Introduction
Nicotine is an alkaloid found in tobacco leaves.
Nicotine stimulates the brain to secrete dopamine, temporarily
clears the head, and relieves fatigue. Smoking prevalence and the
associated health risks are higher in psychiatric patients.
However, smoking prevention has been a neglected issue in
psychiatry (1). It has been
experimentally demonstrated that nicotine increases neuronal firing
rates and induces the release of dopamine (2,3). In
addition, nicotine metabolism occurs in the liver by cytochrome
P450, and 70-80% of nicotine is metabolized into cotinine (4,5).
Nicotine is rapidly removed from the central nervous system for 1-3
h, while cotinine is known to remain in the body for >20-30 h
(6). There are a number of studies
available on nicotine addiction (4-6);
however, the effects of cotinine on the human body have not yet
been fully elucidated. Lithium carbonate
(Li2CO3) has been used as a mood stabilizer
in bipolar disorder. Of note however, the effective dose range of
Li2CO3 is 0.6-1.0 mM and >1.5 mM as
lithium in serum, which is close to the addiction level of
Li2CO3 (7).
Nicotine intake in conjunction with the administration of the mood
stabilizer, Li2CO3, may have a synergistic
effect on brain cells, although this remains to be determined.
The author of the present study recently found that
platinum nano colloids promoted the autoxidation of dopamine
(8). The platinum nano colloids and
dopamine increased intracellular reactive oxygen species generation
and suppressed the proliferation of human glioblastoma cells
(8). On the other hand,
2-phenylethylamine did not suppress the growth of human
glioblastoma cells, regardless of the combined use with platinum
nano colloids. Thus, the administration of platinum nano colloids
affected the growth of human glioblastoma cells, depending on the
type of coexisting neurotransmitters (8).
The present study investigated the effects of
nicotine and its metabolite, cotinine, in conjunction with the use
of Li2CO3, on the proliferation of U-251MG
human glioblastoma cells, and compared these to those of
acetylcholine and dopamine. The U-251MG cell line was used as a
replacement for glial cells, which comprise the majority of brain
cells.
Materials and methods
Cells and cell culture
The human glioblastoma cell line, U-251MG, was
obtained from the JCRB Cell Bank (cat. no. IFO50288). The U-251MG
cells are glial fibrillary acidic protein-positive glioblastoma
multiforme derived from a grade III-IV astrocytoma in the brain of
a 75-year-old patient, which was established as a cell line by
Bigner et al (9) in 1981. The
U-251MG cells express an α7 nicotinic acetylcholine receptor
(10). The U-251MG cells were
cultured in Eagle's minimum essential medium with L-glutamine
(FUJIFILM Wako Pure Chemical Corporation) supplemented with 10%
fetal bovine serum (Equitech-Bio. Inc.) and
penicillin-streptomycin-amphotericin B suspension (FUJIFILM Wako
Pure Chemical Corporation) at 37˚C with 5% CO2.
Cell proliferation assay
The U-251MG cells were seeded at 2,000 cells/well in
a 96-well culture plate (Sumitomo Bakelite Co., Ltd.) as
independent wells with n=5 and pre-incubated for 24 h at 37˚C with
5% CO2. A solution of (-)-nicotine (FUJIFILM Wako Pure
Chemical Corporation) or (-)-cotinine (FUJIFILM Wako Pure Chemical
Corporation) was applied to each well at 0-6 mM and incubated for 4
days at 37˚C with 5% CO2. During the 4 days of
incubation, the U-251MG cells were in the logarithmic growth phase.
The medium was then replaced with 5%
2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium
solution (WST-8; Dojindo Laboratories, Inc.) diluted with an
Eagle's minimum essential medium and incubated for 1.5 h at 37˚C
with 5% CO2. The absorbance was then measured at λ=450
nm using a multi-spectrophotometer (Viento; Dainippon Sumitomo
Pharma, Co. Ltd.). The amount of formed formazan is proportional to
the number of viable cells, as intracellular mitochondrial
dehydrogenase reduces WST-8 to yellowish-orange formazan (11). Acetylcholine chloride (Nacalai
Tesque, Inc.) and dopamine hydrochloride (Nacalai Tesque, Inc.)
were also examined for comparison with nicotine.
Under the administration of
Li2CO3, cell proliferation was examined
following exposure to nicotine and cotinine. The U-251MG cells were
seeded at 2,000 cells/well in a 96-well culture plate and
pre-incubated for 24 h at 37˚C. In the experiment described above,
a solution of 3.4 mM nicotine or 6 mM cotinine, which was found to
decrease cell viability to ~70-80%, was applied to each well in
combination with 0-1.5 mM Li2CO3 (Nacalai
Tesque, Inc.) and incubated at 37˚C with 5% CO2.
Following 4 days of incubation, cell proliferation was measured
using WST-8 assay. A solution of 6 mM acetylcholine and 60 µM
dopamine was applied for comparison with nicotine.
To clarify the mechanisms of action of nicotine as
regards cell proliferation, mecamylamine, a non-competitive
antagonist of nicotinic acetylcholine receptors, was used in the
experiments. The U-251MG cells were seeded at 2,000 cells/well in a
96-well culture plate and pre-incubated for 24 h at 37˚C. A
solution of 20 µM mecamylamine hydrochloride (Cayman Chemical
Company) was first applied to each well and incubated at 37˚C with
5% CO2. Following a 5-h incubation, the medium was
aspirated. Subsequently, 3.6 mM nicotine were applied to each well
in combination with 20 µM mecamylamine hydrochloride and incubated
at 37˚C with 5% CO2. After 4 days of incubation, cell
proliferation was measured using WST-8 assay as described
above.
Determination of mitotic catastrophe
in cells
For the evaluation of mitotic catastrophe,
multinucleated cells with two or more nuclei were counted using
nuclear staining (12). The U-251MG
cells were treated with 3.4 mM nicotine or 6 mM cotinine in
combinaion with or without 1 mM Li2CO3.
Following 4 days of incubation for 24 h at 37˚C, the U-251MG cells
were fixed with 4% paraformaldehyde at pH7.4 (FUJIFILM Wako Pure
Chemical Corporation), and their cell nuclei were stained with
Hoechst 33342 (Dojindo Laboratories, Inc.). Mitotic catastrophe in
the U-251MG cells was observed and counted using a fluorescent
microscope CKX-53 (Olympus Corporation) at an excitation/emission
of 330-385 nm/420 nm and a magnification of x200. The ratio of
cells undergoing mitotic catastrophe (%) was calculated for
quantitative analysis on five fluorescence micrographs of each
treatment.
Measurement of intracellular reactive
oxygen species generation
The Nitroblue-tetrazolium (NBT) reduction assay was
used to determine the production of superoxide anion radicals
(O2·-) in cells (13,14). The
U-251MG cells were seeded at 6,000 cells/well in a 96-well culture
plate as independent wells with n=5 and pre-incubated for 24 h at
37˚C with 5% CO2. A solution of 3.4 mM nicotine or 6 mM
cotinine was applied to each well in combination with or without 1
mM Li2CO3 followed by incubation for 1 day at
37˚C with 5% CO2. The medium was then replaced with 0.2%
nitrotetrazolium blue chloride (NBT, MilliporeSigma) solution and
incubated at 37˚C with 5% CO2. The 0.2% NBT solution was
prepared at use, dissolved with an Eagle's minimum essential
medium, and filtered through a 0.22-µm filter. Following a 5-h
incubation, the absorbance of the NBT-formazan was measured at
λ=620 nm using the multi-spectrophotometer (Viento). The U-251MG
cells were fixed with 4% paraformaldehyde at pH7.4 and observed
under a phase contrast microscope (CKX-53; Olympus Corporation) at
x400 magnification. The amount of formed NBT-formazan is
proportional to the amount of superoxide anion radicals
(O2·-) in cells. Cell proliferation was
examined using WST-8 assay (as described above) in the same
treatment groups, which confirmed that there were no differences
between the groups (data not shown).
Statistical analysis
The data for cell proliferation, mitotic catastrophe
and intracellular reactive oxygen species are expressed as the mean
± SD, as independent wells with n=5. The data were analyzed using
one-way ANOVA followed by Dunnett's test. By contrast, the data for
mecamylamine and NBT assay were analyzed using a Student's t-test
with KaleidaGraph 4.5J software (HULINKS Inc.). A value of
P<0.05 was considered to indicate a statistically significant
difference. In the present study, experiments were repeated at
least two times.
Results
Proliferation of human glioblastoma
cells
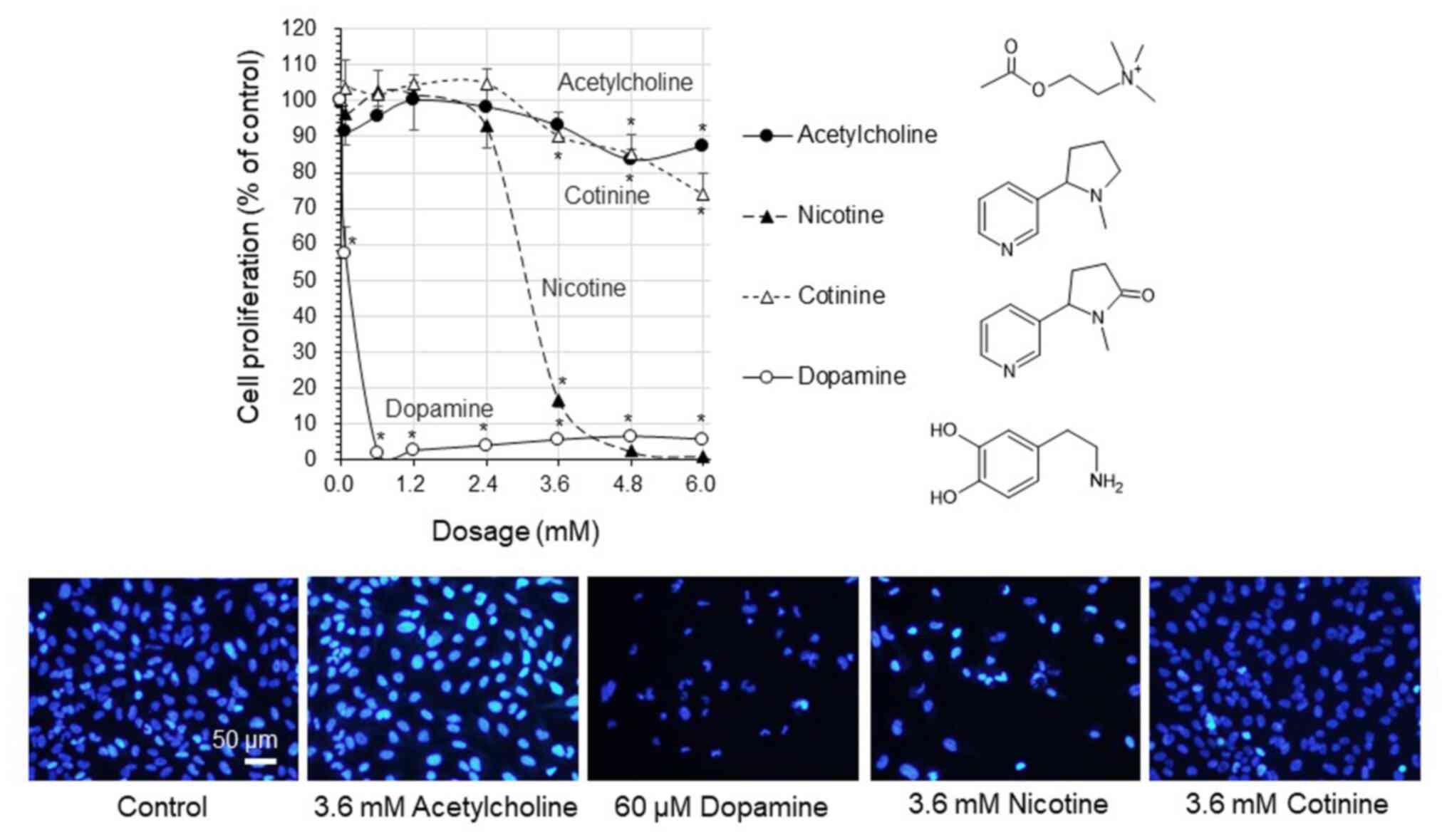
The proliferation of the human glioblastoma U-251MG
cells was markedly decreased to <16.6% at concentrations >2.4
mM nicotine (Fig. 1). Moreover,
following exposure to cotinine, cell proliferation gradually
decreased in a concentration-dependent manner to 73.8% with 6 mM
cotinine. There was almost no decrease in cell proliferation
following exposure to 0-6 mM acetylcholine (a neurotransmitter that
interacts with nicotinic acetylcholine receptors). Cell
proliferation was maintained at 83.5%, even at acetylcholine
concentrations ≥4.8 mM. By contrast, cell proliferation was
significantly decreased to <6.3% with dopamine at concentrations
>60 µM. The microscopic observation clearly indicated that cell
proliferation was not inhibited by acetylcholine and cotinine,
whereas this decreased by dopamine and nicotine.
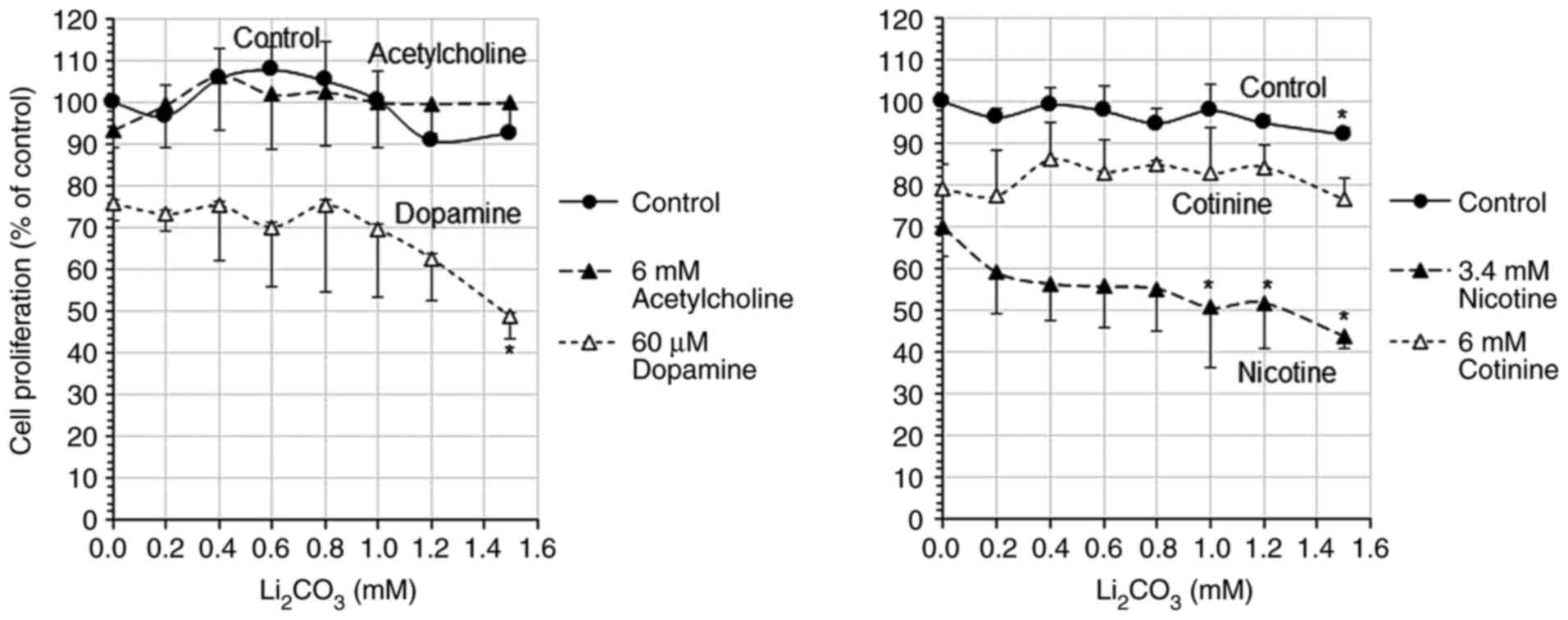
Following exposure to 0-1.5 mM
Li2CO3, cell proliferation decreased from
75.8 to 48.6% in the cells treated with 60 µM dopamine, and from
69.8 to 43.9% in the cells treated with 3.4 mM nicotine in a
concentration-dependent manner (with the increasing
Li2CO3 concentration) (Fig. 2). By contrast, the cells treated with
6 mM acetylcholine or 6 mM cotinine exhibited a proliferation of
93.4-106.2 and 76.6-86.0%, respectively, which did not decrease
even following exposure to high concentrations of
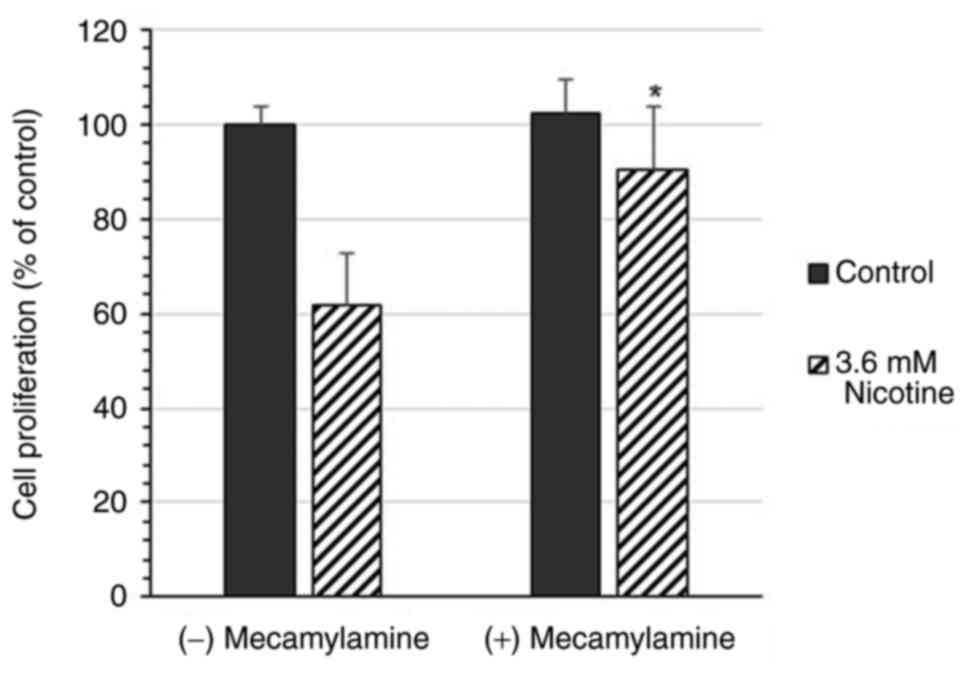
Li2CO3. Exposure to 20 µM mecamylamine, a
non-competitive antagonist of nicotinic acetylcholine receptors,
restored the proliferation of the cells treated with 3.6 mM
nicotine from 61.7 to 90.5% (Fig.
3).
Mitotic catastrophe in cells
The ratio of cells undergoing mitotic catastrophe
was 3.5% in the controls; however, this increased to 5.2% in the
cells exposed to 1 mM Li2CO3 (Fig. 4). In the cells treated with 3.4 mM
nicotine, the ratio of cells undergoing mitotic catastrophe was
significantly increased to 10.4, and to 14.4% in the cells treated
with 3.4 mM nicotine in combination with 1 mM
Li2CO3. Exposure to cotinine at 6 mM slightly
increased the ratio of cells undergoing mitotic catastrophe to
4.9%, and the ratio remained relative unaltered following the
administration of 1 mM Li2CO3.
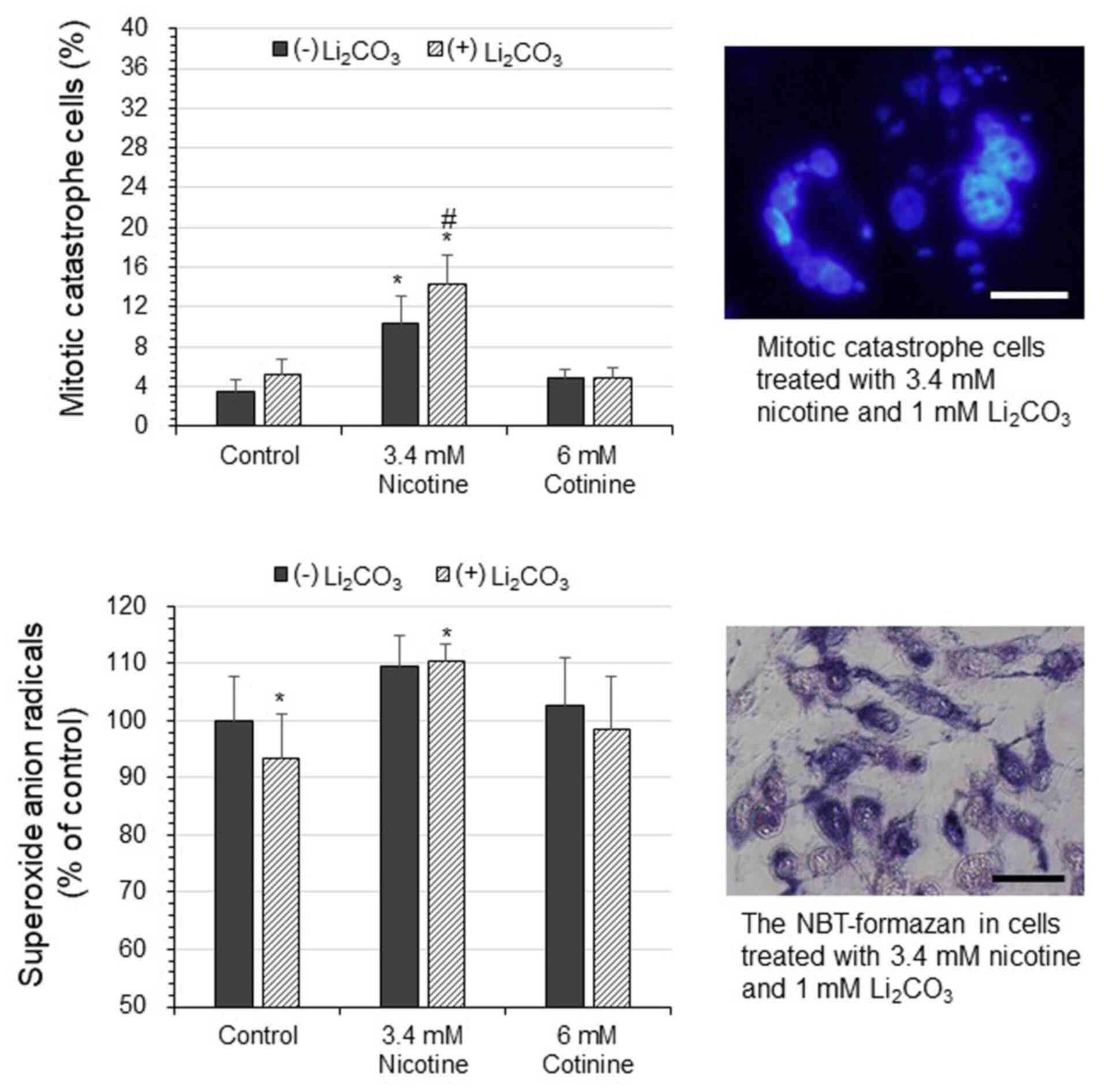 | Figure 4Mitotic catastrophe and intracellular
reactive oxygen species in human glioblastoma U-251MG cells under
Li2CO3 administration. The U-251MG cells were
treated with 3.4 mM nicotine or 6 mM cotinine in combination with
or without 1 mM Li2CO3. After 4 days of
incubation, the cell nuclei were stained with Hoechst 33342. The
mitotic catastrophe cells (%) ratio was calculated on five
fluoresce micrographs of each treatment. The data are presented as
the mean ± SD, n=5; *P<0.05 (vs. control without
Li2CO3), #P<0.05 (vs. without
Li2CO3). The absorbance was measured at an
excitation/emission of 330-385 nm/420 nm (magnification, x200).
Scale bar, 20 µm. In the other experiments, the U-251MG cells were
treated as mentioned above, and after 1 day of incubation,
superoxide anion radicals were measured using NBT assay. The data
are presented as the mean ± SD, n=5; *P<0.05 (vs.
control without Li2CO3). Magnification, x400;
scale bar, 40 µm. Li2CO3, lithium carbonate;
NBT, nitroblue-tetrazolium. |
Intracellular reactive oxygen species
generation
The amount of O2·- (% of
control) was slightly decreased to 93.4% in the cells treated with
1 mM Li2CO3 (Fig.
4). In the cells treated with 3.4 mM nicotine, the amount of
O2·- was 109.4%, and this was increased to
110.4% in the cells treated with 3.4 mM nicotine in combination
with 1 mM Li2CO3. Moreover, the cells treated
with 6 mM cotinine exhibited almost no increase in the amount of
O2·- at 102.8%, which was almost the same as
the control even under the administration of 1 mM
Li2CO3.
Discussion
The present study examined the effects of nicotine
and its metabolite, cotinine, on the proliferation of U-251MG cells
exposed to Li2CO3, and compared these effects
to those of acetylcholine and dopamine. The results indicated that
the proliferation of U-251MG human glioblastoma cells was markedly
decreased at high concentrations of nicotine. Moreover, cell
proliferation was gradually decreased in a concentration-dependent
manner with cotinine. Previous studies have demonstrated the actual
blood nicotine and cotinine levels of smokers. Blood nicotine
concentrations have been shown to vary from 25 to 444 nmol/l, and
the average was found to be 203 nmol/l in 206 females and 126 males
(15). In human serum samples from
smokers, the concentrations of nicotine and cotinine have been
found to be 49-92 and 885-2111 nmol/l, respectively (16). Of note, the concentrations of
nicotine and cotinine used in the present study were significantly
higher than the blood levels of actual smokers, which allowed for
the clarification of the differences in the biological effects of
nicotine and cotinine. Cotinine is the major metabolite of nicotine
(17), with a half-life of 10 to 30
h, while nicotine has a half-life of 0.5 to 2 h (18). Riah et al (19) reported that nicotine was 100-fold
more toxic than cotinine, and 10-fold more rapid than cotinine at
producing respiratory arrest in mice. Similarly, the results of the
present study demonstrated that the cytotoxicity of nicotine
outweighed that of cotinine. There was almost no decrease in cell
proliferation following the exposure of the cells to acetylcholine
(a neurotransmitter that interacts with nicotinic acetylcholine
receptors). The microscopic examination clearly revealed that
acetylcholine and cotinine did not inhibit cell proliferation,
whereas dopamine and nicotine decreased cell proliferation. It was
also observed that cells treated with dopamine or nicotine
underwent mitotic catastrophe. In the present study, following
exposure to 0-1.5 mM Li2CO3, cell
proliferation decreased in the dopamine- and nicotine-treated cells
in a concentration-dependent manner (with the increasing
concentrations of Li2CO3). By contrast, the
cells treated with acetylcholine or cotinine did not exhibit a
decrease in proliferation, even at high concentrations of
Li2CO3. de Sousa et al (20) demonstrated that lithium treatment
increased mitochondrial electron transport complex I activity in
leukocytes of subjects with bipolar disorder. The lithium
carbonate, Li2CO3, has been used as a mood
stabilizer in bipolar disorder at a dose range of 0.6-1.0 mM and
>1.5 mM as lithium in serum, which is close to the addiction
level of Li2CO3 (7). In the present study, following exposure
to 0-1.5 mM Li2CO3, cell proliferation
decreased in the dopamine- and nicotine-treated cells in a
concentration-dependent manner (with the increasing concentrations
of Li2CO3). By contrast, cells treated with
acetylcholine or cotinine did not exhibit a decrease in growth,
even with high concentrations of Li2CO3.
These results suggest that under Li2CO3
administration, the increase in nicotine and dopamine levels due to
smoking may increase health risks through cytotoxicity.
It is known that nicotinic acetylcholine receptors
are activated by acetylcholine or exogenous ligands, such as
nicotine (21). Nicotine
administration increases the firing ratio of substantia nigra pars
compacta neurons and induces striatal dopamine release (2,3). It has
been found that dopamine is an essential endogenous regulator of
malignant glioma growth (22), and
dopamine increases apoptosis by inducing mitochondrial dysfunction
(22). The findings of the present
study are consistent with these findings in that dopamine induced a
decrease in cell proliferation.
The physiological effects of nicotine are mediated
mainly by nicotinic acetylcholine receptors, expressed in human
brain cells and on the surface of the pancreas, colon, bladder,
airway epithelia, etc. (23-25).
The U-251MG cells express an α7 nicotinic acetylcholine receptor
(10). The role of nicotinic
acetylcholine receptors can be assessed by the co-incubation of
nicotine and a non-competitive nicotinic acetylcholine receptor
antagonist (26). In the present
study, mecamylamine, a non-competitive antagonist of nicotinic
acetylcholine receptors, restored cell proliferation which had been
reduced by nicotine, suggesting that a decrease in cell
proliferation was induced by nicotine via nicotinic acetylcholine
receptors.
The ratio of cells undergoing mitotic catastrophe
was significantly increased in the cells treated with nicotine in
combination with Li2CO3. Cotinine slightly
increased the ration of cells undergoing mitotic catastrophe, and
the ratio was almost the same under the administration of
Li2CO3. It has been reported that 10 µM
nicotine-induced mitochondrial fragmentation and cell growth
inhibition in a nicotinic acetylcholine receptor-dependent manner
in NT2/D1 human multipotent embryonic carcinoma cells (27). Pastor et al (28) indicated that
Li2CO3 exerted a cytotoxic effect in Chinese
hamster repair-deficient mutant AA8 CHO cells, mitotic
abnormalities such as multipolar anaphases and lagging chromosomes,
leading to micronuclei. Cell proliferation represents an increase
in cell number resulting from regulated cell growth and cell
division. The results of the present study suggested that nicotine
caused mitotic catastrophe, which was amplified under
Li2CO3 administration, and the suppression of
cell proliferation was attenuated when nicotine was metabolized to
cotinine.
As regards the biological effects of nicotine, there
are reports that nicotine increases oxidative stress, or
conversely, nicotine reduces oxidative stress. In the mitochondrial
and microsomal compartments of the rat brain, nicotine-induced
oxidative stress is accompanied by increasing levels of
thiobarbituric acid-reactive substances and 4-hydroxynonenal
specific glutathione-S-transferase activity (29). However, Cormier et al reported
that nicotine bound to complex I of the mitochondrial respiratory
chain and inhibited NDH-ubiquinone reductase activity, resulting in
decreased levels of O2·- in rat brain
mitochondria (30). In the present
study, the amount of O2·- (% of control) was
slightly decreased in the cells treated with
Li2CO3. In the cells treated with nicotine in
combination with Li2CO3, the amount of
O2·- was increased by 10%. Moreover, cotinine
led to almost no increase in the amount of
O2·-, the levels of which were almost the
same as those of the control, even under the administration of
Li2CO3. These results suggest that nicotine
increases the amount of O2·- regardless of
Li2CO3 administration, and that when nicotine
is metabolized into cotinine, the increase in
O2·- generation is attenuated. The formation
of O2·- was slightly reduced with 1 mM
Li2CO3 in the controls, probably due to the
reducing ability of lithium. The suppression of cell proliferation
by nicotine may involve both nicotine-induced cytotoxicity and the
increase in reactive oxygen species, which is not canceled by
Li2CO3.
Overall, the results of the present study suggested
that nicotine in combination with Li2CO3
suppressed the proliferation of human glioblastoma cells,
accompanied by mitotic catastrophe and O2·-
generation. Increased mitotic catastrophe and
O2·- production suggest that cellular
dysfunction has occurred. The cytotoxicity of cotinine is lower
than that of nicotine. However, nicotine is metabolized into
cotinine by the hepatic cytochrome, P450 2A6 enzyme (31). Smoking is one of the factors that
impair hepatic function (32). Under
lithium carbonate administration, it is advisable to refrain from
smoking as much as possible to avoid the health risks associated
with nicotine.
In conclusion, the present study demonstrated that
nicotine significantly decreased cell proliferation via nicotinic
acetylcholine receptors, and cell proliferation was further
suppressed by the combined administration of
Li2CO3 in U-251MG human glioblastoma cells.
As opposed to nicotine, the suppression of cell proliferation was
less observed in the cells treated with cotinine. As regards
endogenous neurotransmitters involved in nicotinic acetylcholine
receptors, acetylcholine exerted no cytotoxic effects, whereas
dopamine suppressed cell proliferation under
Li2CO3 administration. These findings provide
cellular biological insight into the risks of smoking under
Li2CO3 administration. However, since the
present study was an in vitro study, no conclusions could be
drawn about the association between psychiatric patients and
smoking by extrapolating these data, and thus, further in
vivo studies are warranted.
Acknowledgements
The present study was conducted using devices at the
Center for Molecular Biology and Genetics and the Radioisotope
Experimental Facility of Mie University.
Funding
Funding: The present study supported by a research support grant
from Mie University (Tsu, Japan).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
SK was involved in the conceptualization,
methodology, investigation and writing of the study. SK confirms
the authenticity of all the raw data. The author has read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Glassman AH: Cigarette smoking:
Implications for psychiatric illness. Am J Psychiatry. 150:546–553.
1993.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Balfour DJ and Fagerstrom KO: Pharmacology
of nicotine and its therapeutic use in smoking cessation and
neurodegenerative disorders. Pharmacol Ther. 72:51–81.
1996.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lichtensteiger W, Hefti F, Felix D,
Huwyler T, Melamed E and Schlumpf M: Stimulation of nigrostriatal
dopamine neurones by nicotine. Neuropharmacology. 21:963–968.
1982.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hukkanen J, Jacob P III and Benowitz NL:
Metabolism and disposition kinetics of nicotine. Pharmacol Rev.
57:79–115. 2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Henningfield JE and Zeller M: Nicotine
psychopharmacology: Policy and regulatory. Handb Exp Pharmacol.
511-534:2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Katner SN, Toalston JE, Smoker MP, Rodd
ZA, McBride WJ and Engleman EA: Time-course of extracellular
nicotine and cotinine levels in rat brain following administration
of nicotine: Effects of route and ethanol coadministration.
Psychopharmacology (Berl). 232:551–560. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Young W: Review of lithium effects on
brain and blood. Cell Transplant. 18:951–975. 2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kato S: Effects of platinum-coexisting
dopamine with X-ray irradiation upon human glioblastoma cell
proliferation. Hum Cell. 34:1653–1661. 2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bigner DD, Bigner SH, Ponten J, Westermark
B, Mahaley MS, Ruoslahti E, Herschman H, Eng LF and Wikstrand CJ:
Heterogeneity of Genotypic and phenotypic characteristics of
fifteen permanent cell lines derived from human gliomas. J
Neuropathol Exp Neurol. 40:201–229. 1981.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shulepko MA, Bychkov ML, Lyukmanova EN and
Kirpichnikov MP: Recombinant analogue of the human protein SLURP-1
inhibits the growth of U251 MG and A172 glioma cells. Dokl Biochem
Biophys. 493:211–214. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ishiyama M, Tominaga H, Shiga M, Sasamoto
K, Ohkura Y and Ueno K: A combined assay of cell viability and in
vitro cytotoxicity with a highly water-soluble tetrazolium salt,
neutral red and crystal violet. Biol Pharm Bull. 19:1518–1520.
1996.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Milanovic D, Firat E, Grosu AL and
Niedermann G: Increased radiosensitivity and radiothermosensitivity
of human pancreatic MIA PaCa-2 and U251 glioblastoma cell lines
treated with the novel Hsp90 inhibitor NVP-HSP990. Radiat Oncol.
8(42)2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Baehner RL and Nathan DG: Leukocyte
oxidase: Defective activity in chronic granulomatous disease.
Science. 155:835–836. 1967.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Choi HS, Kim JW, Cha YN and Kim C: A
quantitative nitroblue tetrazolium assay for determining
intracellular superoxide anion production in phagocytic cells. J
Immunoassay Immunochem. 27:31–44. 2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Russell MA, Jarvis M, Iyer R and
Feyerabend C: Relation of nicotine yield of cigarettes to blood
nicotine concentrations in smokers. Br Med J. 280:972–976.
1980.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yasuda M, Ota T, Morikawa A, Mawatari K,
Fukuuchi T, Yamaoka N, Kaneko K and Nakagomi K: Simultaneous
determination of nicotine and cotinine in serum using
high-performance liquid chromatography with fluorometric detection
and postcolumn UV-photoirradiation system. J Chromatogr B Analyt
Technol Biomed Life Sci. 934:41–45. 2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bowman ER, Turnbull LB and Mc KH Jr:
Metabolism of nicotine in the human and excretion of pyridine
compounds by smokers. J Pharmacol Exp Ther. 127:92–95.
1959.PubMed/NCBI
|
|
18
|
Benowitz NL, Kuyt F, Jacob P III, Jones RT
and Osman AL: Cotinine disposition and effects. Clin Pharmacol
Ther. 34:604–611. 1983.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Riah O, Dousset JC, Courriere P, Stigliani
JL, Baziard-Mouysset G and Belahsen Y: Evidence that nicotine
acetylcholine receptors are not the main targets of cotinine
toxicity. Toxicol Lett. 109:21–29. 1999.PubMed/NCBI View Article : Google Scholar
|
|
20
|
de Sousa RT, Streck EL, Zanetti MV,
Ferreira GK, Diniz BS, Brunoni AR, Busatto GF, Gattaz WF and
Machado-Vieira R: Lithium increases leukocyte mitochondrial complex
I activity in bipolar disorder during depressive episodes.
Psychopharmacology (Berl). 232:245–250. 2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Karlin A: Structure of nicotinic
acetylcholine receptors. Curr Opin Neurobiol. 3:299–309.
1993.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lan YL, Wang X, Xing JS, Lou JC, Ma XC and
Zhang B: The potential roles of dopamine in malignant glioma. Acta
Neurol Belg. 117:613–621. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Egleton RD, Brown KC and Dasgupta P:
Nicotinic acetylcholine receptors in cancer: Multiple roles in
proliferation and inhibition of apoptosis. Trends Pharmacol Sci.
29:151–158. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ginzkey C, Stueber T, Friehs G, Koehler C,
Hackenberg S, Richter E, Hagen R and Kleinsasser NH: Analysis of
nicotine-induced DNA damage in cells of the human respiratory
tract. Toxicol Lett. 208:23–29. 2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Keiger CJ, Case LD, Kendal-Reed M, Jones
KR, Drake AF and Walker JC: Nicotinic cholinergic receptor
expression in the human nasal mucosa. Ann Otol Rhinol Laryngol.
112:77–84. 2003.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Papke RL, Sanberg PR and Shytle RD:
Analysis of mecamylamine stereoisomers on human nicotinic receptor
subtypes. J Pharmacol Exp Ther. 297:646–656. 2001.PubMed/NCBI
|
|
27
|
Hirata N, Yamada S, Asanagi M, Sekino Y
and Kanda Y: Nicotine induces mitochondrial fission through
mitofusin degradation in human multipotent embryonic carcinoma
cells. Biochem Biophys Res Commun. 470:300–305. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Pastor N, Kaplan C, Dominguez I, Mateos S
and Cortes F: Cytotoxicity and mitotic alterations induced by
non-genotoxic lithium salts in CHO cells in vitro. Toxicol In
Vitro. 23:432–438. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Bhagwat SV, Vijayasarathy C, Raza H,
Mullick J and Avadhani NG: Preferential effects of nicotine and
4-(N-methyl-N-nitrosamine)-1-(3-pyridyl)-1-butanone on
mitochondrial glutathione S-transferase A4-4 induction and
increased oxidative stress in the rat brain. Biochem Pharmacol.
56:831–839. 1998.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cormier A, Morin C, Zini R, Tillement JP
and Lagrue G: In vitro effects of nicotine on mitochondrial
respiration and superoxide anion generation. Brain Res. 900:72–79.
2001.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Miyazawa M, Kawauchi Y, Okuno Y and Oda Y:
The novel assay method for nicotine metabolism to cotinine using
high performance liquid chromatography. Chem Pharm Bull (Tokyo).
59:295–297. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Rutledge SM and Asgharpour A: Smoking and
liver disease. Gastroenterol Hepatol (NY). 16:617–625.
2020.PubMed/NCBI
|