Introduction
Angiomatous meningioma (AM) is a relatively rare
subtype of WHO grade I meningioma, constituting 3.24% of grade I
meningiomas and 2.1% of all meningiomas (1-2).
Angiomatous meningioma (AM) is a relatively rare variant of WHO
grade I meningioma, which features a predominance of blood vessels
over meningioma cells. The vascular channels may be small- or
medium-sized, thin-walled or thick, and most are small with
markedly hyalinized walls. AM is usually characterized by the onset
of slow progressive symptoms and the main symptoms result from
compression of the adjacent structures. Headache and epilepsy are
the initial clinical manifestations. AMs are similar to other types
of meningiomas in that they are more likely to be caused by
radiation than by sex hormone levels in women. The current
treatment for this disease is mainly complete surgical resection
supplemented by radiotherapy, and the prognosis is good. We
recently encountered a relatively rare case of AM in a 45-year-old
woman. In our case, we not only observed the typical AM
histological pattern but also a large number of cells with bizarre,
large, deep staining and unevenly distributed nuclei. Our report
may be the first to describe the presence of bizarre nuclei in
AM.
Case reports
A 45-year-old female patient presented to the
hospital because of intermittent headache for more than one year.
The patient had pain and discomfort on the left side of the head,
lasting from a few minutes to 1 h, accompanied by nausea and
vomiting in severe cases, which were relieved after rest. There was
no dizziness, blurred vision, body convulsions or sensory
disorders, language disability, or abnormality during physical
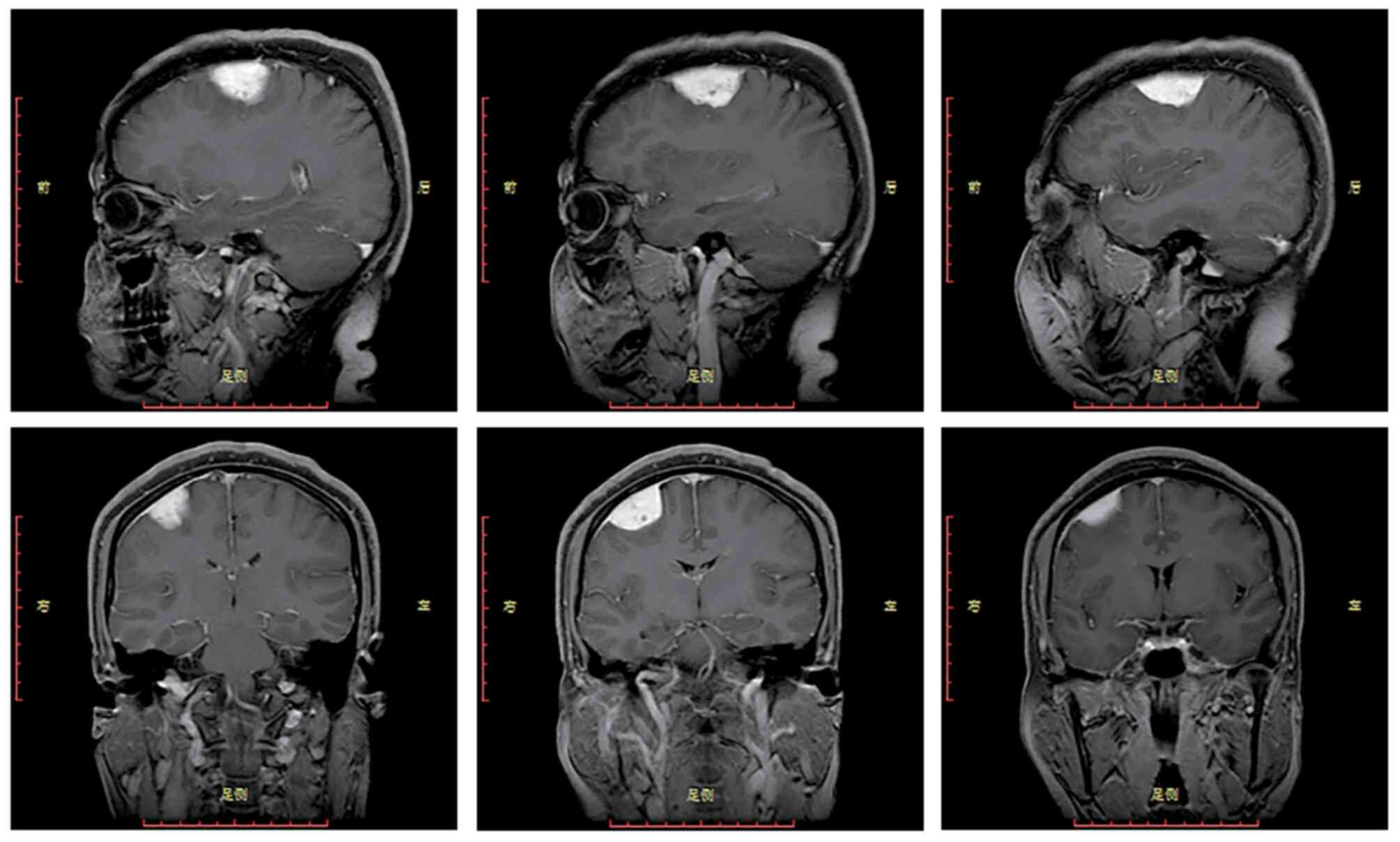
examination upon admission. Magnetic resonance imaging (MRI) of the
brain detected a soft tissue mass attached to the meninges in the
right frontal lobe (Fig. 1), which
was subsequently resected. The CT examination of this patient was
performed at another hospital, and the patient had already been
discharged when the results of the pathological examination
appeared, so I did not review these CT images at the time of the
patient's diagnosis or treatment. The tumour was excised and
submitted for histological examination.
At the macroscopic level, the formalin-fixed
surgical specimen of the tumour tissue and dura mater tissue was
2.0x2.0x0.8 cm in size. The tissues were grey-white in colour and
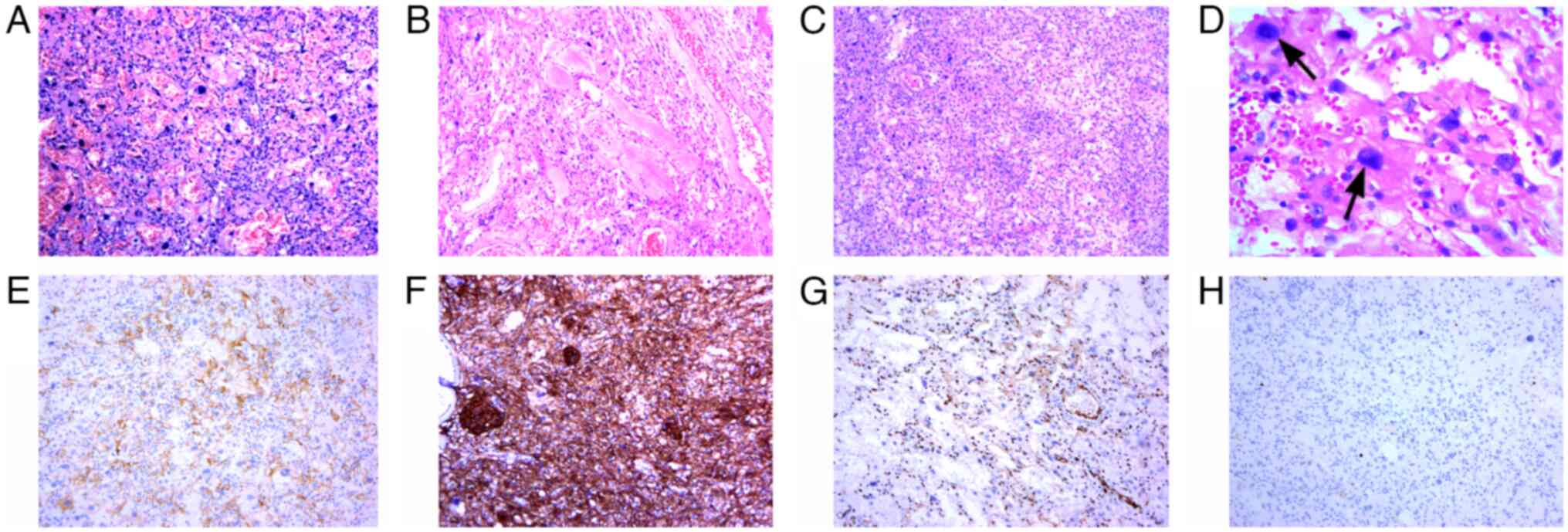
smooth. Microscopically, dense small vessels (Fig. 2A) and thick-walled large vessels
(Fig. 2B) were interwoven to form
a vascular network, most of which were hyalinized, and some tumour
cells could be seen within the vascular network. The whirlpool-like
structure of meningioma was faintly seen in the focal area
(Fig. 2C). In addition to the
translucent blood vessels and meningeal cells seen above, a large
number of cells with bizarre, large, deeply staining and unevenly
distributed nuclei were also seen (Fig. 2D) (10% neutral formalin was fixed
for 24 h and H&E staining at room temperature for 10 min, light
microscope, slicing thickness of 4 µm). No mitotic figures were
observed in any cells.
Immunohistochemically, the meningeal epithelial
cells were strongly positive for Vimentin and SSTR2A (Fig. 2E), and EMA (Fig. 2F) was focally positive. PR was
partially positive. CD34 and ERG (Fig.
2G) showed positive staining on vascular endothelial cells and
negative staining on tumour cells. GFAP, Olig-2 and HMB45 were
negative. The MIB-1(Ki-67) (Fig.
2H) labelling index was less than 3% (SSTR2A, EMA, ERG and
Ki-67 staining at 37˚C for 5 min, light microscope, slicing
thickness of 4 µm). The cells with bizarre nuclei had the same
immunophenotype as meningeal epithelial cells. This was diagnosed
as angiomatous meningioma with bizarre nuclei, WHO grade I.
Discussion
Angiomatous meningioma (AM) is a relatively rare
subtype of WHO grade I meningioma, constituting 3.24% of grade I
meningiomas and 2.1% of all meningiomas (1). Hasselblatt et al defined AM as
any meningioma whose vascular component exceeds 50% of the total
tumour area, and AM is divided into two histological subtypes: the
macrovascular subtype (diameter of >50% of all vessels larger
than 30 µm) and the microvascular subtype (diameter of >50% of
all vessels smaller than 30 µm) (2). In our case, we not only observed the
typical AM histological pattern but also a large number of cells
with bizarre, large, deeply staining and unevenly distributed
nuclei. These cells with bizarre nuclei showed a similar pattern of
immunoreactivity as meningeal epithelial cells, including SSTR2A
(somatostatin receptor 2A) positivity, which is a prominent
immunomarker of meningioma (3).
Although the presence of a large number of cells with bizarre
nuclei in this case increases tumour cell atypia, the cells did not
differ with regard to proliferative activity and mitotic imaging.
Therefore, according to the published revised WHO 2016 guidelines
(4), our case did not meet grade
II meningioma and even grade III meningioma criteria. Therefore, we
ultimately diagnosed the patients as having ‘AM with bizarre
nuclei, WHO grade I’ by exclusion of the other types.
AM with bizarre nuclei has been very rarely
reported. We performed a systematic search of Medline and PubMed
and found few reported cases of AM and even fewer reports of
angiomatous meningioma with bizarre nuclei. In 2004, Hasselblatt
et al systematically analysed the clinicopathological
characteristics of 38 consecutive AM patients (2). In 2013, Liu et al (5) retrospectively studied the clinical
presentation, neuroimaging results, and treatment follow-up of 27
AM patients, and in 2016, Ben Nsir et al conducted the
largest multicentre long-term follow-up study of 58 AM patients
(6). AM with bizarre nuclei is not
mentioned in the above literature. Therefore, our report may be the
first to describe the presence of bizarre nuclei in AM. The
presence of cells with bizarre nuclei in AM does not bear clinical
consequences; that is, they are not a feature associated with grade
II or grade III meningiomas. This manifestation of nuclear atypia
and pleomorphism may be due to ‘degenerative changes’ in
preexisting, long-established vascular lesions, similar to those
seen in degenerative schwannomas and symplastic haemangioma, rather
than being considered an indicator of malignancy. Differential
diagnoses of AM include the following: 1) vasogenic tumour,
especially angiosarcoma; 2) hemangioblastoma; and 3) others, such
as solitary fibrous tumours (SFTs) and malignant melanoma. These
tumours can be well identified by immunophenotype.
The most common aberration in grade I meningiomas is
monosomy of chromosome 22, with resultant loss of the
neurofibromatosis 2 (NF2) gene on chromosome 22q (7). This aberration is frequently the only
copy number change in WHO grade I meningiomas (8). However, Abedalthagafi et al
demonstrated that AM is distinct from other meningiomas, bearing
numerous chromosomal polysomies and lacking mutations
characteristic of other meningioma subtypes. In addition,
chromosomal alterations usually involve chromosome 5(9).
There are no established screening guidelines for
meningioma. Gross total resection is still the treatment of choice,
including dural attachment and infiltrated bone (10). The patient was followed up for more
than 2 years, and no recurrence was found.
In conclusion, to the best of our knowledge, the
present report is the first to describe the presence of bizarre
nuclei in AM. This manifestation of nuclear atypia and pleomorphism
was related to degenerative changes and a long clinical history.
Nevertheless, knowledge of such histological changes certainly may
aid in the diagnosis of AM.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YS and WY participated in the study design. YS wrote
the manuscript. XL provided all clinical data of the patient and
performed the histologically stained of the specimens. YS and WY
confirm the authenticity of all the raw data. WY and XL revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mehdorn HM: Intracranial meningiomas: A
30-year experience and literature review. Adv Tech Stand Neurosurg.
139–184. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hasselblatt M, Nolte KW and Paulus W:
Angiomatous meningioma: A clinicopathologic study of 38 cases. Am J
Surg Pathol. 28:390–393. 2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Menke JR, Raleigh DR, Gown AM, Thomas S,
Perry A and Tihan T: Somatostatin receptor 2a is a more sensitive
diagnostic marker of meningioma than epithelial membrane antigen.
Acta Neuropathol. 130:441–443. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
Classification of Tumors of the Central Nervous System: A summary.
Acta Neuropathol. 131:803–820. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Liu Z, Wang C, Wang H, Wang Y, Li JY and
Liu Y: Clinical characteristics and treatment of angiomatous
meningiomas: A report of 27 cases. Int J Clin Exp Pathol.
6:695–702. 2013.PubMed/NCBI
|
|
6
|
Ben Nsir A, Chabaane M, Krifa H, Jeme H
and Hattab N: Intracranial angiomatous meningiomas: A 15-year,
multicenter study. Clin Neurol Neurosurg. 149:111–117.
2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Goutagny S and Kalamarides M: Meningiomas
and neurofibromatosis. J Neurooncol. 99:341–347. 2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Choy W, Kim W, Nagasawa D, Stramotas S,
Yew A, Gopen Q, Parsa AT and Yang I: The molecular genetics and
tumor pathogenesis of meningiomas and the future directions of
meningioma treatments. Neurosurg Focus. 30(E6)2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Abedalthagafi MS, Merrill PH, Bi WL, Jones
RT, Listewnik ML, Ramkisson SH, Thorner AR, Dunn IF, Beroukhim R,
Alexander BM, et al: Angiomatous meningiomas have a distinct
genetic profile with multiple chromosomal polysomies including
polysomy of chromosome 5. Oncotarget. 5:10596–10606.
2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Proctor DT, Ramachandran S, Lama S and
Sutherland GR: Towards molecular classification of meningioma:
Evolving treatment and diagnostic paradigms. World Neurosurg.
119:366–373. 2018.PubMed/NCBI View Article : Google Scholar
|