1. Introduction
Treatment of cancer by standard methods, surgery,
radiation, and chemotherapy, is less effective in advanced-stage
disease and causes numerous side effects. Consequently, researchers
are in the quest to explore the possibility of developing more
effective, less toxic therapy. Recently immunotherapy has emerged
as a sound approach that includes immune checkpoint inhibitors,
T-cell transfer therapy, monoclonal antibodies, vaccines, and
immune system modulators. The most studied type of immunotherapy is
T-cell transfer therapy or adoptive cell transfer (ACT). ACT is the
collection and the use of patients' immune cells to treat their
cancer. Currently, there are a few types of ACT-based therapies,
tumor-infiltrating lymphocytes (TIL), engineered T cell receptor
(TCR), natural killer (NK) cells, iNKT cells, Chimeric antigen
receptors (CAR) T-cell (1), and
γδT cells (2). TIL uses T cells
around or in a patient's tumor tissues. These T cells are
collected, and the best that recognizes and kills the tumor
ex-vivo is selected, expanded, and adoptively transferred
back to the patient to eliminate tumor cells. TCR or transduced
T-cell is the genetic engineering of T-cells to express new
specific TCR to recognize tumors ex vivo. NK cells therapy
depends on the immune system's activation against abnormal cells.
Unlike TLs, NK cell receptors interact with target cells
independent of antigen processing and presentation. γδ T cells are
T cells that express a unique TCR composed of one γ-chain and one
δ-chain (3,4). In CAR T cell therapy, the T
lymphocytes undergo modification with a receptor based on a
recognition sequence of an antibody, called CAR, a non-MHC
restricted receptor, to attach to specific proteins (antigens) on
cancer cells' surface ex-vivo. The T cells in CAR therapy
have an improved ability to attack and kill the cancer cells
compared to T cells in TIL therapy (5). In these therapies, the lymphocyte
undergoes modification via plasmids or viral vectors, such as
adenovirus, retrovirus, or lentivirus (6).
CAR T therapy showed promising success in treating
malignant blood diseases such as acute lymphoblastic leukemia (ALL)
and diffused-large B cell lymphoma (DLBCL) in children and young
adults. Therefore, the FDA authorized cluster of differentiation 19
(CD19) specific CAR T cell therapies for these diseases.
Tisagenlecleucel (Kymriah™) against ALL and DLBCL for
children/young adults, Axicabtagene ciloleucel (Yescarta™) against
adult non-Hodgkin lymphoma (NHL) and DLBCL, Brexucabtagene
autoleucel (Tecartus™) for relapsed or refractory (R/R) mantle cell
lymphoma (MCL) treatments (7-10),
and most recently, Breyanzi (lisocabtagene maraleucel) for the
treatment of relapsed or refractory (R/R) large B cell lymphomas
(LBCL) in adult patients (11).
However, these CAR T therapies have limited success
in solid tumors. CAR T cells treatment directed against antigens
such as vascular endothelial growth factor receptor 2 (VEGF-R2),
CD171, folate receptor alpha, disialoganglioside GD2, human
epidermal growth factor receptor 2, mesothelin, EGFRvIII, or
carbonic anhydrase IX, in patients with solid tumors failed to
produce similar beneficial outcomes as seen in blood-related
malignancies (12).
Translating successful CAR T-cell therapies to solid
tumors requires overcoming several barriers, including identifying
an ideal tumor-associated antigen to target and overcome antigen
expression heterogeneity, addressing the tumor-suppressive
microenvironment, and employing a preclinical model that faithfully
represents the disease. The review collected data using PubMed,
Google Scholar and other publicly available databases and discusses
the design and evolution of CARs and the challenges facing CAR
therapies in solid tumors. Also, it discusses the advantages and
disadvantages of preclinical animal models emphasizes the
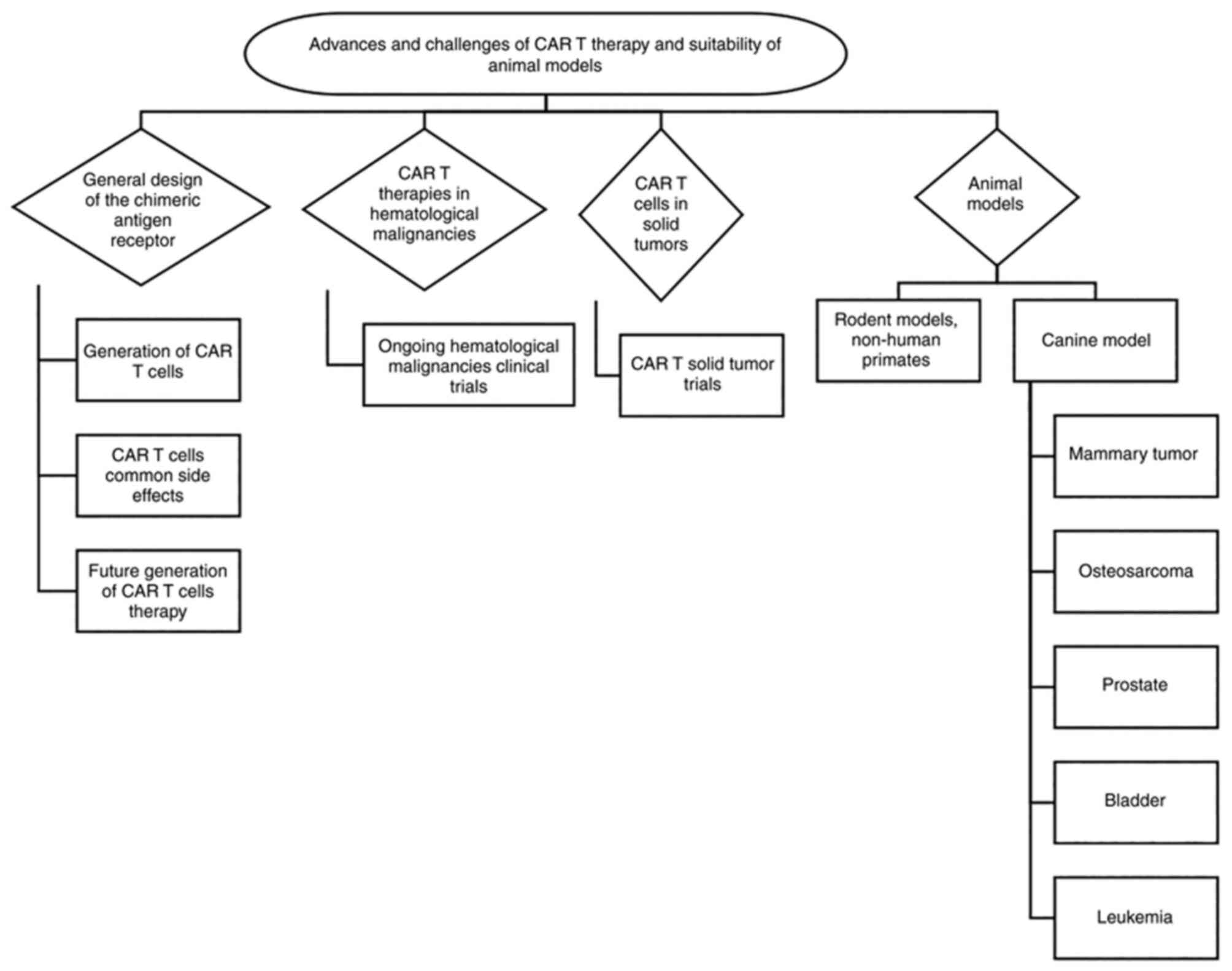
advantages of using the canine model (Fig. 1).
2. General design of the CARs
The discovery of the CARs started around the 1980s.
Several factors are essential for CAR T cell therapy to be
effective, such as recruitment, activation, expansion, and
persistence of bioengineered T cells at the tumor site. Even though
~41 years have passed since the first CAR T cell's creation, some
essential components of its structure remained the same (13). However, these components have
undergone numerous modifications to enhance CAR T therapeutic
capabilities over the years. The structure consists of four
components: the ectodomain (the domain of a membrane protein
outside the cytoplasm) a hinge, the transmembrane domain, and the
intracellular signaling endodomain. Each domain has a specific
function and optimal molecular design. The extracellular domain,
the target-binding domain, is usually a single-chain variable
fragment (scFv) of the antigen-binding region of a monoclonal
antibody's light and heavy chain. It recognizes any antigen and
binds targets with high affinity. The hinge connects the
extracellular antigen-binding domain to the intracellular signaling
domains and regulates the extracellular domain flexibility,
facilitating the migration and binding capacity to tumor cell
receptors. The length and composition of the hinge can affect
antigen binding and signal through the CAR. Generally, the hinge
domain consists of amino acid sequences from CD8, CD28, IgG1, or
IgG4. The transmembrane domains anchor the CAR in the T cell
membrane. It consists of a hydrophobic alpha helix that spans the
membrane, such as CD3ζ, CD28, CD4, or CD8α. The primary function of
the transmembrane domain is to stabilize the CAR. The endodomain
domain (intracellular signaling domain) comprises of the activation
domain, a TCR-derived CD3ζ-derived immunoreceptor tyrosine-based
activation motifs, and intracellular costimulatory domains derived
from CD28 or 4-1BB (CD137) (14,15).
The first CAR generations with CD3-ζ transmembrane domains suffered
detachment from the surface of T cells. Consequently, CAR T
structure is subjected to modification with a well-balanced
transmembrane domain composed of the CD4, CD8, or CD28 molecules
(16). Antigen-specific T cell
activation, in nature, requires three signals to gain full
functionality that enables proliferation, differentiation, and
survival. Co-stimulation plays a vital role in the CAR T-cell
functionality as it triggers the T-cell immune response against
foreign antigens. The absence of co-stimulation can enter T cells
in a state of anergy, leading to its unresponsiveness to antigen
binding (17). Unfortunately,
cancer cells promote co-stimulatory-ligand deficient environments
generating unfavorable antitumor responses. Therefore, CAR T is
designed with various costimulatory molecules to overcome the tumor
cell suppressing environment. The conserved region of a CD3-ζ
domain, the immunoreceptor tyrosine-based activation motifs
(ITAMs), carries out signaling transduction pathways on CAR T cells
to build sufficient T cell activation (18).
Also, CARs function without relying on the major
histocompatibility complex (MHC), allowing it to target various
antigens without antigen presentation for activation since
activated with the single-chain Fv domain interaction with the
targeted TAA (19) The MHC
independence is an essential feature of CAR design since the tumor
microenvironment consistently down-regulates the MHC complexes.
3. Generations of CAR T cells
Although CAR T therapy can lead to long-lasting
remissions for some patients with very advanced malignant disease,
it can cause severe and fatal side effects such as cytokines storm
and neurological problems, including termer, delirium, and
seizures. Therefore, scientists modified CAR T cells to create safe
and more effective therapy by building on the CAR T cell's original
components and information gained from clinical trials. These
include:
CAR 1st generation
It consists of a single-chain variable fragment
(scFv) ectodomain and a TCR-derived signaling CD3-ζ constant region
representing the endodomain fragment. These 1st generation CAR
cannot maintain the CAR stable on the T cell membrane and T cell
activation for a considerable amount of time (20).
CAR second and third generations
The second and the third generation compared to the
1st generation were modified to enhance the receptor cohesion
toward the lymphocyte surface, thus allowing optimal functionality.
As a result, these CARs generations have one (2nd generation) or
two (3rd generation) costimulatory signals that augment T cell
proliferation, differentiation, and survival despite the effect of
tumor-suppressing environments (17).
CAR 4th generation
The fourth generation compared to 2nd and 3rd
generation CAR, create a robust immune attack to eliminate the
tumor before they re-generate or mutate. The 4th generation CAR T
cells redirected for universal cytokine killing (TRUCK), has the
same structure and physiology as the 2nd and the 3rd CAR
generations with a slight genotypic difference (20). These TRUCKs contain a nuclear
factor of the activated T cells (NFAT), codifying a transgenic
cytokine. NFATs are found in T cells and play a crucial role in
cytokine expression. TRUCKs deliver a considerable amount of IL-12
on the tumor site stimulating T cells and recruiting other
immunological cells to target tumor cells not recognized by the
(svFc) fragment of a CAR (21).
CAR 5th generation
The 5th generation have the same structure as the
second generation of CARs, but they contain a truncated cytoplasmic
IL-2 receptor β-chain domain with a binding site for the
transcription factor STAT3. The antigen-specific activation of this
receptor simultaneously triggers TCR (through the CD3ζ domains),
costimulatory (CD28 domain), and cytokine (JAK-STAT3/5) signaling
required physiologically to drive full T cell activation and
proliferation.
4. CAR T-cell therapies common side
effects
CAR-based therapy's common side effects are the
body's immunological defense impulses triggered by the T cell
artificial receptor. These autoimmune consequences can affect the
patient's prognosis and disease outcomes. The most common side
effects include.
Cytokine release syndrome (On-target
on-tumor toxicity)
One of the most frequent setbacks in using CAR T
therapies is releasing proinflammatory cytokines into the body or
cytokine release syndrome (CRS) due to excessive antigen-CAR T cell
engagement. These cytokines are small proteins that act as cell
messengers to help direct the body's immune response. Increased
cytokine levels lead to chronic inflammation throughout the body,
which can be harmful and interfere with several body functions. CRS
is characterized by increased serum levels of cytokines, fever,
diarrheas, hypotension, hypoxemia, low blood pressure, and organ
dysfunctions. Most patients have a mild CRS form, but it may be
severe or life-threatening in some individuals due to organ
failure. The severity of CRS depends upon the disease burden.
Generally, splitting the initial dose and strictly monitoring the
vital parameters can mitigate the risk. Also, treating specific
symptoms to lower the immune response, such as tocilizumab and
siltuximab, interferes with IL-6 or corticosteroids to help reduce
inflammatory and immune response (22).
Immune effector cell-associated
neurotoxicity syndrome (ICANS)
Although CAR T neurotoxicity is the most common side
effect, its pathophysiology is not entirely understood. Recent
studies suggested that blood-brain barrier disfunction (BBB) causes
CAR T cells' infiltration into the cerebrospinal fluid (23). Symptoms include confusion,
myoclonus, seizures, delirium, aphasia, memory loss, and coma
(8,9,22).
Neurotoxic issues are reported in patients within the first two
months of CAR T treatment lasting between 6-17 days, depending on
the type of blood cancer treated and the specific drug-infused
(24). Trials studying GD2 in
treating neuroblastoma with high-affinity GD2 specific CAR T and
ERBB2 with ERBB specific CAR T for metastatic colorectal cancer
found it to cause severe neurotoxicity and multi-organ failure,
respectively (25,26).
On-target toxicities (On-target
off-tumor toxicity)
On-target off-tumor effect arises in patients with
target antigens expressed on both tumors and healthy tissues. The
condition was first noticed in patients who experienced uncommon
reductions of healthy B-lymphocytes, B-cell aplasia, in trials
utilizing a CD-19 specific CAR T cell due to the binding of the
engineered T cells to both CD-19 malignant and healthy B cell
(27,28). Similarly, low-level ERBB2, CAIX,
and CEACAM5 expression on healthy lung, liver, and gastrointestinal
epithelia resulted in deadly toxicities in these organs (25,29).
Thus, it is crucial to know the background expression of the target
antigen in healthy tissues to determine whether its levels are over
the threshold that may cause toxicity and the potential
severity.
Off-target toxicity
Off-target toxicity occur when CAR T cells attack an
antigen other than those for which the CAR T was meant to bind or
activate themselves independently from their specificity. The risk
of off-target toxicity occurs due to the inherited CAR T makeup
(23). For example, patients
treated with CAR T-anti-HER2/neu. CAR T-anti-HER2/neu carries
IgG1-derived CH2CH3 domain as an extracellular spacer which can
interact with the Fc receptor expressed on innate immune cells and,
as a result, lead to antigen-independent activation (29).
5. Future generations of CAR T therapy
Even though treatment with CAR-T cells has produced
remarkable clinical responses with specific subsets of B cell
leukemia or lymphoma, a number of challenges (mentioned above)
limit the therapeutic efficacy of CAR-T cells in solid tumors and
hematological malignancies. However, researchers are working
restlessly to overcome these limitations by pursuing various new
CAR concepts and models to generate the next generation of CAR
therapies. These concepts include:
The bispecific adaptor platform
Among numerous platforms to improve CAR T therapy,
the adaptor CAR platforms have received much attention and immense
research. The platform separates the tumor-targeting and signaling
moieties of conventional CARs resulting in a system consisting of
an adaptor CAR or Universal CAR and soluble, tumor-specific adaptor
molecules. The universal CAR construct contains cytoplasmic
activation domains in conventional CAR and an extracellular
single-chain variable fragment (scFv) that recognizes fluorescein
(anti-FITC CAR T cell. The bispecific adapter molecule comprises
fluorescein linked to a tumor-specific ligand. Such an adaptor
brings the CAR T cell to the tumor cell triggering CAR T-cell
activation and the subsequent destruction of the cancer cell-the
omission of the bispecific adapter prevents CAR T-cell engagement
with the cancer cell and the tumor cell killing. A cocktail of
orthogonal fluorescein-linked bispecific adapters in which each
fluorescein-linked adapter is attached to a unique tumor-specific
ligand capable of binding one of the cancer cell's antigens could
be prepared (30,31). Developing this platform improves
conventional CAR T cells' flexibility, tumor specificity, and
controllability (32).
Dual CAR T-cells
Despite the great successes with Tisagenlecleucel
and Axicabtagene, Anti-CD19 chimeric CAR T cell, therapy in
leukemia, up to 60% of patients relapse due to CD19 antigen loss. A
new approach to overcoming antigen loss targets more than one
antigen on cancer cells, such as autologous CD19/CD22 CAR T cell
therapy, which demonstrated to be safe and had anti-leukemic
activity in patients with relapsed/refractory B-ALL (33).
Dominant-negative receptor CAR T
cells
In addition to the target antigen scFv,
dominant-negative receptor CAR T cells are transduced with an
additional co-inhibitory receptor that controls inhibitory signals
sent by the tumor milieu to the T cell. Those receptors include
PD-1 and TGF-βRII (34,35). Other upregulated receptors when the
T cell is exhausted, and potential candidates for this type of
method are CTLA-4, TIM-3, and TIGIT.
Off-the-shelf CAR T cells
These Off-the-shelf CAR T cells are a third-party,
healthy donor-derived alternative. Because the preparation of
autologous CAR T cells takes time, the patient needs to be stable
to withdraw their T cells by leukapheresis; pre-made CAR T cells
offer a ready-to-use therapy for advance stage cancer patients.
6. CAR T therapies in hematological
malignancies
The FDA gave authorization for five CART therapies
up to date. The first four therapies utilize slightly different
methods of genetic engineering to transform the patient's T cells
into CAR-T cells. However, all therapies produced CAR T cells that
bind to the cluster differentiation 19 (CD19) protein on the B-cell
surface.
The first approved CAR-T therapy is tisagenlecleucel
(Kymriah; Novartis), approved in August 2017. In this therapy, the
T cells are induced by a vector that encodes a second-generation
CAR with scFv, derived from the CD19-specific monoclonal antibody
FMC63 and the costimulatory domain from 4-1BB and CD3ζ. The therapy
is indicated to treat acute lymphoblastic leukemia, the most common
cause of cancer-related deaths among children in the USA age 25 or
younger (36).
The second FDA-approved CAR T therapy is
Axicabtagene ciloleucel (Yescarta™), developed by Kite, a Gilead
Science, Inc company, in October 2017. In this therapy,
patient-derived T cells are transduced using a gamma-retroviral
vector expressing a second-generation CAR that targets CD19.
Yescarta is created from CD3+ enriched autologous T
cells, while Kymriah is generated from autologous CD4/CD8 T-cell.
The therapy works similarly to Kymriah but is indicated for
treating adults with certain non-Hodgkin lymphomas, including
diffuse large B-cell lymphoma (37).
The third FDA-approved CART therapy is
brexucabtagene autoleucel (Tecartus), on July 24, 2020, developed
by Kite Pharma to treat relapsed or refractory (R/R) mantle cell
lymphoma (MCL), which is a form of non-Hodgkin lymphoma occurring
in cells from the ‘mantle’ zone of the lymph node. It is aggressive
cancer that primarily affects men 60 years and over. Tecartus is
similar to Yescarta in generation and CAR structure. It is the
first and only CAR-T cell therapy for adult patients suffering from
R/R mantle cell lymphoma (38).
In February 2021, the FDA approved the fourth CAR T
therapy, Lisocabtagene maraleucel (Breyanzi®; Bristol Myers
Squibb). Breyanzi® is indicated for adult patients with
relapsed or refractory large B-cell lymphoma, including diffuse
large B-cell lymphoma (DLBCL) not otherwise specified (including
DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma,
primary mediastinal large B-cell lymphoma, and follicular lymphoma
grade 3B after two or more lines of systemic therapy.
However, these treatments caused two potentially
fatal side effects: neurologic toxicity and cytokine release
syndrome (CRS). CRS occurred in 94% of patients; 13% experienced
symptoms that required aggressive treatment or were considered
life-threatening in the phase II ZUMA-1 trial (11,39).
Recently, in March 2021, FDA approved the first
B-cell maturation agent (BCMA)-directed CAR T cell therapy,
idecabtagene vicleucel (Abecma®) developed by Bristol
Myers Squibb. It is indicated for relapsed or refractory multiple
myeloma treatment after four or more prior lines of therapy
(40). BCMA is a member of the
tumor necrosis factor superfamily and only expressed by some B
cells, normal plasma cells, and malignant plasma cells and not
expressed by hematopoietic stem cells and normal essential
non-hematopoietic tissues (41).
Ongoing hematological malignancies
clinical trials
Currently, numerous trials used CAR T cells against
different hematological malignancies: A Phase I clinical trial
(NCT03778346) against Refractory/Recurrent Multiple Myeloma using
BCMA-7x19 CAR-T cells by Wenzhou Medical University. The CAR-T cell
targets BCMA antigens and expresses IL-7 and CCL19. This design
provides superior T cells differentiation, migration, expansion,
and tumor killing. Both patients enrolled achieved complete
remission (CR) and very good partial response (VGPR) with a
response of over 12 months. Side effects included Grade 1 cytokine
release syndrome one month after the first infusion. A Phase II
clinical trial evaluated the efficacy and safety of anti-CD19 CAR-T
cells alone or in combination with anti-B cell maturation antigen
CAR-T cells therapy against relapsed/refractory multiple myeloma.
The disease targeted immunoglobulin D (IgD) multiple myeloma, a
rare subtype with a worse prognosis. A total of 7 patients enrolled
in the trial. Six achieved stringent complete remissions (CR), and
one with extracellular disease achieved minimal response (MR) 60
days after the first infusion.
Clinical trials conducted by Kite Pharma, Inc., the
developers of Yescarta™, are currently underway to demonstrate
safety and clinical benefits to patients with R/R Indolent
Non-Hodgkin Lymphoma (iNHL). ZUMA-5 is a Phase II multicenter trial
in which participants receive an infusion of axi-cel CAR-T cells
(2x106 cells/kg). The participants included 124 patients
with follicular lymphoma (FL) and 22 with marginal zone lymphoma
(MZL). Out of the evaluated 104 patients, the ORR was 92%, with a
CR of 76% after a 17.5-month follow-up. FL patients (n=84)
responded with an ORR of 94% and CR of 80% compared to the MZL
patients (n=20) with 85% ORR and a 60% CR.
Three different clinical trials ELIANA
(NCT02435849), ENSIGN (NCT02228096), and B2101J (NCT01626495),
tested Kymriah™ (Novartis Pharmaceuticals Corp.) in CD19-positive
R/R B cell acute lymphoblastic leukemia. The patients of all three
trials experienced a minimum of 69-95% overall remission rates
(ORR) with durable remission. A Phase I clinical trial using m971
anti-CD22 CAR-T cells targeting R/R B-cell ALL patients previously
received an infusion of CD19 CAR-T cells. Even though CD19 CAR T
has impressive results treating ALL patients, some patients
relapse. The trial consisted of two cohorts of patients with R/R
Large B cell lymphoma (n=9) and patients with R/R B-cell ALL (n=6)
that undergo allogeneic hematopoietic stem cell transplant.
Patients that experienced R/R Large B cell lymphoma received an
infusion of 1x106 (n=3) and 3x106 cells/kg
(n=6), while all R/R B-cell ALL received 1x106 cells/kg.
Large B cell lymphoma patients experienced ORR of 78% and CR of
56%. Five of the R/R B-cell ALL patients were minimal disease
negative in the 28 days, while all subjects except one experienced
relapse. Flow cytometry analysis showed that ALL patients
downregulate CD22, promoting relapse.
7. CAR T cells in solid tumors
T cell therapy's potential to induce successful
immunological responses in patients with solid tumors has been
demonstrated in immune checkpoint therapy (42) and TIL and TCR therapies in
melanoma, sarcoma, cholangiocarcinoma, and breast cancer in a few
patients (43), suggesting T cells
can eliminate solid tumors under adequate condition. However, few
CAR-T cell therapy attempts have been reported in glioblastoma and
neuroblastoma (44,45). The Key challenges posed to CAR T
cell therapy success in solid tumors can be described in three
steps: finding, entering, and surviving in the tumor. These
challenges include the lack of tumor-specific target antigens and
tumor cell heterogeneity, CAR T cell trafficking/infiltration
towards tumor sites, T cell inhibitory signals in solid tumors,
physical barriers in the solid tumor microenvironment, and the
immunosuppressive microenvironment (26,46,47).
Antigen selection and heterogeneity in
solid tumors
Target selection in solid tumors is a major hurdle
in implementing CAR T-cell therapy against solid tumors. Also, in
contrast with hematological malignancies, where the surface antigen
expression is uniform and intense, solid tumor cells rarely express
uniformly one specific antigen, and even when present, the levels
may be quite variable (47). The
antigen is also more common to be enriched on tumors and at low
levels on healthy tissues, increasing the potential risk of
significant on-target off-tumor toxicity. Almost all currently
targeted TAAs for solid tumors display this heterogeneity,
including CEA, ERBB2, EGFR, GD2, mesothelin, MUC1, and PSMA. The
lack of antigen specificity and the acceptance of low levels of the
target antigen on normal tissues have led to a number of
catastrophic events. A patient with metastatic colon cancer died
after receiving an infusion of CAR T cells targeted to the HER2
(ERBB2) antigen (48). Another
patient died from encephalitis when infused with a high-affinity
anti-GD2 CAR for neuroblastoma (49). CAR targets used for the treatment
of solid malignancies include:
Prostate-specific membrane antigen (PSMA).
PSMA is a Glutamate carboxypeptidase 2, a type II membrane protein
highly expressed on most prostate-cancer cells and tumor-associated
neovasculature of numerous solid tumors (50).
Mesothelin (MSLN). MSLN is a protein present
in malignant pleural mesothelioma, ovarian, pancreatic, and lung
cancers. Also, mesothelin is expressed on non-transformed
peritoneal, pleural and pericardial mesothelial cells (51).
Fibroblast activation protein-α (FAP). FAP is
a type-II transmembrane serine protease expressed almost
exclusively in pathological conditions including fibrosis,
arthritis, and cancer, where explicitly expressed on
cancer-associated stromal cells present in epithelial cancers
(52).
Epidermal growth factor receptor (EGFR). EGFR
is a transmembrane protein that serves as receptors for numerous
epidermal growth factor families of extracellular protein ligands.
Different human tumors, including non-small cell lung cancer
(NSCLC), breast, head, neck, gastric, colorectal, esophageal,
prostate, bladder, renal, pancreatic, and ovarian cancers, express
EGFR. EGFR signaling causes increased proliferation, decreased
apoptosis, and enhanced tumor cell motility and
neo-angiogenesis.
Carcinoembryonic antigen (CEA). CLA are
glycosylphosphatidylinositol (GPI) cell-surface-anchored
glycoproteins, characterized as members of the CD66 cluster of
differentiation. These proteins serve as functional colon carcinoma
L-selectin and E-selectin ligands (53). Currently, CEA-targeted CAR T cell
is used to treat patients with liver metastases that are positive
for CEA expression.
The human epidermal (HER2). HER2 is a
receptor tyrosine-protein kinase member of the human epidermal
growth factor receptor (HER/EGFR/ERBB) family. HER2 is expressed on
epithelial cells in the gastrointestinal, respiratory,
reproductive, and urinary tract, and it is amplification or
over-expression on breast cancer denote aggressive types of breast
cancer (54).
CAR T trafficking in solid tumors
In hematological malignancies, infused CAR T Cells
and tumor cells co-circulate in the blood and have a higher
propensity to migrate to similar areas such as bone marrow and
lymph nodes. On the other hand, CAR T cells in solid tumors
encounter a number of hurdles, including difficulty migrating to
and adequately penetrating the tumor, binding to receptors, and
completing their cytotoxic function. Chemokines, such as CXCL12 and
CXCL5, secreted by the tumor inhibit T-cell migration into the
tumor. In some instances, the chemokine receptors on T cells do not
adequately match the tumors' chemokine signature, resulting in
little migration to the tumor site. For example, it has been shown
that T cells genetically modified to express CXCR2 migrate towards
tumor cells expressing CXCL1. Chemokines secreted by the tumor's
stroma, the chemokine repertoire in the tumor location, and the
local ‘normal’ cytokine milieu also affect the CAR T cell movement
and migration. Furthermore, solid tumor stroma sends chemokines
signals that mismatch the chemokine-receptors on T cells' surface,
resulting in dysregulation and cancer progression (55).
T cell inhibitory signals in solid
tumors
Endogenous suppressive signal and their upregulation
reduce CAR T cells' therapeutic ability. Intrinsic inhibitory T
cells and upregulation inhibitory receptors CTLA-4/PD-1 may cause T
cell exhaustion and prevent T cell persistence by interacting with
ligands overexpressed on tumor cells.
Physical barriers in the solid tumor
microenvironment
Physical barriers generated by excessive
tumor-stromal density favors tumor progression and aggressiveness.
The physical barriers that affect CAR T cell function in solid
tumors include:
Hypoxia. Abnormal vascularization and rapidly
growing tumor cells limit the amount of oxygen (hypoxia) in the
tumor. Hypoxia impacts CAR-T cells' attributes by decreasing CAR-T
cells' expansion ability, blocking their differentiation into
effector memory cells, and enriching the cultures with T cells with
a central memory cell phenotype (56). Also, abnormal hypoxia-derived tumor
vessels affect T cell adhesion and extravasation towards the solid
tumor. Additionally, abnormalities of blood vessels, known as high
endothelial venules (HEV), compromise immune cell trafficking to
the tumor (47,57).
Extracellular matrix. Peritumoral
extracellular matrix (ECM) collagen fibers limit T cell access to
tumors, and it is known that tumors with high collagen density
present lower levels of infiltrating T cells.
Tumor vasculature. The tumor's core exhibits
immature vessel formation, leading to low permeability (46).
Fibroblasts. Other non-immune cells that
enhance tumorigenesis are stromal cells, such as cancer-associated
fibroblast (CAF) (47). The cells
are involved in the secretion of pro-tumorigenic molecules
contributing to tumor vasculature and anti-inflammatory reaction to
immune cells (47,57). In addition, fibroblast
differentiation can express activation makers that support matrix
degradation and remodeling (46).
Tumor microenvironment
The immunosuppressive nature of the tumor
microenvironment plays an essential role in tumor survival,
metastatic progression, and influences immunotherapies' outcomes
(57). Numerous suppressive immune
cells and molecular factors in the tumor microenvironment can block
CAR T cell's antitumor immune function. These immune cells include
immune suppressor cells, such as Tregs, myeloid-derived suppressor
cells, and tumor-associated macrophages. In contrast, molecular
factors include cytokines and soluble factors associated with
immunosuppression, such as TGF-β and IL-10, promoting T cell anergy
by indirect contact. Another factor known to condition the
antitumor effect of T cells in solid tumors is soluble factors such
as transforming growth factor B (TGF-β) and vascular endothelial
growth factors (VEGF) secreted mainly by stromal and tumor cells
(47). TGF-β can also be secreted
by regulatory T cells (Tregs), platelets, macrophages, and
fibroblasts to suppress T cell proliferation and effect function
(25). Evidence suggests that it
promotes Treg maturation and modulate CD8+ effector cell
function (26,58).
CAR T solid tumors trials
The accomplishments surrounding CAR T-cell-based
therapies hinge on their success in hematological diseases;
however, for the reasons mentioned above, much work is needed to
sure their success in solid tumors (59).
The CAR T cells' persistence in the stromal
micro-environment was the main setback in two clinical trials
targeting neuroblastoma and ovarian tumors. Neuroblastoma CARs were
generated with the use of EBV-specific cytotoxic T lymphocytes
(EBV-CTLs) and activated T cells (ATCs) targeting GD2(45). Although both engineered T cells
were found to circulate the system at higher concentrations
demonstrating improved functionality for CAR-T cell therapy
purposes, only three out of eleven patients with active disease
completed remission (45).
Furthermore, few clinical trials used CAR T-EGFR to
treat biliary tract cancers (BTC), cholangiocarcinomas, and
gallbladder carcinomas that express EGFR. The results reported that
out of 19 patients, one achieved complete remission and ten stable
diseases, concluding that CAR T-EGFR treatment was a safe and
promising strategy for EGFR-positive advanced biliary tract cancers
(60) Also, trials targeted
carcinoembryonic antigens (CEA), utilizing CAR T-CEA. CEA is
overexpressed in lung, gastrointestinal, and breast cancers and is
used as a tumor marker for cancer patients' diagnosis and prognosis
(61) In this Phase I trial, a
total of 8 patients with CEA-positive liver metastases were
included, of which 4 have more than ten metastatic foci in the
liver. Patients received treatment with anti-CEA CAR T cells via
hepatic arterial infusions. In addition to CAR T cell infusion,
half of the patients received IL-2 cytokine. The trial results
indicated that patients experienced no fatal side effects or
adverse unpredictable outcomes and that patients tolerated very
well the anti-CEA CAR-T therapy with or without IL2 administration
(62).
8. Animal models
Preclinical animal testing requires using a relevant
animal model that truly represents the human disease and can elicit
a biological response similar to what would happen in humans.
However, the preclinical model used in testing the safety and
efficacy of CAR T cell therapy fell short to adhere to the standard
due to variability in cross-species reactivity to non-human target
antigens and, therefore, difficult to identify potential adverse
events in humans and often offer a false sense of safety.
Rodent models
Before testing new therapeutic approaches in human
patients for clinical trial purposes, safety and efficacy are
usually assessed pre-clinically in animal models such as mice,
zebrafish, among others. Rodent models have been critical for
understanding pathways, identifying tumor-target antigens, and
understanding the tumor physiology and the microenvironment
(63). However, despite rodent
models' role in preclinical trials, which led to numerous
breakthroughs in modern medicine, it has a number of limitations.
For example, among drugs that showed strong efficacy and inhibited
tumor growth in mice, only 11% are approved for human use by FDA.
Furthermore, side effects seen in humans were not observed in mice
(64).
Also, rodent models do not appropriately portray the
complex microenvironment relationship between the immune cells and
tumor cells (65). These animals
do not develop spontaneous tumors. Their living condition, which is
pathogen-free, impacts their immune system flora (64). Thus, rodents do not produce
‘normal’ immune cell lines found in humans or animals exposed to
natural environments, resulting in the same immune milieu between
them and identical gene sequence composition. Therefore, studies
using animals with none functioning immune systems have limited
translational impact. In the case of toxicities involving immune
system signaling, brain swelling after CAR T cells therapy is not
detectable in studies using immunodeficient mice. All these
mentioned factors make rodent models less trustworthy and raise
questions regarding whether their contribution is sufficient to use
them as preclinical models.
Non-human primate model
Of all the animal models mentioned, the one that
more accurately resembles the human genetic composition are the
non-human primates. Although similar, these models are not adequate
for comparative studies since they experience low spontaneous
cancer rates (64), high
maintenance, and ethical regulation surrounding these models.
Taraseviciute et al studied how neurotoxicities can affect
the non-primate model, rhesus macaque, after transferring
autologous CD20-specific CAR T cells. The group demonstrated that
CD20 CAR and non-CAR T cells infiltrate and accumulate in the
cerebrospinal fluid (CSF) and brain parenchyma, causing high levels
of proinflammatory cytokines in the CSF and pan-encephalitis
(66).
Canine model
Unlike the rodent models, dogs develop spontaneous
tumors that resemble human disease in morphology, molecular
aspects, and genetic behavior (67). Also, dogs have intact immune
systems with considerable similarities to humans' immune milieu
because dogs and humans cohabitate in the same household,
therefore, sharing the same environmental risk factors (64). Furthermore, the genetic diversity
displayed by different dog breeds provides an ideal tool that
enriches the preclinical studies by providing similar challenges
seen in humans' studies from different ethnic groups. Also, cancer
is the number one cause of death in dogs (63). All hematological malignancies and
solid tumors in dogs are similar to human diseases. These included
mammary tumors (breast), osteosarcoma, prostate, bladder cancer,
and leukemia.
Canine mammary tumors
Studies revealed that spontaneous invasive mammary
carcinomas are closely similar in pathology, epidemiology, and
immunohistochemical characterization with human breast cancers
(68). Commonly overexpressed
estrogen and progesterone hormone receptors, the conglomeration of
similar tumor-infiltrating lymphocyte ratios, and homologous cancer
risk factors such as obesity and age are similar between humans and
canines' tumors (64,69) Clinical outcomes after tumor
progression are closely related to these two species. Furthermore,
molecular markers such as the nuclear protein Ki-67, the p53 tumor
suppressor gene, and the BCRA genes provide valuable information
regarding both species' prognosis status (70). Clinical trials using canine CAR T
therapy in canine mammary tumors are not initiated yet. However,
CAR T cell therapies' benefits in humans breast cancer have been
explored over the last years. The following trials are ongoing and
centered on improving the safe dose and uncovering the different
effects (good and bad). Phase I trials are ongoing targeting
HER2+ breast cancer (NCT04650451 and NCT03740256) in
patients with advanced-stage III (NCT04650451) or metastatic (stage
IV) (NCT04650451 and NCT03740256) cancer with no other treatment
option available using BPX-603 and HER2 specific CAR T cells,
respectively. City of Hope Medical Center conducted a trial using
HER2 specific CAR T cells targeting HER2+ breast cancer
cells (NCT03696030) in patients with brain or leptomeningeal
metastases. Two trials (NCT02414269 and NCT02792114) at the
Memorial Sloan Kettering Cancer Center are ongoing targeting
Mesothelin in patients with metastatic (stage IV) breast cancer
that spread to the pleura (iCasp9M28z CAR T-cells-Phase I/II) and
HER2-cells (Mesothelin CAR T cells-Phase I), respectively. Tmunity
Therapeutics using CART-TnMUC1 (NCT04025216) in patients with
triple-negative and ER-low, HER2-breast cancer with TnMUC1 positive
cells. Minerva Biotechnology Corporation conducts a trial targeting
MUC1* (NCT04020575) utilizing huMNC2-CAR44 CAR T cells in patients
with metastatic (stage IV) breast cancer. Fred Hutchinson Cancer
Research Center conducts a phase I trial on triple-negative and
ER-low breast cancer (NCT02706392), targeting ROR1 positive cells.
Lastly, patients that received a minimum of two therapies for
advanced cancer expressing GD2 antigen are carried on by Baylor's
College of Medicine (NCT03635632) using a C7R-GD2 CAR T cell.
Canine osteosarcoma
Canine develops osteosarcoma (OSA) at a much higher
rate than humans (71), serving as
a remarkable model for developing treatments and overcoming the
numerous challenges in solid tumor therapies. There are a number of
similarities between the canine and humans concerning this disease.
The tumor location, the pattern of metastasis, genetic drivers of
the disease, and response to therapy are similar in both species.
Canine OSA is a spontaneous, naturally occurring disease as in
humans. Canine OSA has aggressive biology and an increased rate of
metastasis, and the animal often dies within six months, and almost
96% of dogs with OSA perish from the disease. Canine trials or
in-vitro experiments related to osteosarcoma are scarce in
the literature. Mata et al (65) developed a CAR-T cell targeting HER2
overexpressing tumor cells in-vitro. Canine and
human-derived transmembrane and signaling domains were tested on
tumor cells, demonstrating little to no difference in tumor
suppression (65) On the other
hand, Baylor College of Medicine is conducting a Phase I clinical
trial (NCT03635632) in human patients with relapsed or refractory
osteosarcoma with increased expression of GD2 antigen utilizing
C7R-GD2 CAR-T cells. The National Cancer Institute (NCI) has
completed a Phase I clinical targeting GD2 positive solid tumors
with anti-GD2 CART cells in children and young adults that suffer
osteosarcoma (NCT02107963), no final data has been posted yet.
Canine prostate cancer
Canines are a few animal models that develop
spontaneous prostate cancer as humans (72,73).
Both dogs and humans share similar risk factors, including advanced
age, low mortality rates, clinical outcomes, and prostate gland
functionality, suggesting that these animals may be ideal models
for future clinical trials (72-74).
Unfortunately, a lack of prostate cancer screening in canine
augments the malignancy's mortality rate and aggressiveness, thus
not allowing proper treatment strategies (74,75).
On the other hand, human screening methods have strengthened over
the last few years, enabling rapid diagnosis (76). ACT therapy for prostate cancer has
been developed mainly in humans. CAR-T cells against TCRγ chain
alternative reading frame protein (TARP), prostate stem cell
antigen (PSCA), and prostate-specific membrane antigen (PSMA) were
developed and used to suppress tumor growth in vitro
(77-80).
Phase I clinical trials are currently conducted in patients with
castrate-resistance prostate cancer targeting PSMA with doses of
CART-PSMA-TGFβRDN, LIGHT-PSMA CART P-PSMA-101 CART cells
(NCT03089203, NCT04053062, and NCT04249947). The City of Hope
Medical Center conducted another trial against metastatic
castration-resistance prostate cancer, targeting the PSCA antigen's
overexpression with anti-PSCA-4-1BB/TCRζ-CD19 CART cells
(NCT03873805). Phase I/II clinical trial (NCT02744287), sponsored
by Bellicium Pharmaceuticals, PSCA-CART (BPX-601), is currently
used to treat patients with previously treated advanced tumors,
including metastatic prostate and metastatic castrate-resistance
prostate cancer. Finally, the First Affiliated Hospital of Chengdu
Medical College targeted EpCAM positive prostate cancer with an
EpCAM-specific CART cell (NCT03013712), a second-generation CAR
(CD28/CD3ζ) targeting PSMA.
Canine bladder cancer
Invasive Urinary bladder cancer (InvUC), Invasive
transitional cell carcinoma (TCC), and invasive urothelial
carcinoma (UC) are three different subtypes of bladder cancer
spontaneously developed in canines that resemble ‘humans’
malignancies (79,81,82).
Similarities in clinical outcomes, histological features, and
progression sites make canines straightforward compared to humans
(79). Canine trials or CAR-T
generations are not seen in literature, but human clinical trials
are currently under investigation. A Phase I/II clinical trial,
conducted by Shenzen Geno-Immune Medical Institute (NCT03185468),
is currently evaluating the safety and efficacy of a 4SCART-PSMA
CART cell against PSMA-expressing bladder cancer.
Canine leukemias
As mentioned above, preclinical trials driven with
canine models could represent an enormous step in adoptive T cell
therapy development. Unfortunately, preclinical trials using canine
models are scarce in the scientific literature. The few clinical
trials available are primarily performed in B cell lymphomas.
Panjwani conducted a trial in patients with B cell lymphomas,
targeting the CD20 antigen. The study concluded the need for stable
CAR T cell expression and that further studies must be performed
(83). Nonetheless, the second
trial showed stable CAR T transduction using lentiviral vectors
(84). Their CD20-BB-ζ CAR T cell,
alongside cytokines IL7 and IL5, proved to be durable and
antigen-specific against DLBCL. Non-Hodgkin's Lymphoma (NHL) is the
most common cancer in dogs, and the most common sub-type is Diffuse
Large B-Cell Lymphoma (DLBCL). While combinations of chemotherapy
agents lead to clinical remission in ~75% of dogs, most dogs
relapse within six to nine months of standard treatment, a
statistic that has remained unchanged for the past 30 years. An
urgent need exists for new therapies for canine lymphoma.
Furthermore, evaluating these new therapies in pet dogs with
naturally occurring cancer may also provide vital information to
advance novel therapies for individuals.
9. Conclusions
The remarkable progress that adoptive immunotherapy
has experienced these past years, especially in blood-related
cancers, provides optimism for CARs therapy. Trials of CAR T in
leukemia and lymphomas had shown positive outcomes, with some cases
experiencing mild side effects. Notwithstanding, trials conducted
in solid tumors represent a daunting task to achieve. Tumor
microenvironment, CARs tracking and duration, and the various
toxicities experienced by a number of patients represent
significant setbacks that need addressing. The animal model that
faithfully resembles humans is another milestone in this endeavor.
Up to date, all preclinical studies of CAR T safety and efficacy
are conducted in mice, including syngeneic, transgenic, and
xenograft, and humanized mouse models to represent humans' immune
responses and diseases to test the safety and efficacy of CART
therapy. However, these models fell short in representing the
disease and its adverse effect. The dog's importance is recently
recognized as a preclinical model for cancer CAR T therapy because
of its human physiology, immune responses, and disease
similarities. The development of reagents and the use of the dogs
in clinical trials will help advance the CAR T therapy field for
both species.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SIM conceived and outlined the review. XERC, WL and
SIM wrote the manuscript and edited it. Data authentication is not
applicable. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang Z, Guo Y and Han W: Current status
and perspectives of chimeric antigen receptor modified T cells for
cancer treatment. Protein Cell. 8:896–925. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Pilones KA, Aryankalayil J and Demaria S:
Invariant NKT cells as novel targets for immunotherapy in solid
tumors. Clin Dev Immunol. 2012(720803)2012.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Gentles AJ, Newman AM, Liu CL, Bratman SV,
Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al: The
prognostic landscape of genes and infiltrating immune cells across
human cancers. Nat Med. 21:938–945. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Zhao Y, Niu C and Cui J: Gamma-delta (γδ)
T cells: Friend or foe in cancer development? J Transl Med.
16(3)2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Chmielewski M and Abken H: TRUCKs: The
fourth generation of CARs. Expert Opin Biol Ther. 15:1145–1154.
2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Izsvák Z, Hackett PB, Cooper LJN and Ivics
Z: Translating sleeping beauty transposition into cellular
therapies: Victories and challenges. Bioessays. 32:756–767.
2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zheng PP, Kros JM and Li J: Approved CAR T
cell therapies: Ice bucket challenges on glaring safety risks and
long-term impacts. Drug Discov Today. 23:1175–1182. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Maude SL, Laetsch TW, Buechner J, Rives S,
Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers
GD, et al: Tisagenlecleucel in Children and Young Adults with
B-Cell Lymphoblastic Leukemia. N Engl J Med. 378:439–448.
2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Neelapu SS, Locke FL, Bartlett NL, Lekakis
LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T,
Lin Y, et al: Axicabtagene ciloleucel CAR T-cell therapy in
refractory large B-cell lymphoma. N Engl J Med. 377:2531–2544.
2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Schuster SJ, Bishop MR, Tam CS, Waller EK,
Borchmann P, McGuirk JP, Jäger U, Jaglowski S, Andreadis C, Westin
JR, et al: Tisagenlecleucel in adult relapsed or refractory diffuse
large B-cell lymphoma. N Engl J Med. 380:45–56. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mullard A: FDA approves fourth CAR-T cell
therapy. Nat Rev Drug Discov. 20(166)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tokarew N, Ogonek J, Endres S, von
Bergwelt-Baildon M and Kobold S: Teaching an old dog new tricks:
Next-generation CAR T cells. Br J Cancer. 120:26–37.
2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Charrot S and Hallam S: CAR-T Cells:
Future perspectives. Hemasphere. 3(e188)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kuwana Y, Asakura Y, Utsunomiya N,
Nakanishi M, Arata Y, Itoh S, Nagase F and Kurosawa Y: Expression
of chimeric receptor composed of immunoglobulin-derived V resions
and T-cell receptor-derived C regions. Biochem Biophys Res Commun.
149:960–968. 1987.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ramos CA and Dotti G: Chimeric antigen
receptor (CAR)-engineered lymphocytes for cancer therapy. Expert
Opin Biol Ther. 11:855–873. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dotti G, Gottschalk S, Savoldo B and
Brenner MK: Design and development of therapies using chimeric
antigen receptor-expressing T cells. Immunol Rev. 257:107–126.
2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sharpe AH and Abbas AK: T-cell
costimulation-biology, therapeutic potential, and challenges. N
Engl J Med. 355:973–975. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zhang C, Liu J, Zhong JF and Zhang X:
Engineering CAR-T cells. Biomark Res. 5(22)2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Fesnak AD, June CH and Levine BL:
Engineered T cells: The promise and challenges of cancer
immunotherapy. Nat Rev Cancer. 16:566–581. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chmielewski M, Hombach AA and Abken H: Of
CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered
with an inducible cytokine to modulate the tumor stroma. Immunol
Rev. 257:83–90. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pegram HJ, Lee JC, Hayman EG, Imperato GH,
Tedder TF, Sadelain M and Brentjens RJ: Tumor-targeted T cells
modified to secrete IL-12 eradicate systemic tumors without need
for prior conditioning. Blood. 119:4133–4141. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Hunter BD and Jacobson CA: CAR T-Cell
associated neurotoxicity: Mechanisms, clinicopathologic correlates,
and future directions. J Natl Cancer Inst. 111:646–654.
2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sun S, Hao H, Yang G, Zhang Y and Fu Y:
Immunotherapy with CAR-Modified T Cells: Toxicities and overcoming
strategies. J Immunol Res. 2018(2386187)2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yáñez L, Sánchez-Escamilla M and Perales
MA: CAR T cell toxicity: Current management and future directions.
Hemasphere. 3(e186)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Fucà G, Reppel L, Landoni E, Savoldo B and
Dotti G: Enhancing chimeric antigen receptor T-cell efficacy in
solid tumors. Clin Cancer Res. 26:2444–2451. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ma S, Li X, Wang X, Cheng L, Li Z, Zhang
C, Ye Z and Qian Q: Current progress in CAR-T cell therapy for
solid tumors. Int J Biol Sci. 15:2548–2560. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Davila ML and Brentjens RJ: CD19-Targeted
CAR T cells as novel cancer immunotherapy for relapsed or
refractory B-cell acute lymphoblastic leukemia. Clin Adv Hematol
Oncol. 14:802–808. 2016.PubMed/NCBI
|
|
28
|
Dotti G: The other face of chimeric
antigen receptors. Mol Ther. 22:899–900. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Hombach A, Hombach AA and Abken H:
Adoptive immunotherapy with genetically engineered T cells:
Modification of the IgG1 Fc ‘spacer’ domain in the extracellular
moiety of chimeric antigen receptors avoids ‘off-target’ activation
and unintended initiation of an innate immune response. Gene Ther.
17:1206–1213. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lee YG, Marks I, Srinivasarao M, Kanduluru
AK, Mahalingam SM, Liu X, Chu H and Low PS: Use of a single CAR T
cell and several bispecific adapters facilitates eradication of
multiple antigenically different solid tumors. Cancer Res.
79:387–396. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Darowski D, Kobold S, Jost C and Klein C:
Combining the best of two worlds: Highly flexible chimeric antigen
receptor adaptor molecules (CAR-adaptors) for the recruitment of
chimeric antigen receptor T cells. mAbs. 11:621–631.
2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Arndt C, Fasslrinner F, Loureiro LR,
Koristka S, Feldmann A and Bachmann M: Adaptor CAR platforms-next
generation of T Cell-based cancer immunotherapy. Cancers (Basel).
12(1302)2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D,
Han X, Liu Y, Zhang W, Wang C, et al: Bispecific CAR-T cells
targeting both CD19 and CD22 for therapy of adults with relapsed or
refractory B cell acute lymphoblastic leukemia. J Hematol Oncol.
13(30)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chen N, Morello A, Tano Z and Adusumilli
PS: CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one
approach for solid tumor immunotherapy. Oncoimmunology.
6(e1273302)2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kloss CC, Lee J, Zhang A, Chen F,
Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y and June CH:
Dominant-Negative TGF-β receptor enhances PSMA-targeted human CAR T
cell proliferation and augments prostate cancer eradication. Mol
Ther. 26:1855–1866. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Makita S, Yoshimura K and Tobinai K:
Clinical development of anti-CD19 chimeric antigen receptor T-cell
therapy for B-cell non-Hodgkin lymphoma. Cancer Sci. 108:1109–1118.
2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Maude SL, Frey N, Shaw PA, Aplenc R,
Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et
al: Chimeric antigen receptor T cells for sustained remissions in
leukemia. N Engl J Med. 371:1507–1517. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Reagan PM and Friedberg JW: Axicabtagene
ciloleucel and brexucabtagene autoleucel in relapsed and refractory
diffuse large B-cell and mantle cell lymphomas. Future Oncol.
17:1269–1283. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
FDA approves second CAR T-cell therapy.
Cancer Discov. 8:5–6. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Mullard A: FDA approves first
BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discov.
20(332)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Novak AJ, Darce JR, Arendt BK, Harder B,
Henderson K, Kindsvogel W, Gross JA, Greipp PR and Jelinek DF:
Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A
mechanism for growth and survival. Blood. 103:689–694.
2004.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ribas A and Wolchok JD: Cancer
immunotherapy using checkpoint blockade. Science. 359:1350–1355.
2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Guedan S, Ruella M and June CH: Emerging
cellular therapies for cancer. Annu Rev Immunol. 37:145–171.
2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Brown CE, Alizadeh D, Starr R, Weng L,
Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J,
Simpson J, et al: Regression of glioblastoma after chimeric antigen
receptor T-cell therapy. N Engl J Med. 375:2561–2569.
2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Louis CU, Savoldo B, Dotti G, Pule M, Yvon
E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al: Antitumor
activity and long-term fate of chimeric antigen receptor-positive T
cells in patients with neuroblastoma. Blood. 118:6050–6056.
2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Vignali D and Kallikourdis M: Improving
homing in T cell therapy. Cytokine Growth Factor Rev. 36:107–116.
2017.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Martinez M and Moon EK: CAR T cells for
solid tumors: New strategies for finding, infiltrating, and
surviving in the tumor microenvironment. Front Immunol.
10(128)2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Morgan RA, Yang JC, Kitano M, Dudley ME,
Laurencot CM and Rosenberg SA: Case report of a serious adverse
event following the administration of T cells transduced with a
chimeric antigen receptor recognizing ERBB2. Mol Ther. 18:843–851.
2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Richman SA, Nunez-Cruz S, Moghimi B, Li
LZ, Gershenson ZT, Mourelatos Z, Barrett DM, Grupp SA and Milone
MC: High-Affinity GD2-Specific CAR T cells induce fatal
encephalitis in a preclinical neuroblastoma model. Cancer Immunol
Res. 6:36–46. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Chang SS: Overview of prostate-specific
membrane antigen. Rev Urol. 6 (Suppl 10):S13–S18. 2004.PubMed/NCBI
|
|
51
|
Hassan R and Ho M: Mesothelin targeted
cancer immunotherapy. Eur J Cancer. 44:46–53. 2008.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Fitzgerald AA and Weiner LM: The role of
fibroblast activation protein in health and malignancy. Cancer
Metastasis Rev. 39:783–803. 2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Hammarström S: The carcinoembryonic
antigen (CEA) family: Structures, suggested functions and
expression in normal and malignant tissues. Semin Cancer Biol.
9:67–81. 1999.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Slamon DJ, Godolphin W, Jones LA, Holt JA,
Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al:
Studies of the HER-2/neu proto-oncogene in human breast and ovarian
cancer. Science. 244:707–712. 1989.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Berahovich R, Liu X, Zhou H, Tsadik E, Xu
S, Golubovskaya V and Wu L: Hypoxia selectively impairs CAR-T cells
in vitro. Cancers (Basel). 11(602)2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Oliver AJ, Lau PKH, Unsworth AS, Loi S,
Darcy PK, Kershaw MH and Slaney CY: Tissue-Dependent tumor
microenvironments and their impact on immunotherapy responses.
Front Immunol. 9(70)2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Newick K, O'Brien S, Moon E and Albelda
SM: CAR T cell therapy for solid tumors. Annu Rev Med. 68:139–152.
2017.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Kershaw MH, Westwood JA, Parker LL, Wang
G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S,
Rogers-Freezer L, et al: A phase I study on adoptive immunotherapy
using gene-modified T cells for ovarian cancer. Clin Cancer Res.
12(20 Pt 1):6106–6115. 2006.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Guo Y, Feng K, Liu Y, Wu Z, Dai H, Yang Q,
Wang Y, Jia H and Han W: Phase I study of chimeric antigen receptor
modified T cells in patients with EGFR-positive advanced biliary
tract cancers. Clin Cancer Res. 24:1277–1286. 2018.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Lee JH and Lee SW: The roles of
carcinoembryonic antigen in liver metastasis and therapeutic
approaches. Gastroenterol Res Pract. 2017(7521987)2017.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Katz SC, Burga RA, McCormack E, Wang LJ,
Mooring W, Point GR, Khare PD, Thorn M, Ma Q, Stainken BF, et al:
Phase I hepatic immunotherapy for metastases study of
intra-arterial chimeric antigen receptor-modified T-cell therapy
for CEA+ liver metastases. Clin Cancer Res.
21:3149–3159. 2015.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Paoloni M and Khanna C: Translation of new
cancer treatments from pet dogs to humans. Nat Rev Cancer.
8:147–156. 2008.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Park JS, Withers SS, Modiano JF, Kent MS,
Chen M, Luna JI, Culp WTN, Sparger EE, Rebhun RB, Monjazeb AM, et
al: Canine cancer immunotherapy studies: Linking mouse and human. J
Immunother Cancer. 4(97)2016.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Mata M, Vera JF, Gerken C, Rooney CM,
Miller T, Pfent C, Wang LL, Wilson-Robles HM and Gottschalk S:
Toward immunotherapy with redirected T cells in a large animal
model: Ex vivo activation, expansion, and genetic modification of
canine T cells. J Immunother. 37:407–415. 2014.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Taraseviciute A, Tkachev V, Ponce R,
Turtle CJ, Snyder JM, Liggitt HD, Myerson D, Gonzalez-Cuyar L,
Baldessari A, English C, et al: Chimeric antigen receptor T
cell-mediated neurotoxicity in nonhuman primates. Cancer Discov.
8:750–763. 2018.PubMed/NCBI View Article : Google Scholar
|
|
67
|
van Steenbeek FG, Hytönen MK, Leegwater PA
and Lohi H: The canine era: The rise of a biomedical model. Anim
Genet. 47:519–527. 2016.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Abadie J, Nguyen F, Loussouarn D, Pena L,
Gama A, Rieder N, Belousov A, Bemelmans I, Jaillardon L, Ibisch C
and Campone M: Canine invasive mammary carcinomas as models of
human breast cancer. Part 2: Immunophenotypes and prognostic
significance. Breast Cancer Res Treat. 167:459–468. 2018.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Queiroga FL, Raposo T, Carvalho MI, Prada
J and Pires I: Canine mammary tumours as a model to study human
breast cancer: Most recent findings. In Vivo. 25:455–465.
2011.PubMed/NCBI
|
|
70
|
Buishand FO, Kik M and Kirpensteijn J:
Evaluation of clinico-pathological criteria and the Ki67 index as
prognostic indicators in canine insulinoma. Vet J. 185:62–67.
2010.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Siobhan S, Dunning MD, de Brot S,
Grau-Roma L, Mongan NP and Rutland CS: Comparative review of human
and canine osteosarcoma: Morphology, epidemiology, prognosis,
treatment and genetics. Acta Vet Scand. 59(71)2017.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Leroy BE and Northrup N: Prostate cancer
in dogs: Comparative and clinical aspects. Vet J. 180:149–162.
2009.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Sun F, Báez-Díaz C and Sánchez-Margallo
FM: Canine prostate models in preclinical studies of minimally
invasive interventions: Part I, canine prostate anatomy and
prostate cancer models. Transl Androl Urol. 6:538–546.
2017.PubMed/NCBI View Article : Google Scholar
|
|
74
|
McEntee M, Isaacs W and Smith C:
Adenocarcinoma of the canine prostate: Immunohistochemical
examination for secretory antigens. Prostate. 11:163–170.
1987.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Sorenmo KU, Goldschmidt M, Shofer F,
Goldkamp C and Ferracone J: Immunohistochemical characterization of
canine prostatic carcinoma and correlation with castration status
and castration time. Vet Comp Oncol. 1:48–56. 2003.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Yu H, Pan J, Guo Z, Yang C and Mao L: CART
cell therapy for prostate cancer: Status and promise. Onco Targets
Ther. 12:391–395. 2019.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Hillerdal V, Nilsson B, Carlsson B,
Eriksson F and Essand M: T cells engineered with a T cell receptor
against the prostate antigen TARP specifically kill
HLA-A2+ prostate and breast cancer cells. Proc Natl Acad
Sci USA. 109:15877–15881. 2012.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Morgenroth A, Cartellieri M, Schmitz M,
Günes S, Weigle B, Bachmann M, Abken H, Rieber EP and Temme A:
Targeting of tumor cells expressing the prostate stem cell antigen
(PSCA) using genetically engineered T-cells. Prostate.
67:1121–1131. 2007.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Fulkerson CM, Dhawan D, Ratliff TL, Hahn
NM and Knapp DW: Naturally occurring canine invasive urinary
bladder cancer: A complementary animal model to improve the success
rate in human clinical trials of new cancer drugs. Int J Genomics.
2017(6589529)2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Ma Q, Gomes EM, Lo AS and Junghans RP:
Advanced generation anti-prostate specific membrane antigen
designer T cells for prostate cancer immunotherapy. Prostate.
74:286–296. 2014.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Knapp DW, Ramos-Vara JA, Moore GE, Dhawan
D, Bonney PL and Young KE: Urinary bladder cancer in dogs, a
naturally occurring model for cancer biology and drug development.
ILAR J. 55:100–118. 2014.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Dhawan D, Paoloni M, Shukradas S,
Choudhury DR, Craig B, Ramos-Vara JA, Hahn N, Bonney PL, Khanna C
and Knapp DW: Comparative gene expression analyses identify luminal
and basal subtypes of canine invasive urothelial carcinoma that
mimic patterns in human invasive bladder cancer. PLoS One.
10(e0136688)2015.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Panjwani MK, Smith JB, Schutsky K,
Gnanandarajah J, O'Connor CM, Powell DJ Jr and Mason NJ:
Feasibility and safety of RNA-transfected CD20-specific chimeric
antigen receptor T cells in dogs with spontaneous B cell lymphoma.
Mol Ther. 24:1602–1614. 2016.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Panjwani MK, Atherton MJ, MaloneyHuss MA,
Haran KP, Xiong A, Gupta M, Kulikovsaya I, Lacey SF and Mason NJ:
Establishing a model system for evaluating CAR T cell therapy using
dogs with spontaneous diffuse large B cell lymphoma.
OncoImmunology. 9(1676615)2019.PubMed/NCBI View Article : Google Scholar
|