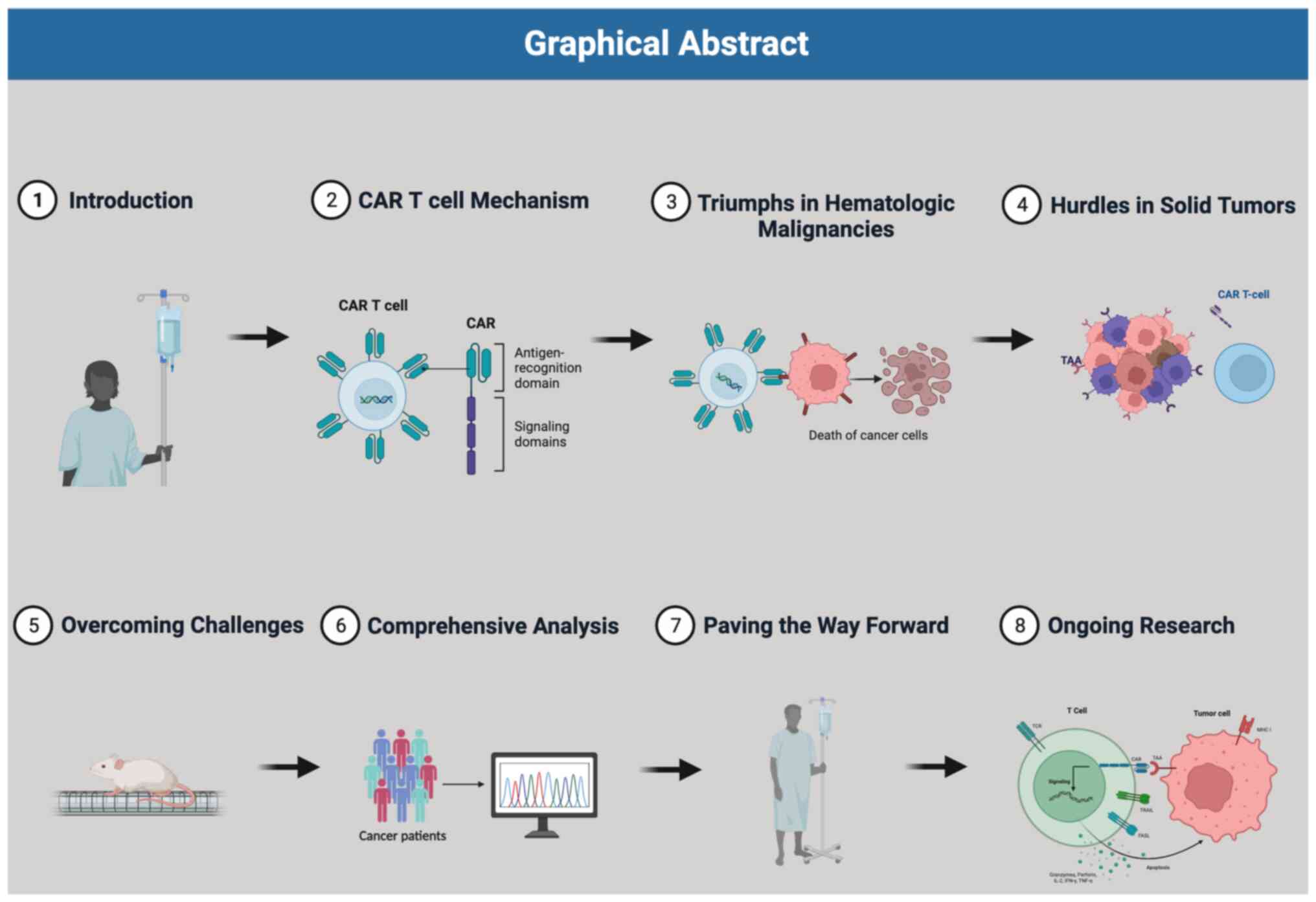
1. Introduction
Cancer poses a substantial worldwide public health
issue, and presently stands as the second primary contributor to
mortality in the United States (1). Projections indicate that by the year
2040, the annual number of new cancer cases is expected to reach a
staggering 29.5 million, with 16.4 million cancer-related deaths
(2). The concept of the connection
between immune cells and cancer was first suggested by Rudolf
Virchow over 150 years ago (3).
This observation laid the groundwork for exploring the potential of
using immune cells as a therapeutic approach. Notably, in the late
19th century, William Coley conducted groundbreaking research where
he injected heat-inactivated bacteria into tumor masses, resulting
in a reduction of tumor size (4).
The process of immune-driven elimination of cancer
cells encompasses a sequence of vital stages (5). Initially, local tissue disruption
caused by stromal remodeling triggers the recruitment of innate
immune system cells such as NK cells, macrophages, and neutrophils
(6). These NK cells recognize the
developing tumors and initiate the process of tumor cell killing
(7). After tumor cell death,
tumor-associated antigens (TAAs) from the deceased cancer cells are
taken up by antigen-presenting cells (APCs) such as dendritic cells
(DCs) (8). The activated DCs then
migrate to draining lymph nodes where they present the tumor
antigens to naïve CD4+ and CD8+ T-cells through major
histocompatibility complex (MHC) class I and II death (8). Additionally, the DCs release
cytokines that regulate T-cell responses and convert naïve CD8+ T
cells into cytotoxic T-cells. As a result, these cytotoxic T-cells
leave the lymphoid organs, enter the bloodstream, infiltrate the
tumor site, and effectively induce tumor cell death (8).
Tumors have developed multiple mechanisms to evade
immune-mediated elimination (9).
They employ inhibitory cytokines such as TGF-β and IL-10, which
activate inhibitory signals, leading to the attenuation of
antitumor immunity (10).
Furthermore, TGF-β plays a role in converting CD4+T cells that
infiltrate the tumor into Foxp3+Tregs, which possess highly
immunosuppressive properties (11). Moreover, most tumors downregulate
the expression of costimulatory molecules necessary for effective
T-cell activation. Additionally, myeloid-derived suppressor cells
(MDSCs) also contribute to inhibiting the antitumor response
(9). Another crucial aspect of T
cell dysfunction in cancer is related to a phenomenon known as T
cell exhaustion, wherein T cells lose their tumor-killing ability
and express inhibitory receptors such as PD1, Tim3, LAG3, CTLA4,
etc (12). As a result of these
strategies, tumors create an immunosuppressive tumor
microenvironment (TME) (9).
Immunotherapy encompasses strategies aimed at
overcoming immune suppression by blocking inhibitory receptors such
as CTLA-4 and PD-1 (immune checkpoint inhibitors) and stimulating
antitumor immune responses through adoptive transfer T cell therapy
(13-15).
The introduction of checkpoint inhibitors, such as ipilimumab
targeting CTLA-4, has revolutionized the field and shown promising
results. Similarly, chimeric antigen receptor (CAR) T cell therapy
has demonstrated remarkable clinical success in treating
hematological malignancies. One significant advantage of CAR T cell
therapy is its independence from antigenic peptide-bound major
histocompatibility complex (MHC) recognition. This is crucial
because tumor cells often evade immune responses by losing
MHC-associated antigen presentation, making traditional T cell
responses less effective. CAR T cells directly target
tumor-specific antigens, bypassing the MHC-related limitations.
In this review, we provide a comprehensive overview
of the development and mechanisms of Chimeric Antigen Receptor
(CAR)-T cell therapy. Additionally, we address the limitations of
CAR T cell therapy in treating solid tumors and explore potential
strategies to manage cytokine release syndrome, a common adverse
effect associated with CAR T cell therapy (Fig. 1).
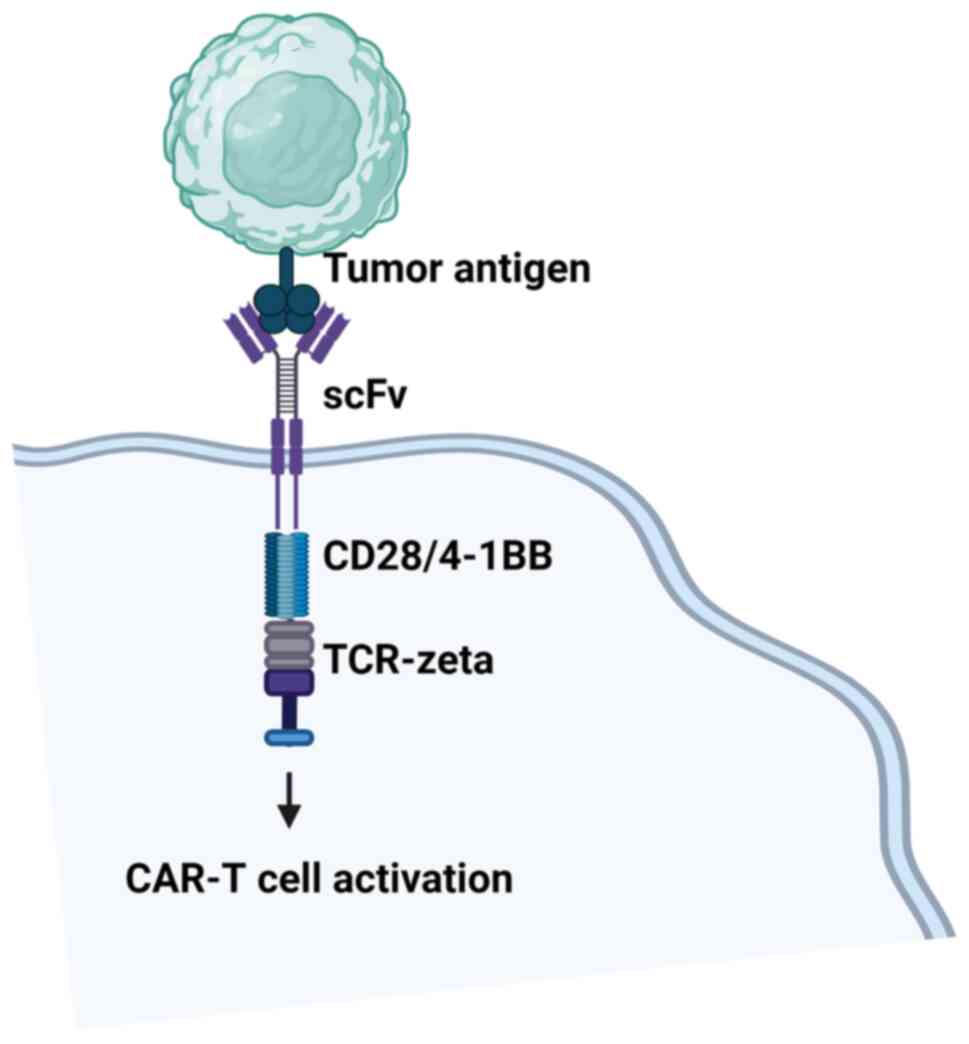
2. CAR design
CARs consist of four distinct parts, each serving
specific functions (16) (Fig. 2). The first part is the
antigen-binding domain, which is the extracellular component
conferring antigen specificity. This domain is formed by connecting
variable heavy (VH) and light (VL) chains of monoclonal antibodies
through a flexible linker, creating a single-chain variable
fragment (scFv) (17). The scFvs
bind to cancer antigens on the cell surface, leading to T cell
activation independent of the major histocompatibility complex
(MHC) (17,18).
Next is the hinge region, linking the
antigen-binding domain to the transmembrane region. Typically
derived from CD28 and CD8, the hinge imparts flexibility,
overcoming steric hindrance, and allowing the antigen-binding
domain to access targeted tumor antigens more effectively (16).
The transmembrane domain spans the T cell membrane's
lipid bilayer, anchoring the CAR to the cell membrane. While its
primary function is anchoring, some evidence suggests it may also
influence CAR T cell function (16). Transmembrane domains are often
derived from natural proteins such as CD3ζ, CD4, CD8α, or
CD28(16).
The fourth component, the intracellular signaling
domain, consists of an activation domain and one or more
costimulatory domains (Fig. 2).
Most CARs utilize CD3ζ-derived immunoreceptor tyrosine-based
activation motifs (ITAMs) for T cell activation, but this signaling
alone is insufficient (19). A
costimulatory signal is essential for optimal T cell function and
persistence (20). Notably, all
FDA-approved CAR T cells include either a CD28 or 4-1BB
costimulatory domain (16).
Upon recognizing specific tumor antigens through
their ScFv, CAR T cells trigger the phosphorylation of ITAM domains
on the CD3ζ chain, initiating signaling through the tyrosine kinase
ζ-associated protein of 70 kDa (ZAP70). Consequently, CAR T cells
become activated, proliferate, release cytokines, undergo metabolic
changes, and exhibit cytotoxicity, unleashing a potent T cell
effector response.
CARs can be categorized based on the number of
signaling domains they contain (Fig.
3) (1) First-generation CARs:
Contain only the CD3ζ activation domain. They show limited
persistence and efficacy (2)
Second-generation CARs: Incorporate a costimulatory domain (e.g.,
CD28 or 4-1BB) in addition to the CD3ζ activation domain. These
CARs exhibit enhanced T cell function and persistence (3) Third-generation CARs: Have multiple
costimulatory domains. The combination of costimulatory domains
aims to improve CAR T cell function and therapeutic efficacy
(4) Four-generation CARs: Also
known as armored CAR T cells, co-express key cytokines, such as
interleukins and chemokines, or suicide genes that can
significantly enhance the efficacy and safety of CAR T therapy
(5) Five-generation CARs: Contain
an extra intracellular domain than their predecessors. The CARs
comprise truncated intracellular domains of cytokine receptors
(e.g., IL-2R chain fragment) with a motif for binding transcription
factors such as STAT-3/5 (21-23).
It should be noted that there weren't widely recognized fourth or
fifth generation CAR T cells. However, researchers might have
explored additional modifications and generations to further
enhance CAR T cell therapy's efficacy, persistence, and safety.
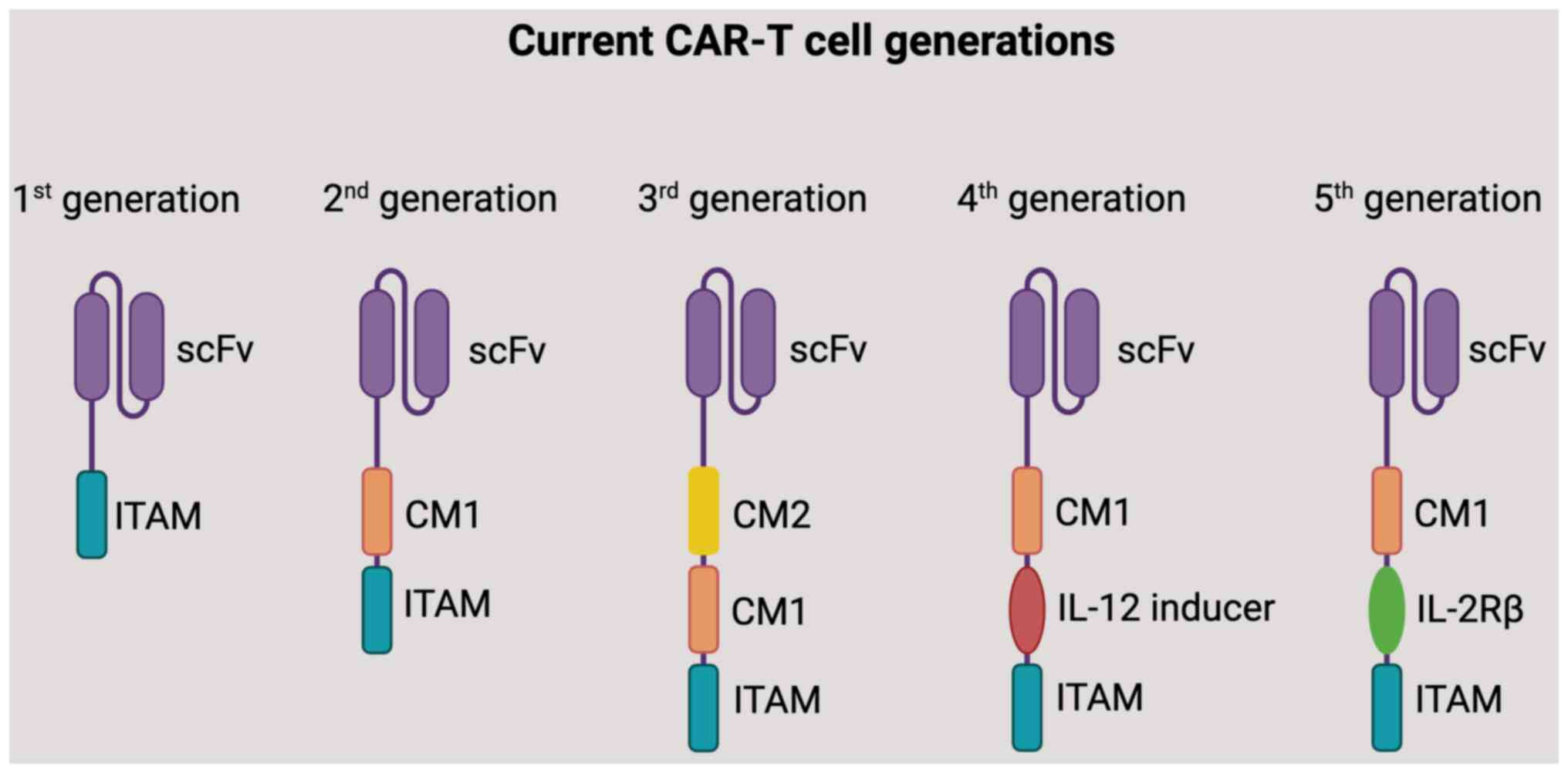 | Figure 3A diagram of five generations of
CARs. CARs are structurally divided into three key regions: i) The
antigen-binding domain, derived from antibodies, facilitates
antigen recognition. This domain typically incorporates a
single-stranded variable fragment sourced from antibodies. ii) The
transmembrane domain provides anchoring support to the plasma
membrane, ensuring stability. iii) The signaling domain triggers
T-cell activation. In first-generation CARs, this domain contains a
CD3ζ-derived signaling module. In second-generation CARs, an
additional co-stimulatory domain is included. Meanwhile,
third-generation CARs feature two co-stimulatory domains, including
CD28, 4-1BB (CD137), CD27 and OX40 (CD134). Furthermore, there are
advanced iterations of CAR T cells: Fourth-generation CAR T cells,
also referred to as TRUCKs, are designed to induce expression of
chemokines such as IL-12, enhancing their therapeutic potential.
Fifth-generation CARs introduce a novel co-stimulatory domain that
activates specific signaling pathways. scFv, single-chain variable
fragment; ITAM, immunoreceptor tyrosine-based activation motif; CM,
co-stimulatory molecule; IL-2Rβ, interleukin-2 receptor β; CAR,
chimeric antigen receptor. |
The choice of antigen-binding domain, costimulatory
domain, and the overall CAR architecture depends on various
factors, including the target tumor antigen, the type of cancer,
and the desired T cell response. Ongoing research aims to optimize
CAR design to enhance specificity, efficacy, and safety in CAR T
cell therapy.
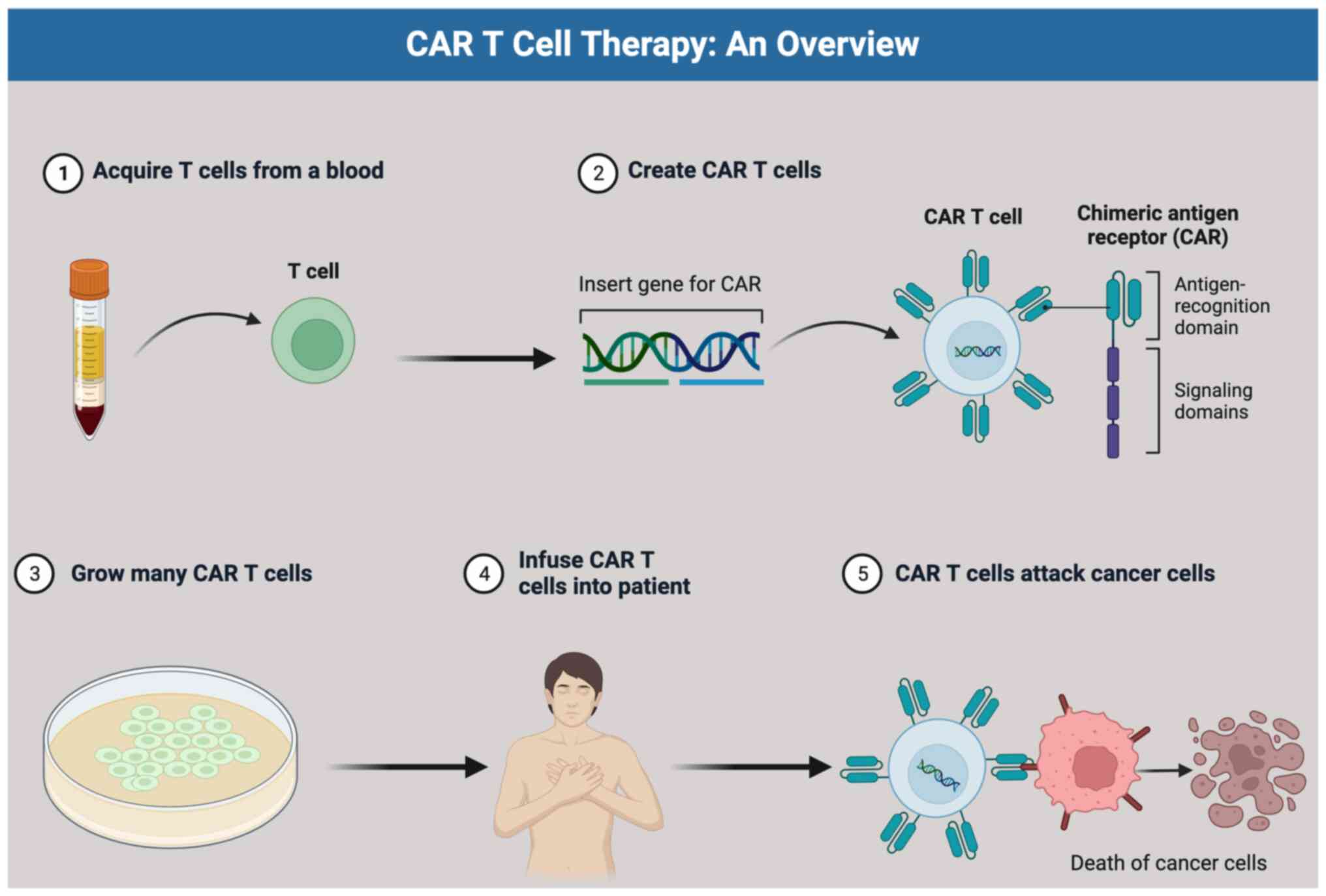
3. Generation of CAR T cells
The selection of the target tumor antigen is a
crucial initial step in the development of CAR T cells (24). The ideal scenario is to have CAR T
cells that solely target tumor cells while sparing normal, healthy
cells. However, a number of tumor antigens are self-antigens, which
means they are also present on normal cells but are often
overexpressed in tumors (24). In
CAR T-cell therapy for CD19- and CD20-positive hematologic
malignancies, CD19 and CD20 have become widely used tumor antigens
(17). These antigens have shown
promising results in treating certain blood cancers.
Various other tumor antigens have been explored for
CAR T cell therapy, and some examples are listed in Table I.
 | Table ITumor antigens targeted for CAR T
cell therapy. |
Table I
Tumor antigens targeted for CAR T
cell therapy.
| Target antigen | Cancer type | (Refs.) |
|---|
| CD22 | B-cell acute
lymphoblastic leukemia | (17) |
| CD30 | Hodgkin
lymphoma | (18-20) |
| CD33 | Acute myeloid
leukemia | (21-23) |
| Estrogen-related
receptor β type 2 | Prostate cancer,
breast cancer | (24,25) |
| Prostate-specific
membrane antigen | Prostate
cancer | (26-28) |
| Carbonic anhydrase
IX | Renal cell
carcinoma | (29) |
| Carcinoembryonic
antigen | Colon cancer | (30) |
| Mesothelin surface
glycoprotein | Malignant pleural
mesothelioma, pancreatic adenocarcinoma, breast cancer, ovarian
cancer | (31-34) |
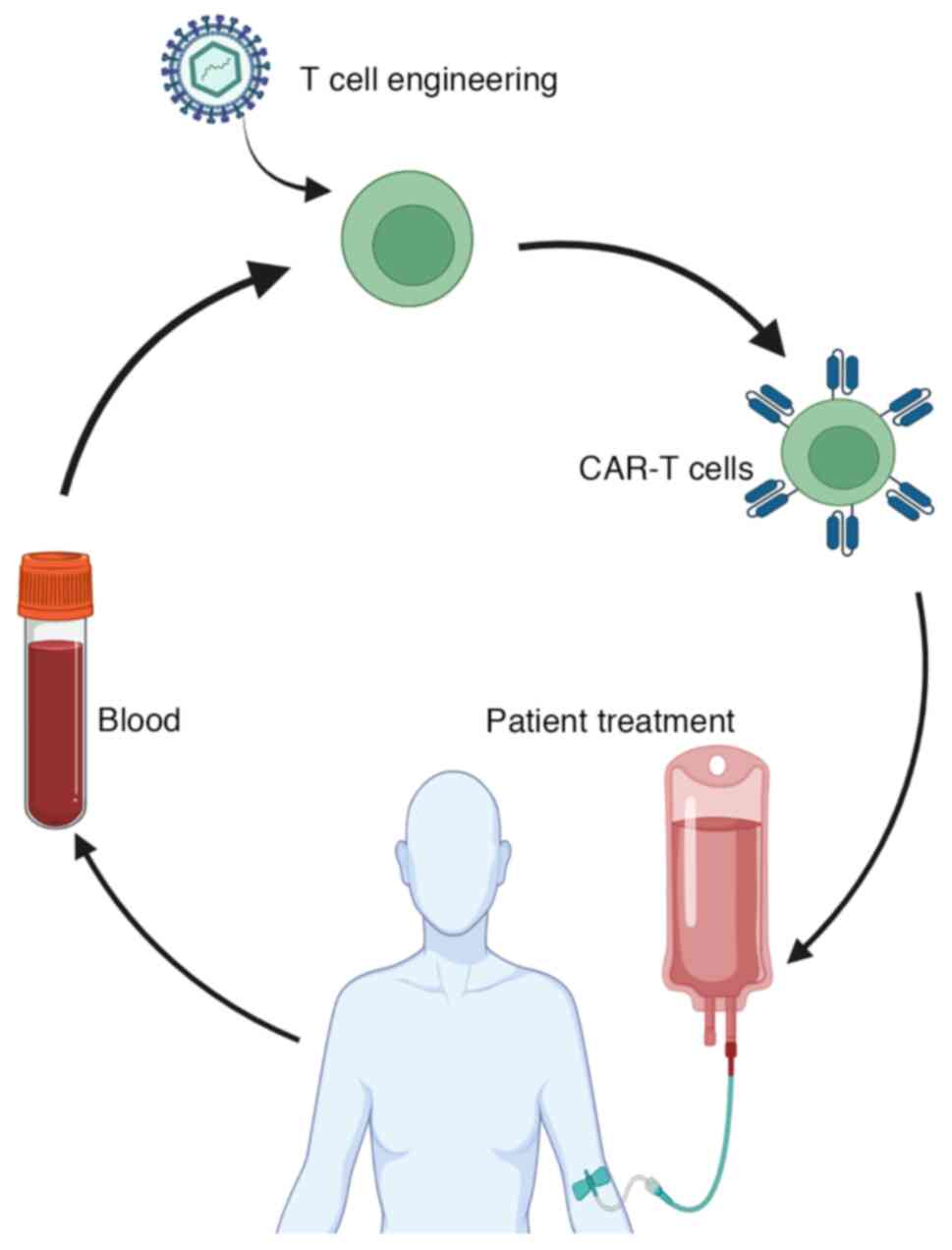
The subsequent step in CAR T cell therapy involves
isolating the patient's T cells through a process called
leukapheresis. During this procedure, the patient's blood is
withdrawn, and leukocytes (white blood cells) are collected, while
the rest of the blood components are returned to the patient. T
cells, including both CD4+ and CD8+ T cells,
are then isolated from the collected leukocytes using specific
antibody bead conjugates, which enriches for T cells.
In the laboratory, the isolated T cells undergo
genetic engineering to express CARs on their surfaces. This process
typically involves the use of retroviral or lentiviral vectors for
transfection (1) Transduction: The
retroviral or lentiviral vectors carry the genetic information
necessary for the CAR's expression. These vectors are engineered to
be non-replicative and safe for use in gene transfer. The T cells
are exposed to the viral particles, and the vectors enter the T
cells (2) Integration: Once inside
the T cells, the viral vectors integrate the genetic material
encoding the CAR into the T cell's genome. This integration ensures
stable and long-term expression of the CAR as the T cells divide
and proliferate (3) CAR
expression: With the CAR's genetic material now integrated into the
genome, the T cells start to express the chimeric antigen receptor
on their cell surfaces. The CAR allows the T cells to recognize and
bind to specific antigens present on cancer cells (4) Expansion: The genetically modified T
cells are then cultured and expanded in the laboratory. This
expansion process helps generate a large population of
CAR-expressing T cells that can be used for the patient's
treatment. By using retroviral or lentiviral vectors, researchers
can efficiently introduce the CAR into the patient's T cells,
ensuring sustained CAR expression and enabling the cells to target
and attack cancer cells when infused back into the patient's body
during CAR T cell therapy. This process has shown significant
promise in treating various hematological malignancies and is being
explored for potential applications in solid tumor therapies as
well.
Once the viral particles carrying the CAR's genetic
material enter the T cells, they integrate the CAR into the T
cell's genome. This integration ensures stable and heritable
expression of the CAR as the T cells divide and proliferate. As the
T cells multiply and expand in culture, the CAR expression is
retained in all the daughter cells, resulting in a large population
of CAR-expressing T cells. This is a crucial step in CAR T cell
therapy as it allows for the production of a sufficient number of
engineered T cells for infusion back into the patient. The
long-lasting expression of the CAR enables the CAR T cells to
recognize and target cancer cells effectively, leading to the
desired antitumor immune response when these modified T cells are
reinfused into the patient for therapy. The ability of CAR
expression to persist as the T cells undergo division and expansion
is essential for the success of CAR T cell therapy in treating
various types of cancers.
Once the CAR T cells have undergone significant
expansion and are ready for therapeutic use, they are sent back to
the hospital for infusion into the patient. However, before the CAR
T cell infusion, a preparatory step known as ‘lymphodepletion’ is
often performed.
Lymphodepletion involves the administration of
chemotherapy or other agents to temporarily suppress the patient's
immune system. This procedure serves several important purposes
(1) Create Space: By reducing the
number of existing immune cells, lymphodepletion creates space for
the infused CAR T cells to expand and exert their antitumor effects
without competition from the patient's endogenous immune cells
(2) Facilitate Persistence: The
temporary suppression of the patient's immune system may help the
infused CAR T cells persist and survive for a more extended period
in the body, increasing the treatment's efficacy (3) Reduce Rejection: Lymphodepletion also
helps reduce the risk of the patient's immune system recognizing
the CAR T cells as foreign and launching an immune response against
them (rejection). This enhances the chances of successful CAR T
cell therapy. Once lymphodepletion is completed, the expanded and
engineered CAR T cells are infused into the patient. These CAR T
cells then target and attack the cancer cells, leveraging the
patient's immune system to fight the disease effectively.
It's important to note that lymphodepletion is not
used in all CAR T cell therapy protocols, and its use may vary
depending on the specific cancer type and the CAR T cell product
being used. The decision to include lymphodepletion is based on the
clinical trial protocol and the medical team's judgment to optimize
the therapy's effectiveness and safety (25) (Fig.
4).
4. Limitation of CAR T cells and
toxicity
The development of resistance to the targeted single
antigen is a significant limitation of CAR T cell therapy. In some
cases, cancer cells in patients treated with CAR T cells either
reduce or completely lose the expression of the target antigen.
This phenomenon is commonly known as antigen escape and has been
observed in patients treated with CD19-targeted CAR T cells for
acute lymphoblastic leukemia (ALL) (26,27).
Similar reduced expression of BCMA has been reported in multiple
myeloma (MM) patients treated with BCMA-targeted CAR T cells
(28,29). Moreover, glioblastoma patients
treated with CAR T cells targeting IL13Ra2 have shown tumor
recurrences with decreased IL13Ra2 expression (30). Of note, the potential for CAR T
cells to suppress cancer stem cells (CSCs) by specifically homing
in on their cell surface markers has been investigated. This
approach holds promise for enhancing the effectiveness of
treatments in individuals with different types of cancer (31).
To overcome this hurdle, one strategy is to target
multiple antigens simultaneously. Clinical trials using
dual-targeted CAR T cells, such as CD19/CD22 or CD19/BCMA, have
shown promising results (32-35).
In solid tumors, tandem CARs have also demonstrated potential in
preclinical models. For example, targeting HER2 and IL13Ra2 in
glioblastoma or HER2 and MUC1 in breast cancer has shown
encouraging outcomes (36,37). By targeting multiple antigens, CAR
T cells have an improved chance of recognizing and attacking cancer
cells, reducing the likelihood of resistance due to antigen escape.
This approach opens up new possibilities for enhancing the
effectiveness of CAR T cell therapy against various types of
cancer.
The immunosuppressive tumor microenvironment (TME)
poses another significant challenge to CAR T cell therapy,
particularly in solid tumors. Within solid tumors, various immune
cells with inhibitory functions, such as myeloid-derived suppressor
cells (MDSCs) and regulatory T cells (Tregs), infiltrate and hinder
CAR T cell activity (38).
Moreover, immune checkpoint pathways, including PD-1, Tim3, Lag3,
and CTLA-4, play a crucial role in suppressing antitumor immunity
and promoting CAR T cell exhaustion (38). As a result, researchers are
actively exploring strategies to combine CAR T cell therapy with
checkpoint blockade, both in hematological malignancies and solid
tumors (39). By blocking these
inhibitory checkpoint pathways, the aim is to enhance the function
and persistence of CAR T cells, enabling them to better combat the
immunosuppressive effects of the TME. This approach holds
significant promise in overcoming the limitations posed by the
immunosuppressive TME and improving the efficacy of CAR T cell
therapy in various cancer types.
Another major limitation of CAR T cells is toxicity
which can be broadly classified under two categories (1) Systemic toxicities: These occur as a
result of the robust activation of T cells, leading to excessive
cytokine production. This phenomenon is known as cytokine-release
syndrome (CRS) and can manifest as severe and potentially fatal
increases in cytokine levels. CRS may also be accompanied by other
complications, such as macrophage activation syndrome (MAS) and
neurotoxicity (2) On-target but
off-tumor toxicities: This type of toxicity occurs when CAR T cells
recognize and attack not only tumor cells but also healthy cells
expressing the same target antigen. This can lead to adverse
effects on normal tissues.
To mitigate the detrimental effects of systemic
toxicities, therapeutic antibodies blocking the IL-6 pathway, such
as tocilizumab or siltuximab, are now being utilized. These
antibodies help reduce the harmful effects of excessive cytokine
release without compromising the antitumor activity of CAR T cells.
Managing toxicity is a critical aspect of CAR T-cell therapy, and
ongoing research and advancements aim to optimize the therapy's
safety and effectiveness (40).
The implementation of ‘off-switches’ in CAR T cell
therapy is an emerging and promising strategy to mitigate toxicity.
These off-switches are designed to selectively block or deactivate
CAR T cells in response to adverse events, providing a way to
rapidly control the therapy's effects when needed. One such example
is the use of inducible cas9, which has shown significant efficacy
in a clinical trial. This approach resulted in the elimination of
over 90% of engineered T cells within just 30 min, offering a swift
and controllable means of attenuating the CAR T cell response when
necessary (41). Another strategy
involves utilizing protease-based small molecule-assisted shutoff
CARs (SMASh-CARs) or switch-off CARs (SWIFF-CARs) (42). These engineered CARs incorporate
specific protease cleavage sites, allowing for external control
over CAR T cell activity through administration of appropriate
small molecules.
However, despite the potential benefits of these
off-switch strategies in reducing toxicity, a challenge remains in
finding a balance between temporary inhibition of CAR T cells and
timely reactivation to resume antitumor activity. Abruptly stopping
therapy can be a concern, particularly if cancer progression occurs
rapidly during the period when CAR T cells are deactivated.
In addition, other approaches have been developed
(1) Tunable CARs: Designing CARs
with tunable activation thresholds can enable the control of CAR T
cell function. These CARs respond to specific signals or
concentrations of antigens, allowing for fine-tuning of CAR T cell
activity based on the tumor burden and potential toxicity (2) Localized delivery: Developing methods
for localized delivery of CAR T cells to the tumor site can
minimize systemic toxicity. By targeting CAR T cells directly to
the tumor, it reduces the risk of damage to healthy tissues and
decreases the likelihood of severe adverse effects (3) Combination therapies: Combining CAR T
cell therapy with other treatments, such as immune checkpoint
inhibitors or targeted therapies, may help modulate CAR T cell
responses and improve safety and efficacy.
To further advance this approach, new and more
sophisticated strategies need to be developed. These strategies
should enable the temporary inhibition of CAR T cell function while
allowing for CAR T cell therapy rescue once the toxicity subsides.
Achieving this delicate balance is essential for CAR T cell therapy
to progress towards becoming a viable first-line treatment option
for both hematological and solid tumors.
Research in these areas is ongoing, and these
innovative strategies hold great promise for overcoming the current
limitations of CAR T cell therapy and moving it closer to becoming
a frontline treatment for both hematological and solid tumors. With
continued advancements in CAR design and safety measures, CAR T
cell therapy has the potential to transform cancer treatment and
significantly improve patient outcomes.
5. Summary
CAR T cell therapy (Fig. 5) has demonstrated remarkable
potential in treating hematologic malignancies. Despite this
promise, its widespread clinical application has been hindered by
several challenges, including target antigen escape, a
tumor-suppressive microenvironment, and adverse reactions. To
overcome these obstacles, it is crucial to gain a comprehensive
understanding of the intricate interactions among engineered T
cells, endogenous immune cells, tumor cells, and other
tumor-associated factors. Such knowledge is paramount for enhancing
the antitumor effects and minimizing the occurrence of adverse
reactions.
Excitingly, recent advancements in genome editing,
proteomics, and metabolomics present an opportunity for adopting
multilayered approaches that address multiple critical aspects in
unison. This multi-faceted strategy holds great promise for further
improving CAR T cell therapy. The next generation of CAR T cells
must also address practical concerns such as the high cost of
treatment and lengthy preparation times. By tackling these issues,
accessibility to this groundbreaking therapy can be significantly
enhanced, making it more accessible to patients in need.
One significant limitation of current CAR T cell
therapy is its potential for severe and occassionally
life-threatening side effects, particularly in the form of CRS and
neurotoxicity. These adverse events are caused by the robust
activation and proliferation of CAR T cells upon encountering their
target antigen. Researchers and clinicians are actively working to
mitigate these limitations through improved patient selection,
improved CAR T cell design, and refined treatment protocols. These
challenges still pose significant hurdles in the broader adoption
and application of CAR T cell therapy.
Overall, a comprehensive approach that combines
cutting-edge technologies with a deep understanding of the complex
interactions within the immune-tumor microenvironment holds the key
to advancing CAR T cell therapy and unleashing its full potential
in the fight against cancer.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by grants from the National
Institutes of Health (grant nos. R01CA221867 and R01CA273002) and
the U.S. Department of Defense (grant no. LC210150).
Availability of data and materials
Not applicable.
Authors' contributions
RP, AFS, BSS and CSS wrote and prepared the original
draft. LW reviewed and edited the manuscript. JS supervised the
study and acquired funding. All authors have read and approved the
final version of the manuscript. Data sharing is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fuller R, Landrigan PJ, Balakrishnan K,
Bathan G, Bose-O'Reilly S, Brauer M, Caravanos J, Chiles T, Cohen
A, Corra L, et al: Pollution and health: A progress update. Lancet
Planet Health. 6:e535–e547. 2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2022. CA Cancer J Clin. 72:7–33.
2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Adams JL, Smothers J, Srinivasan R and
Hoos A: Big opportunities for small molecules in immuno-oncology.
Nat Rev Drug Discov. 14:603–622. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
4
|
Hoption Cann SA, van Netten JP and van
Netten C: Dr William Coley and tumour regression: A place in
history or in the future. Postgrad Med J. 79:672–680.
2003.PubMed/NCBI
|
|
5
|
Fares J, Fares MY, Khachfe HH, Salhab HA
and Fares Y: Molecular principles of metastasis: A hallmark of
cancer revisited. Signal Transduct Target Ther.
5(28)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Miller JF and Sadelain M: The journey from
discoveries in fundamental immunology to cancer immunotherapy.
Cancer Cell. 27:439–449. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kyrysyuk O and Wucherpfennig KW: Designing
cancer immunotherapies that engage T cells and NK cells. Annu Rev
Immunol. 41:17–38. 2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zagorulya M and Spranger S: Once upon a
prime: DCs shape cancer immunity. Trends Cancer. 9:172–184.
2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Thommen DS and Schumacher TN: T Cell
Dysfunction in Cancer. Cancer Cell. 33:547–562. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Briukhovetska D, Dorr J, Endres S, Libby
P, Dinarello CA and Kobold S: Interleukins in cancer: From biology
to therapy. Nat Rev Cancer. 21:481–499. 2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Takeuchi Y and Nishikawa H: Roles of
regulatory T cells in cancer immunity. Int Immunol. 28:401–409.
2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chow A, Perica K, Klebanoff CA and Wolchok
JD: Clinical implications of T cell exhaustion for cancer
immunotherapy. Nat Rev Clin Oncol. 19:775–790. 2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hibino S, Eto S, Hangai S, Endo K,
Ashitani S, Sugaya M, Osawa T, Soga T, Taniguchi T and Yanai H:
Tumor cell-derived spermidine is an oncometabolite that suppresses
TCR clustering for intratumoral CD8(+) T cell activation. Proc Natl
Acad Sci USA. 120(e2305245120)2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Maruhashi T, Sugiura D, Okazaki IM,
Shimizu K, Maeda TK, Ikubo J, Yoshikawa H, Maenaka K, Ishimaru N,
Kosako H, et al: Binding of LAG-3 to stable peptide-MHC class II
limits T cell function and suppresses autoimmunity and anti-cancer
immunity. Immunity. 55:912–924 e8. 2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Spassova I, Ugurel S, Kubat L, Zimmer L,
Terheyden P, Mohr A, Björn Andtback H, Villabona L, Leiter U,
Eigentler T, et al: Clinical and molecular characteristics
associated with response to therapeutic PD-1/PD-L1 inhibition in
advanced Merkel cell carcinoma. J Immunother Cancer.
10(e003198)2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rafiq S, Hackett CS and Brentjens RJ:
Engineering strategies to overcome the current roadblocks in CAR T
cell therapy. Nat Rev Clin Oncol. 17:147–167. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Finck AV, Blanchard T, Roselle CP,
Golinelli G and June CH: Engineered cellular immunotherapies in
cancer and beyond. Nat Med. 28:678–689. 2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jensen MC and Riddell SR: Designing
chimeric antigen receptors to effectively and safely target tumors.
Curr Opin Immunol. 33:9–15. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Brocker T and Karjalainen K: Signals
through T cell receptor-zeta chain alone are insufficient to prime
resting T lymphocytes. J Exp Med. 181:1653–1659. 1995.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Frauwirth KA and Thompson CB: Activation
and inhibition of lymphocytes by costimulation. J Clin Invest.
109:295–299. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Kagoya Y, Tanaka S, Guo T, Anczurowski M,
Wang CH, Saso K, Butler MO, Minden MD and Hirano N: A novel
chimeric antigen receptor containing a JAK-STAT signaling domain
mediates superior antitumor effects. Nat Med. 24:352–359.
2018.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Jan M, Scarfo I, Larson RC, Walker A,
Schmidts A, Guirguis AA, Gasser JA, Słabicki M, Bouffard AA,
Castano AP, et al: Reversible ON- and OFF-switch chimeric antigen
receptors controlled by lenalidomide. Sci Transl Med.
13(eabb6295)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Aspuria PJ, Vivona S, Bauer M, Semana M,
Ratti N, McCauley S, Riener R, de Waal Malefyt R, Rokkam D,
Emmerich J, et al: An orthogonal IL-2 and IL-2Rβ system drives
persistence and activation of CAR T cells and clearance of bulky
lymphoma. Sci Transl Med. 13(eabg7565)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Shirasu N and Kuroki M: Functional design
of chimeric T-cell antigen receptors for adoptive immunotherapy of
cancer: Architecture and outcomes. Anticancer Res. 32:2377–2383.
2012.PubMed/NCBI
|
|
25
|
Gattinoni L, Finkelstein SE, Klebanoff CA,
Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C,
Heimann DM, et al: Removal of homeostatic cytokine sinks by
lymphodepletion enhances the efficacy of adoptively transferred
tumor-specific CD8+ T cells. J Exp Med. 202:907–912.
2005.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Majzner RG and Mackall CL: Tumor Antigen
Escape from CAR T-cell Therapy. Cancer Discov. 8:1219–1226.
2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Maude SL, Teachey DT, Porter DL and Grupp
SA: CD19-targeted chimeric antigen receptor T-cell therapy for
acute lymphoblastic leukemia. Blood. 125:4017–4023. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Brudno JN, Maric I, Hartman SD, Rose JJ,
Wang M, Lam N, Stetler-Stevenson M, Salem D, Yuan C, Pavletic S, et
al: T cells genetically modified to express an Anti-B-Cell
maturation antigen chimeric antigen receptor cause remissions of
poor-prognosis relapsed multiple myeloma. J Clin Oncol.
36:2267–2280. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cohen AD, Garfall AL, Stadtmauer EA,
Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss BM, Dengel K,
Nelson A, et al: B cell maturation antigen-specific CAR T cells are
clinically active in multiple myeloma. J Clin Invest.
129:2210–2221. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Brown CE, Alizadeh D, Starr R, Weng L,
Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J,
Simpson J, et al: Regression of glioblastoma after chimeric antigen
receptor T-Cell therapy. N Engl J Med. 375:2561–2569.
2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Poondla N, Sheykhhasan M, Akbari M, Samadi
P, Kalhor N and Manoochehri H: The Promise of CAR T-Cell therapy
for the treatment of cancer stem cells: A short review. Curr Stem
Cell Res Ther. 17:400–406. 2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yan Z, Cao J, Cheng H, Qiao J, Zhang H,
Wang Y, Shi M, Lan J, Fei X, Jin L, et al: A combination of
humanised anti-CD19 and anti-BCMA CAR T cells in patients with
relapsed or refractory multiple myeloma: A single-arm, phase 2
trial. Lancet Haematol. 6:e521–e529. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D,
Han X, Liu Y, Zhang W, Wang C, et al: Bispecific CAR T cells
targeting both CD19 and CD22 for therapy of adults with relapsed or
refractory B cell acute lymphoblastic leukemia. J Hematol Oncol.
13(30)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zeng W, Zhang Q, Zhu Y, Ou R, Peng L, Wang
B, Shen H, Liu Z, Lu L, Zhang P and Liu S: Engineering Novel
CD19/CD22 Dual-Target CAR T cells for improved anti-tumor activity.
Cancer Invest. 40:282–292. 2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Cordoba S, Onuoha S, Thomas S, Pignataro
DS, Hough R, Ghorashian S, Vora A, Bonney D, Veys P, Rao K, et al:
CAR T cells with dual targeting of CD19 and CD22 in pediatric and
young adult patients with relapsed or refractory B cell acute
lymphoblastic leukemia: A phase 1 trial. Nat Med. 27:1797–1805.
2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Sterner RC and Sterner RM: CAR T cell
therapy: Current limitations and potential strategies. Blood Cancer
J. 11(69)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wilkie S, van Schalkwyk MC, Hobbs S,
Davies DM, van der Stegen SJ, Pereira AC, Burbridge SE, Box C,
Eccles SA and Maher J: Dual targeting of ErbB2 and MUC1 in breast
cancer using chimeric antigen receptors engineered to provide
complementary signaling. J Clin Immunol. 32:1059–1070.
2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kong Y, Tang L, You Y, Li Q and Zhu X:
Analysis of causes for poor persistence of CAR T cell therapy in
vivo. Front Immunol. 14(1063454)2023.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Al-Haideri M, Tondok SB, Safa SH, Maleki
AH, Rostami S, Jalil AT, Al-Gazally ME, Alsaikhan F, Rizaev JA,
Mohammad TAM and Tahmasebi S: CAR T cell combination therapy: The
next revolution in cancer treatment. Cancer Cell Int.
22(365)2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Mohammadi M, Akhoundi M, Malih S,
Mohammadi A and Sheykhhasan M: Therapeutic roles of CAR T cells in
infectious diseases: Clinical lessons learnt from cancer. Rev Med
Virol. 32(e2325)2022.PubMed/NCBI View
Article : Google Scholar
|
|
41
|
Di Stasi A, Tey SK, Dotti G, Fujita Y,
Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG,
Grilley B, et al: Inducible apoptosis as a safety switch for
adoptive cell therapy. N Engl J Med. 365:1673–1683. 2011.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Juillerat A, Tkach D, Busser BW, Temburni
S, Valton J, Duclert A, Poirot L, Depil S and Duchateau P:
Modulation of chimeric antigen receptor surface expression by a
small molecule switch. BMC Biotechnol. 19(44)2019.PubMed/NCBI View Article : Google Scholar
|