Introduction
Paraganglioma is a somewhat rare neuroendocrine
tumor that most often arises from the paraganglion tissues of the
adrenal glands. Only in a tenth of cases, extra-adrenal
localization may occur, of which 85% reside in the abdomen, and
only 3% occur in the head and neck region (1). Usually, these tumors are considered
benign; however, in ~19% of instances, malignant transformation may
occur (2). In the adrenal glands and
abdomen, paraganglioma arises from sympathetic paraganglia. On the
other hand, parasympathetic paraganglioma is almost exclusive to
the head and neck area (3).
According to the anatomic site, the tumor is referred to as vagal
paraganglioma, carotid body paraganglioma (CBP) (or a carotid body
tumor), jugular paraganglioma and tympanicum paraganglioma
(1).
One of the chronic inflammation patterns that occurs
when the complete clearance of antigenic stimuli is not achieved by
the cellular immune system, is granulomatous inflammation, such as
that observed in sarcoidosis (4).
Non-necrotizing epithelioid cell granulomas, such as those observed
in sarcoidosis may rarely be observed in patients who do not meet
the criteria of systemic sarcoidosis (5). This phenomenon is known as sarcoid-like
granulomatous inflammation (SLGI), and it can be difficult to
distinguish between this and systemic sarcoidosis (6,7).
It is crucial to distinguish SLGIs from actual
sarcoidosis, as misdiagnosis may lead to severe consequences
(8). Such phenomena have been
observed in association with various neoplasms, including renal
cell carcinoma, breast, parathyroid, germ cell, gastrointestinal
and salivary gland tumors (5-7,9-12).
However, to the best of our knowledge, the available literature to
date lacks any description regarding such a reaction in a
paraganglionic tumor.
The present study describes a case with the first
reported association of a CBP with SLGI in the absence of any
criteria of systemic sarcoidosis or other SGLI-associated
conditions.
Case report
Patient information
A 54-year-old female patient visited Smart Health
Tower (Sulaimani, Iraq) with the presentation of anterior neck
swelling for a period of 27 years without any other associated
symptoms. She was a married housewife and did not have a history of
smoking.
Clinical findings
Upon examination of the swelling and upon
inspection, a grade 3 multinodular goiter (MNG) was suspected as
the patient had a thick neck mass visible from a distance of 5
meters, and by palpation, the enlargement was noted to be firm. In
addition, a right-side lateral neck mass was observed, which was
firm on palpation and movable side to side, but not vertically.
Diagnostic approach
Routine laboratory investigations mostly revealed
normal findings. However, the patient had decreased thyroid
stimulating hormone levels (<0.4 mIU/l), indicating toxic MNG.
An ultrasonography revealed MNG with retrosternal extension and a
solid lesion on the right side of the neck measuring 40x30x22 mm,
which was suggestive of a CBP. Further imaging diagnosis via a
computed tomography scan also revealed the presence of MNG in both
thyroid lobes, with the largest lobes measuring 75x47 and 87x45 mm
on the right and left side of the thyroid, respectively, and a
lateral solid hypervascular mass between the internal and external
carotid arteries (data not shown).
Therapeutic intervention
Under a general anesthesia, in a supine position,
and via a collar incision, a total thyroidectomy was performed for
the patient while preserving the parathyroid gland, and the other
cervical neck mass was also resected via another longitudinal
incision. A histopathological examination was performed for both
masses. Sections from the thyroid gland revealed multiple nodules
of different sizes composed of hyperplastic thyroid follicles
arranged as large follicles, filled with colloid material, and
associated with cystic changes, hemorrhage, fibrosis, cholesterol
cleft formation and calcifications, indicating MNG (data not
shown).
For histopathological analysis, the sections
(4-µm-thick) were paraffin-embedded and fixed with 10% neutral
buffered formalin at room temperature for 24 h. The sections were
then stained with hematoxylin and eosin (Bio Optica Co.) for 1-2
min at room temperature. They were then examined under a light
microscope (Leica Microsystems GmbH). Sections from the right-side
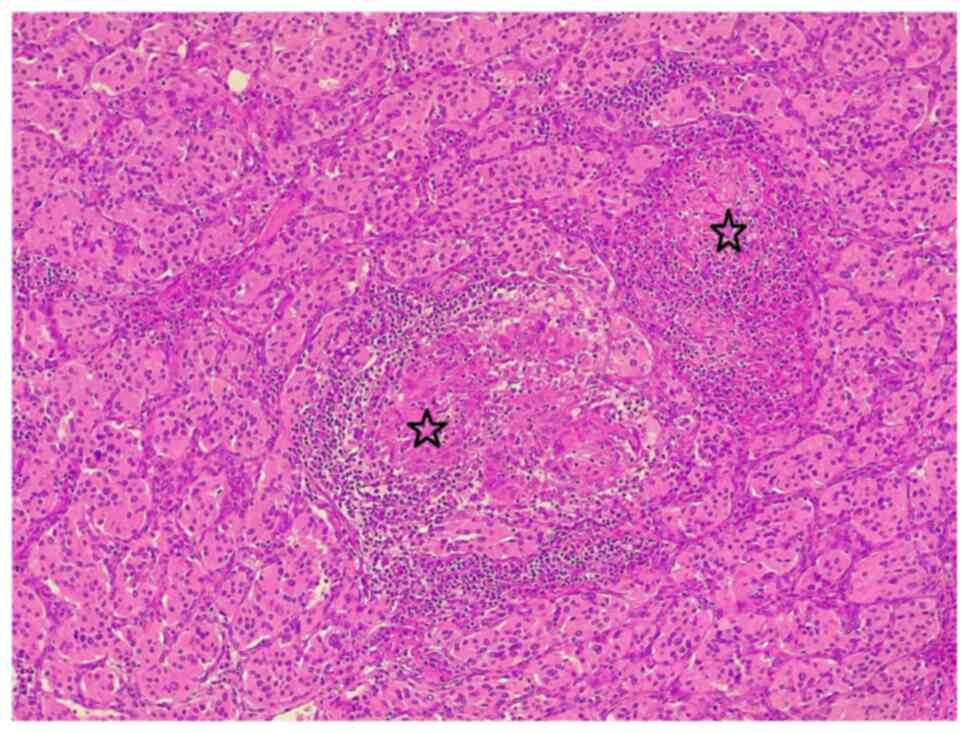
cervical mass revealed a well-demarcated mass composed of nests and
sheets of round-polygonal cells with abundant eosinophilic granular
cytoplasm in a hyalinized vascular stroma, focal areas of atypia
and focal heavy lymphoplasmacytic infiltration, with the presence
of multiple, well-formed, small-medium size, non-necrotizing
epithelioid granulomas associated with multinucleated giant cells,
confirming the diagnosis of CBP with SLGI and focal
lymphoplasmacytic infiltration (Fig.
1).
Follow-up and outcome
The surgery was uneventful, and the patient was
discharged in good health. Following a follow-up period of 6 months
by ultrasound, the patient was in good health without any sign of
recurrence.
Discussion
Non-necrotizing epithelioid cell granuloma,
identical to that observed in systemic sarcoidosis, is a rare, yet
well-reported phenomenon found in association with various primary
neoplasms, infections, drugs (antibiotics, methotrexate, and
non-steroidal anti-inflammatory drugs) and inorganic substances
(11). When this occurs, it is
termed ‘SLGI, sarcoid-like reaction, sarcoid-like granulomatosis,
or sarcoid-like granuloma’ (5-7,9).
The present study describes a previously unpublished association of
CBP and SLGI without any relevant systemic conditions.
Paragangliomas are a family of sympathetic and
parasympathetic paraganglionic tumors. The sympathetic types
originate from within the abdomen and adrenal glands, which are
referred to as abdominal paraganglioma and pheochromocytomas,
respectively. By contrast, the parasympathetic types mainly arise
from the head and neck region (3,13).
The carotid body is a chemoreceptor organ that is
highly vascularized (also reflected in their imaging appearance),
reddish brown in color, and located within the adventitia
posteromedial to the common carotid artery bifurcation. Its role
involves the autonomic regulation of temperature, cardiovascular,
respiratory systems, and acute adaptation to fluctuating
concentrations of oxygen, carbon dioxide and pH. It consists of
sustentacular cells and chief cells derived from the neuroectoderm
and neural crest cells, respectively. Tumors originating from the
chief paraganglia cells are termed CBP (3). Even though it is the most common type
of head and neck paraganglioma, it is still an overall rare
neoplasm that requires a thorough examination for appropriate
diagnosis and management. The tumor can be familial or sporadic,
with the latter being more common (14).
CBPs have been reported to be more prevalent among
females and are most often observed in the third to the sixth
decade of life. The neoplasm usually presents as a non-tender,
slow-growing mass, and rarely does the tumor demonstrate a
bruit/thrill or transmit a carotid pulse. The tumor may encase the
external and internal carotid arteries as it increases in size,
although it does not narrow them. Even though CBP is often
asymptomatic in the initial phase, due to its proximity to the
adjoining vessels and nerves, symptoms may manifest with an
increase in size, such as a hoarseness of voice, odynophagia,
dysphagia and other cranial nerve deficits. Hypertension, sweating
and headaches may also be observed in some patients as a result of
the secretion of vasoactive catecholamines by the CBP (3,15). These
descriptions are in accordance with the case described herein along
with our case, as the patient was a 54-year-old asymptomatic
female. In patients with multiple occurrences, an age <45 years,
bilateralism, a previous history or a current history of CBP, and a
positive family history, performing genetic testing and biochemical
phenotype assay are recommended to aid in the early detection of
complications and the initiation of proper intervention therapies.
Conducting appropriate family screening may also aid in improving
the overall prognosis (14).
Upon a histological examination, CBPs have a
characteristic growth pattern known as ‘zellballen’, an organoid or
well-developed nested growth pattern of the tumor cells with a
stromal component between fragile fibrovascular tissue and
supporting sustentacular cells along the edges of the zellballen or
cell nests. The tumor cells are mostly chief cells with dispersed
chromatin, hyperchromatic, round nuclei and an abundant granular
cytoplasm that may be either basophilic to eosinophilic in color
(12,15). Although their vascularity makes the
condition challenging, surgical removal is the recommended
management approach associated with a good prognosis. In cases of
malignancy and metastatic disease, radiation and chemotherapy do
not appear to be greatly beneficial (15).
A number of granulomatous conditions are able to
mimic sarcoidosis, clinically and histologically, including
sarcoid-like granulomatous reactions, infectious granulomatous
diseases, granulomatous drug reactions, neoplastic disorders,
immunodeficiencies and systemic disorders with granulomatous
characteristics (Rosai-Dorfman disease and Blau syndrome) (8,16).
Distinguishing SLGIs from actual sarcoidosis requires a careful
review of a patient's complaints, demographics, medical history,
clinical examination, laboratory findings, imaging and histological
features. Some clinical characteristics may immediately eliminate
the diagnosis of sarcoidosis due to atypical characteristics, such
as an age <25 years or >45 years, a high fever, acute or
subacute dyspnea and hemoptysis (16).
The first case of SLGI was reported by Wolbach in
1911(17). Since then, this reaction
has been reported in association with various neoplasms. Some
studies have aimed to determine the frequency of these reactions in
different neoplasms, with the reported frequency ranging from 4% in
carcinomas to 20% in lymphomas (5,12).
Sarcoid-like reactions often occur in organs that dendritic cells
can reach, although rarely at the tumor site (6).
In patients with no history of sarcoid disease,
non-necrotizing epithelioid cell granulomas may rarely occur within
tumors. The pathogenesis of SLGIs remains to be fully established.
It has been reported that it may occur as the result of soluble
tumor antigens or cancer-related antigenic factors being shed into
the blood, leading to immunological hypersensitivity and
granulomatous inflammation (7).
Histologically, SLGIs are similar to sarcoidosis
granulomas. They are comprised of well-defined non-caseating
granulomas with the focal accumulation of multinucleated giant
cells and epithelioid cells (5,9).
Usually, no necrosis is observed, although a few patients may be
present with fibrinoid necrosis (12). A similar observation was made in the
case described herein.
The presence of SLGI is an indicator of an
immunological response of macrophages by activated T-lymphocytes to
a neoplasm. Even though the presence of T-lymphocytes should
translate to a protective role and better prognosis, currently, the
prognostic value of SLGIs is not yet well understood (9). It has been reported to be associated
with a good prognosis in various lesions, including gastric cancers
and Hodgkin's disease (12).
However, some studies have reported no prognostic significance in
lung cancers (18,19). In a previous study, in a case of
renal cell carcinoma and SLGI, the patient succumbed due to
metastasis 6 months following ta nephrectomy; however, the authors
argued that the patient's death was more likely influenced by
sarcomatoid features (20). Although
the case described in the present study is of interest, the
authors' were not able to evaluate prognostic significance due to a
limited follow-up period and the fact that the case was the first
of its kind, at least to the best of our knowledge. All the
referenced studies in this report were evaluated for their
credibility before being cited (21).
In conclusion, SLGIs accompanying a paraganglioma is
an extremely rare event that has not previously been reported, at
least to the best of our knowledge. The case described in the
present study is the first to be reported; there was no evidence of
systemic sarcoidosis or tuberculosis. Due to its rarity, it is
difficult to conclude if it confers a better prognosis or not.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AMS and ASM were the surgeons who performed the
operation. SHT was the radiologist who performed the assessment of
the patient's condition. AMA was the pathologist examining the
specimen, and was also a major contributor to the conception of the
study. BJM were FA were major contributors to the conception of the
study, as well as in the literature search for related studies.
FHK, HMR, SHM and DAH were involved in the literature review, in
the writing of the manuscript, in critical revision and in data
organization interpretation. AMS and FHK confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The patient provided written informed consent for
her data to be included in the present case report.
Patient consent for publication
The patient provided written informed consent for
her data and any related images to be published in the present case
report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kakamad FH, Mohammed SH, Fatah ML, Hattam
AS, Qaradakhy AJ and Abdullah AM: Vagal paraganglioma extending to
the brain: A case report with literature review. IJS Short Reports.
7(e52)2022.
|
|
2
|
Ahmed Y, Arif A, Bhatti AM, Nasir SA,
Nofal S, Hamza A and Mughal UJ: Vagal paraganglioma: A rare finding
in a 31-year-old male. Cureus. 13:1–6. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hosalkar RM, Khivasara JS and Swain N:
Carotid body paraganglioma. Ann Maxillofac Surg. 9:423–428.
2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Burhan W, Al Rowaie Z, Rajih E and Akhtar
M: Sarcoid-like granulomatous reaction in renal cell carcinoma:
Report of a case with review of the published reports. Ann Saudi
Med. 33:614–618. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ouellet S, Albadine R and Sabbagh R: Renal
cell carcinoma associated with peritumoral sarcoid-like reaction
without intratumoral granuloma. Diagn Pathol. 7(28)2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Iftikhar A, Cheema MA, Ramachandran P and
Sahni S: Sarcoid-like reaction associated with renal cell
carcinoma-a case report. Respir Med Case Rep. 27:1–4.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Alattia L, Ghildyal A and Gu X: A case
report of sarcoid like-granuloma in renal cell carcinoma. Am J Clin
Pathol. 150(11)2018.
|
|
8
|
Judson MA: Granulomatous sarcoidosis
mimics. Front Med (Lausanne). 8(680989)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Arora K, Divatia MK, Truong L, Shen SS,
Ayala AG and Ro JY: Sarcoid-like granulomas in renal cell
carcinoma: The Houston methodist hospital experience. Ann Diagn
Pathol. 31:62–65. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Mostyka M, Jessurun J and Matrai C:
Sarcoid-like granulomatosis in a patient with breast cancer
mimicking refractory metastatic disease. Int J Surg Pathol.
28:668–671. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Stojsic Z, Brasanac D, Stojiljkovic M,
Babic D, Randjelovic T and Terzic T: Composite carcinoma of the
stomach associated with sarcoid-like granulomas. Pathol Oncol Res.
15:503–510. 2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kandemir NO, Yurdakan G, Bektas S and
Tekin NS: Classic Kaposi sarcoma with sarcoid-like granulomas: A
case report and literature review. Exp Mol Pathol. 87:89–93.
2009.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Mishra P, Padhi S and Behera G: Thyroid
paraganglioma: A case-based systematic review of literature. J
Cancer Res Ther. 16:11–21. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Megías MC, Puyol DR, Rodríguez LF,
Martinez GL and Miguel PM: Pheochromocytoma-paraganglioma:
Biochemical and genetic diagnosis. Nefrología. 36:481–488.
2016.PubMed/NCBI View Article : Google Scholar : (In English,
Spanish).
|
|
15
|
Wieneke JA and Smith A: Paraganglioma:
Carotid body tumor. Head Neck Pathol. 3:303–306. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
El Jammal T, Jamilloux Y, Gerfaud-Valentin
M, Richard-Colmant G, Weber E, Bert A, Androdias G and Sève P:
Challenging mimickers in the diagnosis of sarcoidosis: A case
study. Diagnostics (Basel). 11(1240)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wolbach SB: A new type of cell inclusion,
not parasitic, associated with disseminated granulomatous lesions.
J Med Res. 24:243–258. 1911.PubMed/NCBI
|
|
18
|
Kamiyoshihara MI, Hirai TO, Kawashima O,
Ishikawa SU and Morishita YA: Sarcoid reactions in primary
pulmonary carcinoma: Report of seven cases. Oncol Rep. 5:177–257.
1998.PubMed/NCBI
|
|
19
|
Tomimaru Y, Higashiyama M, Okami J, Oda K,
Takami K, Kodama K and Tsukamoto Y: Surgical results of lung cancer
with sarcoid reaction in regional lymph nodes. Jpn J Clin Oncol.
37:90–95. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Piscioli I, Donato S, Morelli L, Del Nonno
F and Licci S: Renal cell carcinoma with sarcomatoid features and
peritumoral sarcoid-like granulomatous reaction: Report of a case
and review of the literature. Int J Surg Pathol. 16:345–348.
2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Muhialdeen AS, Ahmed JO, Baba HO, Abdullah
IY, Hassan HA, Najar KA, Mikael TM, Mustafa MQ, Mohammed DA, Omer
DA, et al: Kscien's list; A new strategy to discourage predatory
journals and publishers (second version). Barw Med J. 1:1–3.
2023.
|