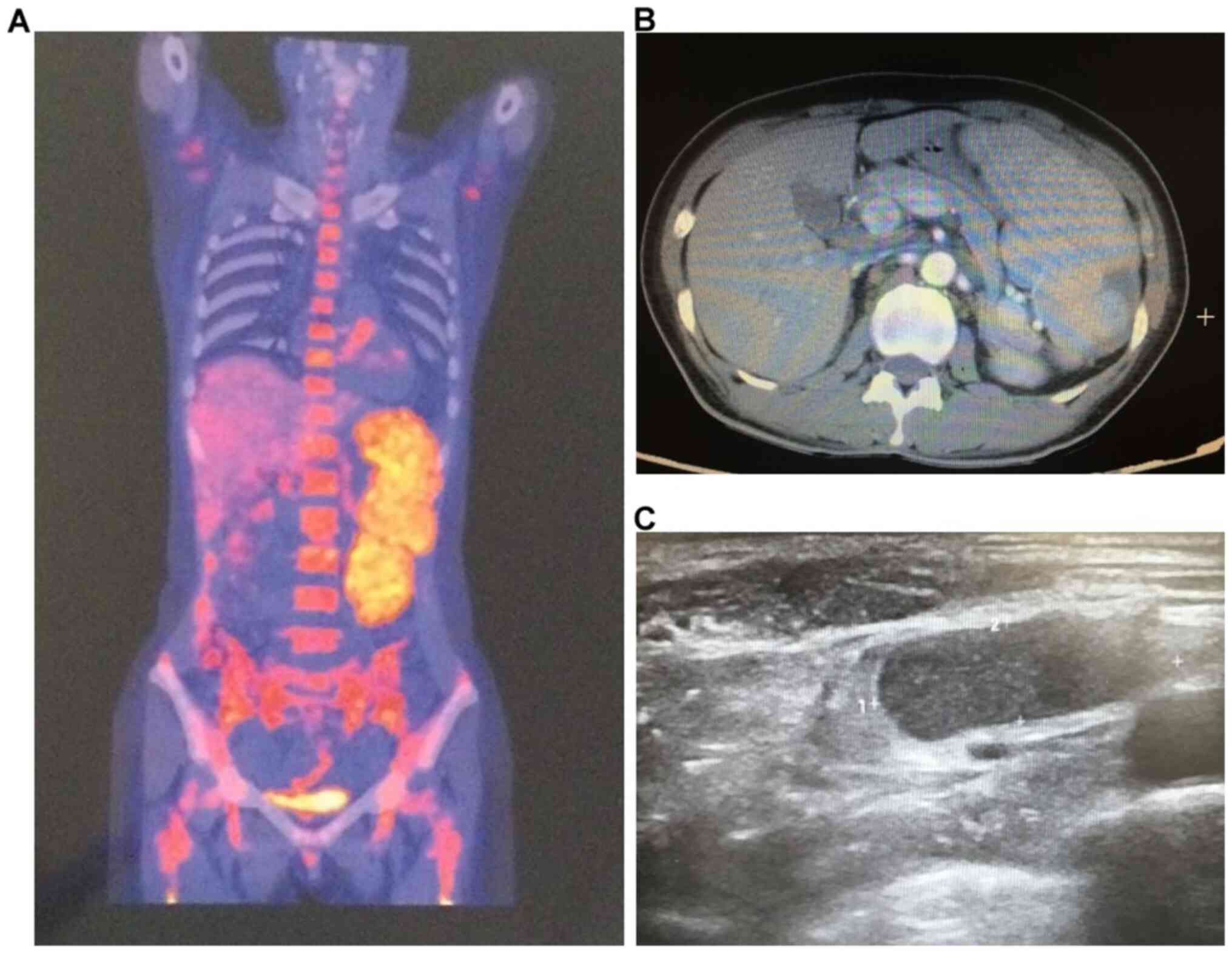
Positron emission tomography-computed tomography (CT) revealed
splenomegaly, high local infarction metabolism, active bone marrow
hyperplasia, and multiple metabolically active lymph nodes around
the mediastinum and aorta (Fig.
1A). B-scan ultrasonography revealed splenomegaly and
mediastinal, neck, and upper clavicle multiple lymphadenopathy
(Fig. 1B). Abdominal CT showed
marked enlargement of the spleen. Thus, lymphoma with splenic and
bone marrow infiltration was considered (Fig. 1C).
 | Table ILaboratory data. |
Table I
Laboratory data.
| Variable | On admission | Reference range,
adults |
|---|
| Hemoglobin (g/l) | 98 | 113-151 |
| White-cell count
(x109/l) | 2.0 | 3.69-9.16 |
| Differential count
(%) | | |
|
Neutrophils | 88.6 | 50-70 |
|
Lymphocytes | 7.8 | 20-40 |
|
Monocytes | 0.6 | 3-10 |
|
Eosinophils | 2.5 | 0.5-5 |
|
Basophils | 0.5 | <1.0 |
| Red-cell count
(x1012/l) | 2.09 | 3.68-5.13 |
| Platelet count
(x109/l) | 59 | 101-320 |
| Albumin (g/l) | 30 | 35-55 |
| Total bilirubin
(µmol/l) | 12.2 | 4.7-24 |
| Direct bilirubin
(µmol/l) | 2.5 | 0-6.8 |
| D-dimer (mg/l) | 0.61 | <0.55 |
| Fibrinogen (g/l) | 1.5 | 1.8-3.5 |
| Epstein-Barr virus
viral capsid antigen IgG antibody | Negative | Negative |
| Epstein-Barr virus
viral nuclear antigen IgG antibody | Negative | Negative |
On physical examination, the patient appeared to be
well and was not anemic or pale. No skin xanthochromia, petechiae,
or ecchymosis was noted. Other findings included arrhythmia without
a pathological murmur, clear pulmonary respiration, a soft abdomen
without tenderness or rebound pain, an intact liver and a spleen
subcostal region of 10 cm. There was no edema in the lower
extremities or significant abnormalities in kidney and liver
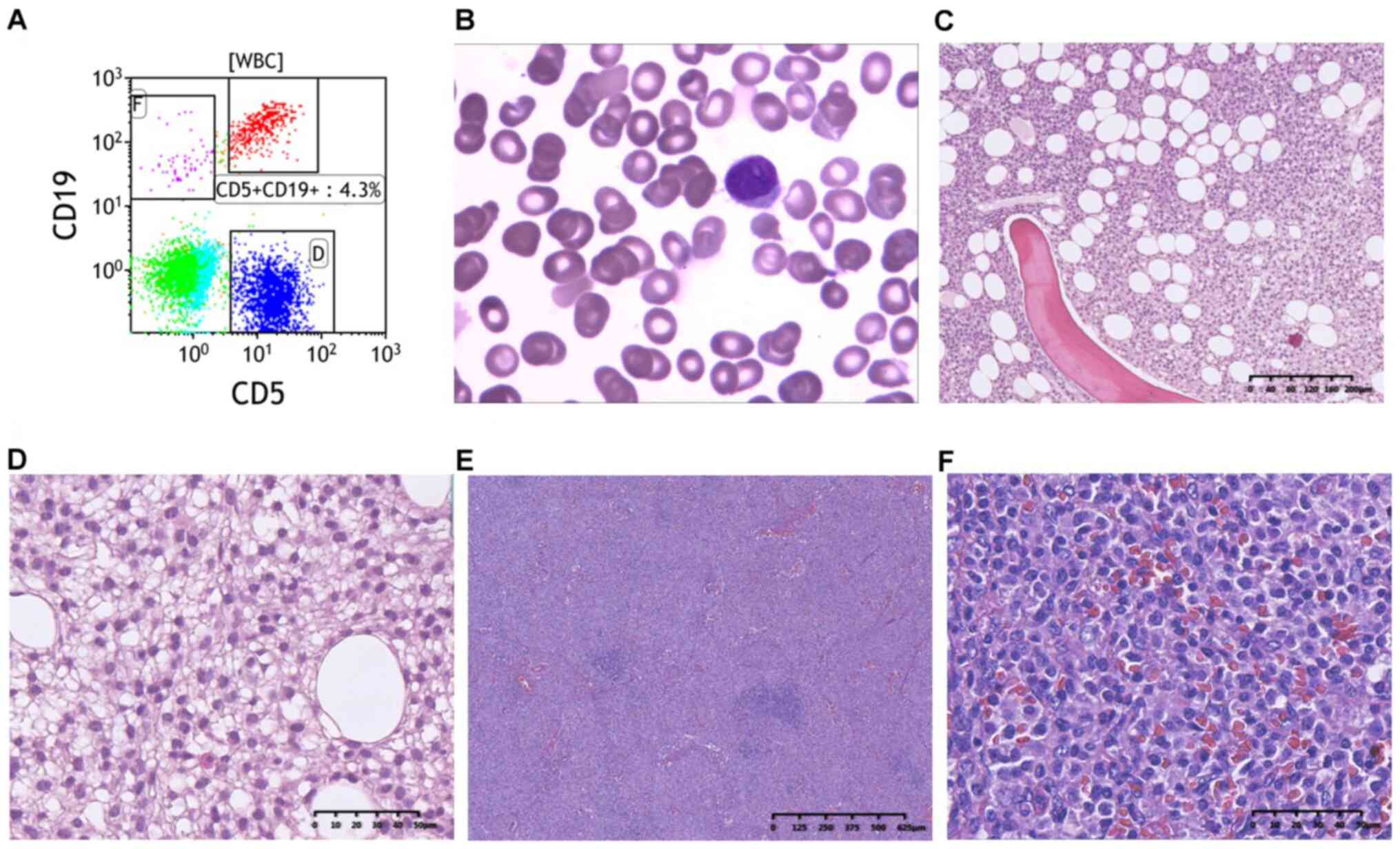
function tests. Flow cytometric immunophenotypic analysis of bone
marrow aspirates showed expression of CD5 and CD19 (Fig. 2A). The typical morphological
features of circulating hairy cells in the bone marrow can be seen
in peripheral blood smear photomicrographs (Fig. 2B). The morphological features
identified following the bone marrow biopsy are shown in Fig. 2C and D. Histological analysis of upper clavicle
lymph node biopsy samples showed a proliferation of small- to
medium-sized lymphoid cells with a vaguely nodular growth pattern
(Fig. 2E). The lymphoid cells had
slightly irregular nuclear contours, and many small vessels were
evident (Fig. 2F). The pathological
diagnosis was challenging.
Cladribine chemotherapy was then administered to the
patient. The patient experienced bone marrow depression and fever
accompanied by agranulocytosis. Anti-inflammatory drugs including
cephalosporin, were administered. The patient's body temperature
returned to normal. The 8-month follow-up revealed that the patient
had recovered well.
This study was approved by the Ethics Committee of
Ruijin Hospital, Shanghai Jiaotong University School of Medicine
(Shanghai, China). Written informed consent was obtained from the
patient for publication of this case report and accompanying images
with preservation of the patient's anonymity.
Pathological findings
The patient's bone marrow had morphologic features
typical of HCL: Circulating small- to medium-sized hairy cells with
abundant clear cytoplasm and oval or indented nuclei. Bone marrow
biopsy showed a diffuse infiltration of lymphoid cells with oval
nuclei, abundant cytoplasm, and prominent borders. Mitotic figures
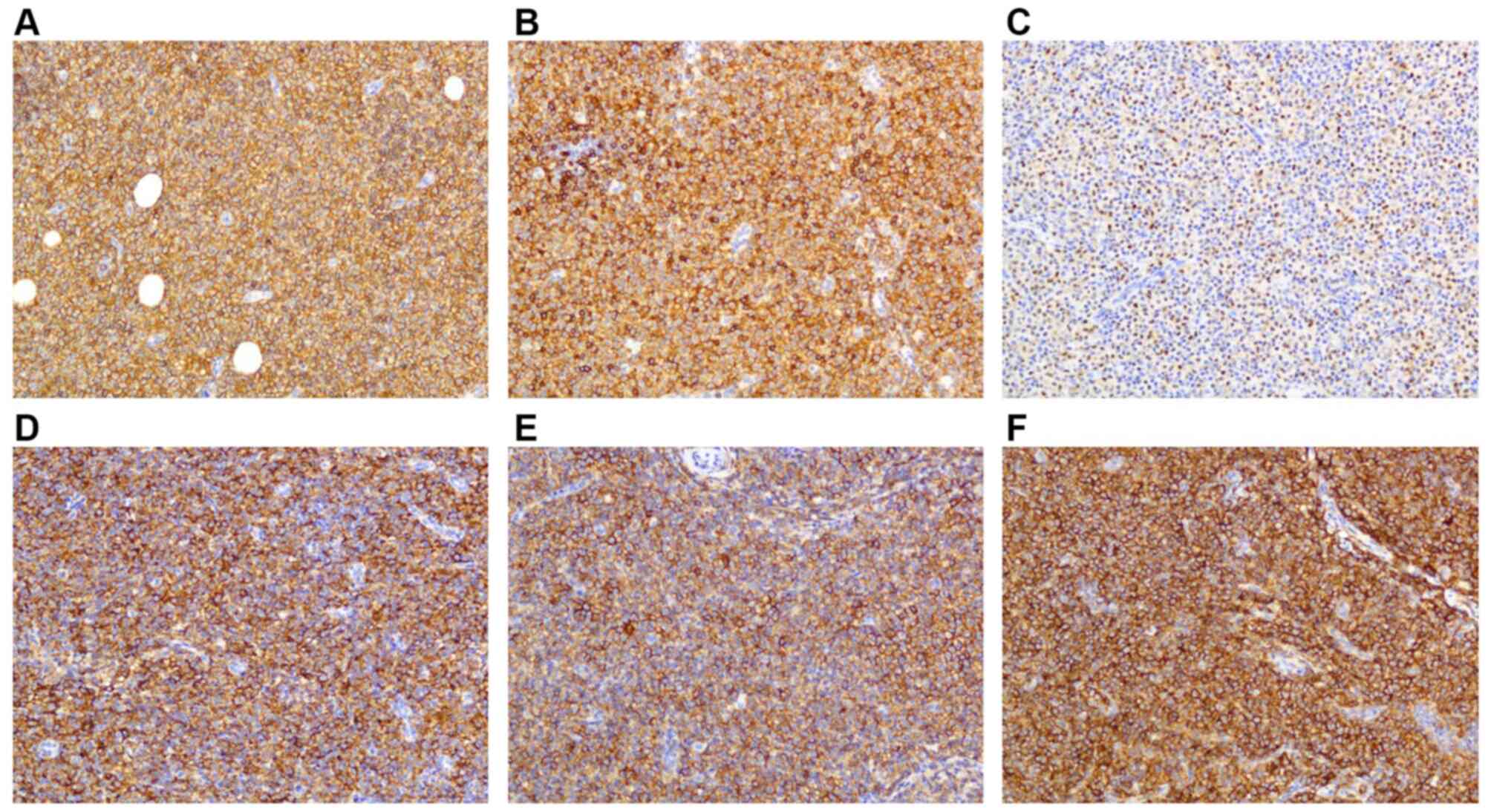
were absent. The B-cell infiltrate was positive for CD5, CD20,
CD79, cyclin D1, Bcl-2, CD25, CD103, CD11C, whereas IgD, CD3, CD10,
CD23, and Mum-1 were not expressed (Fig. 3). Immunohistochemistry of the hairy
cell leukemia cells in the bone marrow biopsy shows strong and
diffuse expression of CD20, CD5, CD103 and partially positive
expression of Annexin A1 (Fig.
4).
Immunohistochemical staining was performed using the
Envision 2-step method with 3,3'-diaminobenzidine as the substrate.
The slides were counterstained with hematoxylin, and CD5, CD11C,
CD20, CD25, CD79, CD103, cyclin D1, Bcl-2, CD25, CD103, CD11C, IgD,
CD3, CD23 and Mum-1 were all prediluted from DAKO. CD10 (DAKO)
dilution of 1:80 was used and appropriate positive controls was
used for all assays.
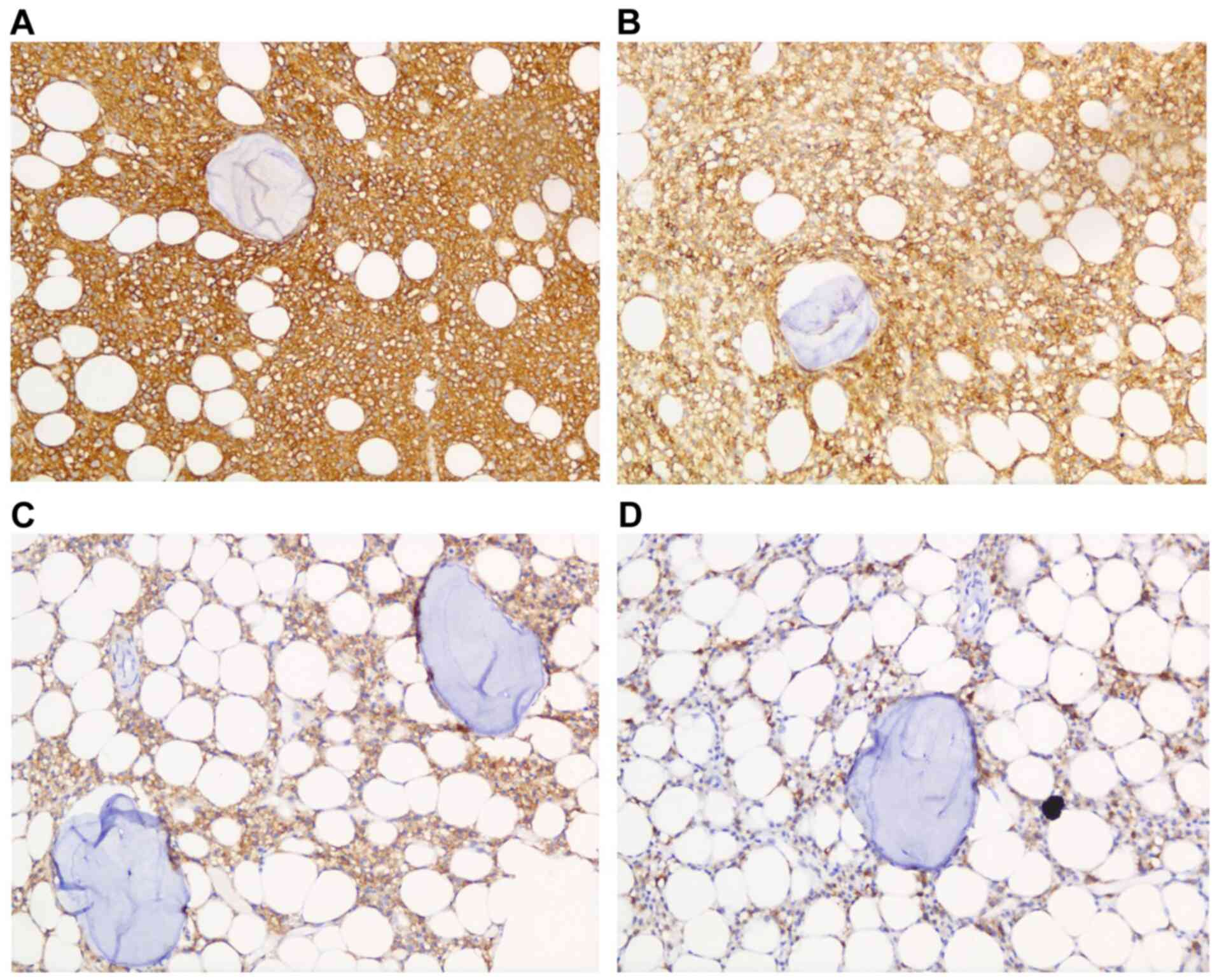
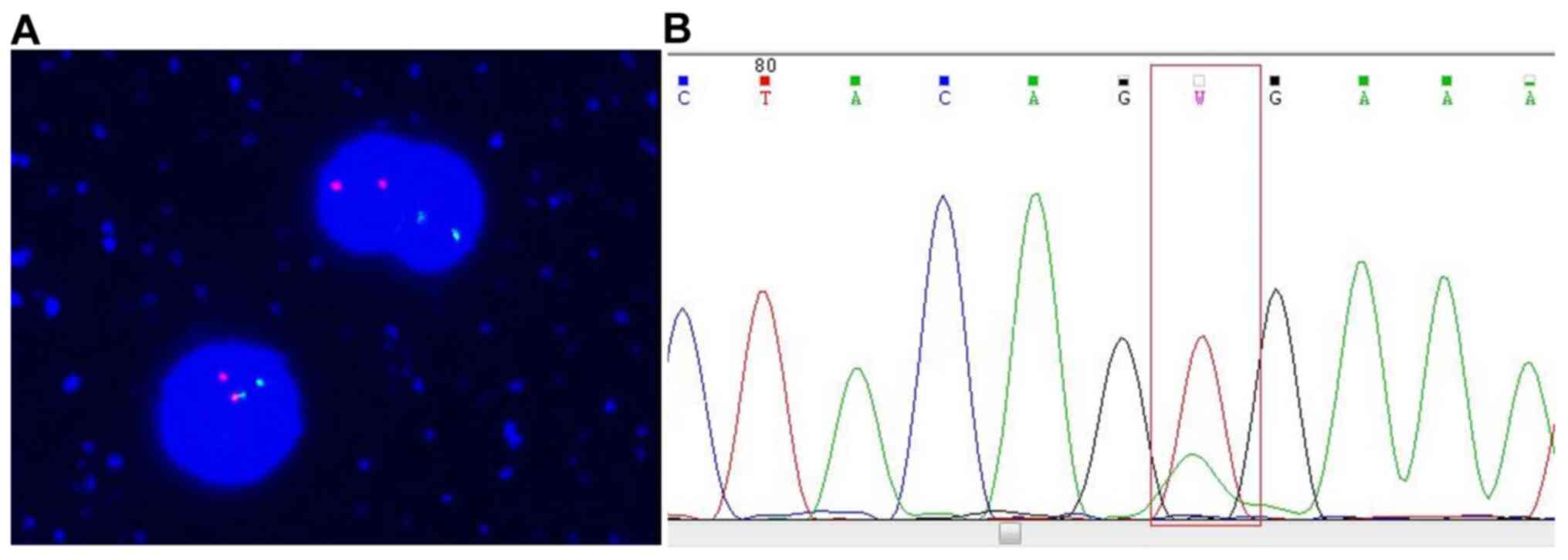
CCND1 t(11;14)(q13;q32) gene fusion and BRAF V600E
mutation were assessed in bone marrow samples via in situ
hybridization and amplification refractory mutation system
polymerase chain reaction (ARMS-PCR), respectively. The BRAF V600E
mutation was detected positively (Fig.
5), whereas the CCND1 t(11;14)(q13;q32) gene fusion was
not. Finally, the overall findings were diagnostic of HCL with
lymph node, peripheral blood, and bone marrow infiltration.
Discussion
This report describes a rare case of HCL involving
the lymph nodes and bone marrow and the immunohistochemical
expression of CD5 and cyclin D1. Given these characteristics, HCL
was initially misdiagnosed as MCL. However, additional analyses
revealed features characteristic of HCL but not MCL, namely, the
absence of the CCND1 gene fusion, the presence of the BRAF
V600E mutation, and the strong expression of CD11C, CD25, and
CD103.
HCL is a chronic malignant hematological malignancy
characterized by dysplastic pallidum-like cells. Bouroncle et
al first described it in 1958(5), and Schrek and Donnelly coined the term
‘HCL’ in 1966 in a report of two patients with leukemia who had
cells identified with numerous hairy processes on the edges of the
peripheral blood cells (6). HCL is
very rare, accounting for only 2% of all lymphoid leukemias. It
mainly affects the elderly, but is not uncommon in young and
middle-aged adults (7). Most
patients present with pancytopenia and splenomegaly, and anemia and
infection have also been reported (8). All the patients with HCL have
different degrees of splenomegaly, and diffuse expansion of the red
pulp can be seen macroscopically. HCL infiltrates the lymph nodes
in a marginal zone or interfollicular pattern, and the nodal
sinuses are often preserved (8). In
the present case, multiple lymph nodes were enlarged and CD5 and
cyclin D1 were strongly co-expressed in the infiltrating lymph
nodes and bone marrow. However, co-expression of CD5 and cyclin D1
in the lymph nodes of patients with HCL has never been previously
reported.
Cortazar et al reported that the
immunophenotype of nodal/extranodal diseases overlapped with that
of other small B-cell lymphomas; proteins expressed by both sets of
diseases included CD5 and cyclin D1(3). MCL is a mature B-cell neoplasm that
accounts for 3-10% of malignant lymphomas. It has an atypical
‘hairy cell-like’ morphology that can easily be confused with that
of other malignant lymphomas with hairy cytoplasmic projections
(9). Its hallmark is CD5 and cyclin
D1 positivity (10,11). As the HCL in our case strongly
expressed CD5 and cyclin D1, it was difficult to differentiate it
from MCL based on immunophenotype alone.
HCL should be differentiated from the HCL-variant,
chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL), and
other indolent B-cell lymphomas. The HCL-variant is a rare disease
that was first identified by Cawley et al (12). Immunophenotypically, HCL cells
usually express the mature B-cell markers CD19 and CD20, as well as
CD11c, CD25, CD103, and CD123. By contrast, the HCL-variant cells
often express CD11c and CD103 but not CD25 or CD123. Moreover, the
BRAF V600E mutation is present in essentially all HCL cases,
but is not present in HCL-variant cases (13-15).
CLL is characterized by mature B lymphocyte clonal proliferative
tumors characterized by lymphocyte aggregation in the peripheral
blood, bone marrow, and spleen. The diagnostic requirements for CLL
are a peripheral blood B lymphocyte count >5x109/l
and CD5 and CD23 expression in B cells. The lymph node in our case
was CD23-negative, which distinguishes our case from CLL.
Immunohistochemical and molecular analyses are
essential for distinguishing HCL from other small B-cell lymphomas.
HCLs co-express CD25, CD103, and CD123. Although common to both HCL
and MCL, cyclin D1 is weakly expressed in the former and strongly
in the latter. CCND1-IGH translocations are present in MCLs but
absent in nearly all HCLs (16,17).
Conversely, the BRAF V600E mutation is present in nearly all HCLs
but is not present in other B-cell lymphomas (18).
In conclusion, diagnosis of HCL requires collective
consideration of cytological, histological, immunohistochemical
data and cytogenetic abnormalities.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LZ performed the histological examination of this
disease, and was a major contributor in writing the manuscript. HX
performed immunohistochemical staining and analysis. JZ and BO
collected the clinical data. CW participated in the design of the
study and assisted in writing the manuscript. All authors read and
approved the final manuscript.
Erhics approval and consent to
participate
This study was approved by the Ethics Committee of
Ruijin Hospital, Shanghai Jiaotong University School of Medicine
(Shanghai).
Patient consent for participation
Written informed consent was obtained from the
patient for publication of this case report and accompanying images
with preservation of the patient's anonymity.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen YH, Tallman MS, Goolsby C and
Peterson L: Immunophenotypic variations in hairy cell leukemia. Am
J Clin Pathol. 125:251–259. 2006.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Troussard X and Cornet E: Hairy cell
leukemia 2018: Update on diagnosis, risk-stratification, and
treatment. Am J Hematol. 92:1382–1390. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Cortazar JM, DeAngelo DJ, Pinkus GS and
Morgan EA: Morphological and immunophenotypical features of hairy
cell leukaemia involving lymph nodes and extranodal tissues.
Histopathology. 71:112–124. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Arcaini L, Zibellini S, Boveri E, Riboni
R, Rattotti S, Varettoni M, Guerrera ML, Lucioni M, Tenore A, Merli
M, et al: The BRAF V600E mutation in hairy cell leukemia and other
mature B-cell neoplasms. Blood. 119:188–191. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Bouroncle BA, Wiseman BK and Doan CA:
Leukemic reticuloendotheliosis. Blood. 13:609–630. 1958.PubMed/NCBI
|
|
6
|
Schrek R and Donnelly WJ: ‘Hairy’ cells in
blood lymphoreticular neoplastic disease and ‘flagellated’ cells of
normal lymph nodes. Blood. 27:199–211. 1966.PubMed/NCBI
|
|
7
|
Thomas MK and Marshall EK: Teenager with
hairy cell leukemia: 30-year follow-up. J Clin Oncol. 27:155–156.
2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sharpe RW and Bethel KJ: Hairy cell
leukemia: Diagnostic pathology. Hematol Oncol Clin North Am.
20:1023–1049. 2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Robier C, Hoefler G, Egger M and Hubmann
E: Atypical ‘hairy cell-like’ presentation of leukemic mantle cell
lymphoma. Clin Chem Lab Med. 19:e34–e36. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
de Boer CJ, van Krieken JH, Kluin-Nelemans
HC, Kluin PM and Schuuring E: Cyclin D1 messenger RNA
overexpression as a marker for mantle cell lymphoma. Oncogene.
10:1833–1840. 1995.PubMed/NCBI
|
|
11
|
Liu Z, Dong HY, Grczyca W, Tsang P, Cohen
P, Stephenson CF, Berger CS, Wu CD and Weisberger J: CD5-Mantle
cell lymphoma. Am J Clin Pathol. 118:216–224. 2003.
|
|
12
|
Cawley JC, Burns GF and Hayhoe FG: A
chronic lymphoproliferative disorder with distinctive features: A
distinct variant of hairy cell leukaemia. Leuk Res. 4:547–559.
1980.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Matutes E, Wotherspoon A and Catovsky D:
The variant form of hairy-cell leukaemia. Best Pract Res Clin
Haematol. 16:41–56. 2003.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Cessna MH, Hartung L, Tripp S, Perkins SL
and Bahler DW: Hairy cell leukemia variant: Fact or fiction. Am J
Clin Pathol. 123:132–138. 2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Xi L, Arons E, Navarro W, Calvo KR,
Stetler-Stevenson M, Raffeld M and Kreitman RJ: Both variant and
IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E
mutation. Blood. 119:3330–3332. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Miranda RN, Briggs RC, Kinney MC, Veno PA,
Hammer RD and Cousar JB: Immunohistochemical detection of cyclin D1
using optimized conditions is highly specific for mantle cell
lymphoma and hairy cell leukemia. Mod Pathol. 13:1308–1314.
2000.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chen D, Ketterling RP, Hanson CA, Colgan
JP, Zent CS and Viswanatha DS: A case of hairy cell leukemia with
CCND1-IGH@ translocation: Indolent non-nodal mantle cell lymphoma
revisited. Am J Surg Pathol. 35:1080–1084. 2011.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kreitman RJ: Hairy cell leukemia-new
genes, new targets. Curr Hematol Malig Rep. 8:184–195.
2013.PubMed/NCBI View Article : Google Scholar
|